

Abstract
First principles of population genetics are used to obtain formulae relating the non-synonymous to synonymous substitution rate ratio to the selection coefficients acting at codon sites in protein-coding genes. Two theoretical cases are discussed and two examples from real data (a chloroplast gene and a virus polymerase) are given. The formulae give much insight into the dynamics of non-synonymous substitutions and may inform the development of methods to detect adaptive evolution.
Keywords: selection, non-synonymous/synonymous ratio, substitution, influenza, chloroplast, adaptive evolution
1. Introduction
Halpern & Bruno [1] devised a model to study the divergence of protein-coding genes based on the Fisher–Wright model of mutation, selection and random genetic drift [2,3]. In the model, each particular codon site in the gene is assigned its own set of amino acid fitnesses, and then the Fisher–Wright model is used to work out the evolutionary rate of the site. The model has seen a resurgence in recent years, and variations of it have been used, for example, to study performance of phylogenetic inference methods [4,5], to study codon usage [6] and to estimate the distribution of selection coefficients in protein-coding genes [7,8]. Perhaps surprisingly, the model has not been used to study the dynamics of the non-synonymous to synonymous rate ratio (also known as ω = dN/dS) of protein-coding genes and its significance in the study of adaptive molecular evolution.
The purpose of this note is to propose a way to define and calculate an equivalent of the classical concept of the non-synonymous to synonymous rate ratio, in the context of the mutation–selection model of Halpern & Bruno [1]. It is hoped that by using first principles of population genetics, we can obtain an expression of ω as a function of the selection coefficients acting at codon sites in the protein-coding gene. This should provide much insight into the evolutionary dynamics of codon sites and it should be of advantage in the building of statistical models to detect adaptive evolution in protein-coding genes.
2. The site-wise mutation–selection model
Consider the evolution of a codon site k in a protein-coding gene in a population with N haploid genomes. Assume the site is currently fixed for codon I (i.e. all N alleles carry I at site k). In the mutation–selection framework [1,8], the substitution rate (the rate at which novel mutant codons J appear and eventually become fixed in the population) is
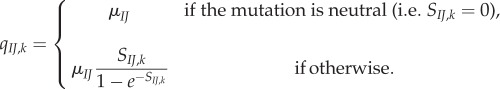 |
2.1 |
Here μIJ is the neutral mutation rate (per generation) from I to J, and SIJ,k = FJ,k – FI,k is the selection coefficient in favour of codon J and FJ,k = 2NfJ,k is the scaled Malthusian fitness of J. Natural selection affects the relative substitution rate. When the mutation is advantageous (SIJ,k > 0), the substitution rate is higher than the neutral rate (qIJ,k > μIJ), but if the mutation is deleterious (SIJ,k < 0), then the substitution rate is reduced (qIJ,k < μIJ). Here, we assume that synonymous substitutions are neutral (SIJ,k = 0), and thus, evolution at site k is determined by 20 amino acid fitnesses. The μIJ can be constructed from standard DNA substitution models (for example, if I = TTT and J = TTC, then 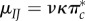 under the HKY substitution model, see [8] for details).
under the HKY substitution model, see [8] for details).
Equation (2.1) describes codon substitution in populations as a continuous-time Markov process. This is sensible if the per generation mutation rate is small compared with the population size (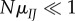 ), so that there is little polymorphism in the population, and at most, two alleles segregate at a site at a time. The proportion of time, πI,k, that site k spends fixed for I (i.e. the stationary frequency of I) is
), so that there is little polymorphism in the population, and at most, two alleles segregate at a site at a time. The proportion of time, πI,k, that site k spends fixed for I (i.e. the stationary frequency of I) is
[8], where 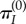 is the frequency for a neutrally evolving sequence (i.e. a pseudo-gene). Thus, the substitution rate at k, averaged over time, is
is the frequency for a neutrally evolving sequence (i.e. a pseudo-gene). Thus, the substitution rate at k, averaged over time, is
where the sum is over all codon pairs I ≠ J. This rate can be partitioned into its non-synonymous and synonymous component rates, ρk = ρN,k + ρS,k, where
and where the indicator function IN = 1 if the substitution is non-synonymous and = 0 if otherwise. Note that the synonymous rate ρS,k varies among sites (for example, if a site is conserved for methionine, then the synonymous rate is zero). For a neutrally evolving sequence, the rates are given by
Note that equation (2.1) gives the instantaneous substitution rate, that is, the rate conditioned on site k being fixed for I at the present time. On the other hand, ρk is the rate at equilibrium, averaged over all codons and weighted by their stationary frequencies.
3. The relative non-synonymous substitution rate
The absolute non-synonymous to synonymous substitution rate ratio at site k is ρN,k/ρS,k. However, because synonymous rates vary over sites, we need to normalize the ratio by the site's synonymous rate ratio, 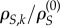 , and then normalize by
, and then normalize by 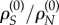 (to correct for the different proportions of synonymous and non-synonymous substitutions at neutrality). This leads to the following definition:
(to correct for the different proportions of synonymous and non-synonymous substitutions at neutrality). This leads to the following definition:
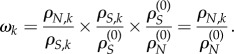 |
3.1 |
Alternatively, we can define ωk as the relative non-synonymous rate ωk = cρN,k where constant c is set so that the ratio is one for neutrally evolving sequences, that is, under the constraint 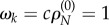 . The obvious solution is
. The obvious solution is 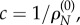 leading to the same definition as above. Note that c has the desirable property of being constant over sites. The reader should not be surprised that the synonymous rate drops out from equation (3.1). When doing statistical inference, synonymous substitutions have information about the neutral mutation rates, and thus inform the value of
leading to the same definition as above. Note that c has the desirable property of being constant over sites. The reader should not be surprised that the synonymous rate drops out from equation (3.1). When doing statistical inference, synonymous substitutions have information about the neutral mutation rates, and thus inform the value of 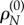 . Similarly, the relative synonymous rate at site k is
. Similarly, the relative synonymous rate at site k is
| 3.2 |
Figure 1a shows an example for the rbcL gene of flowering plants. Fitness values were estimated under the Halpern–Bruno model by Tamuri et al. [9], and we use their values to calculate ωk and γk here. The average rates across sites are 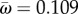 and
and 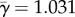 . Note that for many sites, synonymous rates are faster than for a neutrally evolving sequence (i.e. γk > 1). This is owing to the quirky nature of the genetic code coupled with the mutational biases (
. Note that for many sites, synonymous rates are faster than for a neutrally evolving sequence (i.e. γk > 1). This is owing to the quirky nature of the genetic code coupled with the mutational biases (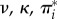 ).
).
Figure 1.

The relative non-synonymous (ωk) and synonymous (γk) substitution rates. (a) Rates for the rbcL chloroplast gene of monocots (flowering plants). (b) Rates for the pb2 gene of influenza A. In (a,b), the fitnesses at each site (FIJ,k) and the mutation parameters (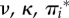 ) were estimated under the Halpern–Bruno model by penalized likelihood (penalty α = 0.01) and are from [9]. Then, equations (3.1) and (3.2) are used to calculate ωk and γk. In (b), 25 adaptive sites (red) were identified where fitnesses are different between viruses evolving in human versus avian (the natural reservoir) hosts [11]. Fitnesses for these sites under each host were estimated without penalty and are from [8]. Then, equation (4.1) is used to calculate
) were estimated under the Halpern–Bruno model by penalized likelihood (penalty α = 0.01) and are from [9]. Then, equations (3.1) and (3.2) are used to calculate ωk and γk. In (b), 25 adaptive sites (red) were identified where fitnesses are different between viruses evolving in human versus avian (the natural reservoir) hosts [11]. Fitnesses for these sites under each host were estimated without penalty and are from [8]. Then, equation (4.1) is used to calculate 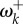 at the host shift. The range of
at the host shift. The range of 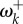 is 0.231–7.64 (the largest values are truncated in the figure).
is 0.231–7.64 (the largest values are truncated in the figure).
4. The non-synonymous rate during adaptive evolution
When the fitnesses of amino acids are constant through time, sites will spend most of the time fixed for the optimal amino acid. Occasionally, suboptimal amino acids may become fixed, and then substituted after a short period of evolutionary time. This means that the non-synonymous rate at sites is reduced compared with the rate for neutrally evolving sequences (i.e. ωk < 1). However, when fitnesses at sites vary over time (for example, after an environment shift or under intense frequency-dependent selection [10]), the non-synonymous rate may be accelerated compared with the rate for neutrally evolving sequences (ωk > 1). We now study the case where fitnesses change as an adaptation to a novel environment.
Consider a site k where the fitness of I is 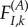 in environment A. The stationary frequencies and instantaneous substitution rates are
in environment A. The stationary frequencies and instantaneous substitution rates are 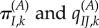 . Now, imagine that the environment shifts (for example, a population of mammals living in a suddenly colder climate, or a virus colonizing a new host, where the intracellular environment in the new host is different from the reservoir host). The fitness of I in the new environment B is now
. Now, imagine that the environment shifts (for example, a population of mammals living in a suddenly colder climate, or a virus colonizing a new host, where the intracellular environment in the new host is different from the reservoir host). The fitness of I in the new environment B is now 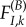 . The probability that the site is currently fixed for I at the moment of the environment shift is
. The probability that the site is currently fixed for I at the moment of the environment shift is 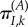 , but the substitution rate is now that of the new environment
, but the substitution rate is now that of the new environment 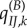 . Thus, the expected absolute and relative non-synonymous rates at the environment shift are
. Thus, the expected absolute and relative non-synonymous rates at the environment shift are
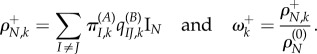 |
4.1 |
If the shift in fitness values is large, then the rate will be much accelerated (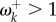 ). This occurs because the site is likely to find itself fixed for a suboptimal amino acid in the new environment, and novel mutations to optimal amino acids will become fixed quickly. However, if the fitness shift is moderate, the rate may still be lower than the neutral rate (
). This occurs because the site is likely to find itself fixed for a suboptimal amino acid in the new environment, and novel mutations to optimal amino acids will become fixed quickly. However, if the fitness shift is moderate, the rate may still be lower than the neutral rate (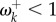 ).
).
Figure 1b shows an example for the pb2 gene of the influenza virus. Fitness values were estimated under the Halpern–Bruno model by Tamuri et al. [9]. A subset of 25 adaptive sites (where fitnesses are different for viruses evolving in human versus avian hosts [8,11]) were identified by Tamuri et al. [11], and their fitnesses estimated by Tamuri et al. [8]. We use the estimates to calculate ωk, γk and 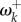 here. The classical lineage of human influenza probably originated from a host shift from an avian to a mammal reservoir in the early-twentieth century [12]. We calculate
here. The classical lineage of human influenza probably originated from a host shift from an avian to a mammal reservoir in the early-twentieth century [12]. We calculate 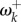 at the putative host shift. The average rate at adaptive sites is
at the putative host shift. The average rate at adaptive sites is 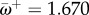 (across all sites
(across all sites 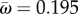 and
and 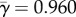 ). Note that for 16 sites for which fitnesses are different between hosts, we find that
). Note that for 16 sites for which fitnesses are different between hosts, we find that 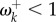 . This indicates that the criterion ωk > 1 to detect adaptive evolution is conservative in this case.
. This indicates that the criterion ωk > 1 to detect adaptive evolution is conservative in this case.
The probability that the site is fixed for I, time t after the environment shift is
| 4.2 |
where 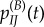 are the transition probabilities obtained using standard Markov theory, i.e. by calculating
are the transition probabilities obtained using standard Markov theory, i.e. by calculating 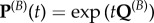 . Thus, the absolute and relative non-synonymous rates, time t after the shift, are
. Thus, the absolute and relative non-synonymous rates, time t after the shift, are
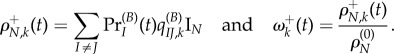 |
The transition probabilities in equation (4.2) are exponential decay functions of time, and so 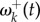 is also an exponential decay. Initially, the value of
is also an exponential decay. Initially, the value of 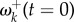 will be high, and as the time goes to infinity,
will be high, and as the time goes to infinity, 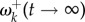 will approach the stationary value given by equation (3.1). In other words, soon after an environment shift, a burst of adaptive substitutions will occur at sites where fitnesses have changed, and substitutions will accumulate until the protein-coding gene reaches a state of adaptive equilibrium. For example, figure 2a shows the decay of
will approach the stationary value given by equation (3.1). In other words, soon after an environment shift, a burst of adaptive substitutions will occur at sites where fitnesses have changed, and substitutions will accumulate until the protein-coding gene reaches a state of adaptive equilibrium. For example, figure 2a shows the decay of 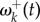 for the 25 adaptive sites in the pb2 gene after a host shift.
for the 25 adaptive sites in the pb2 gene after a host shift.
Figure 2.
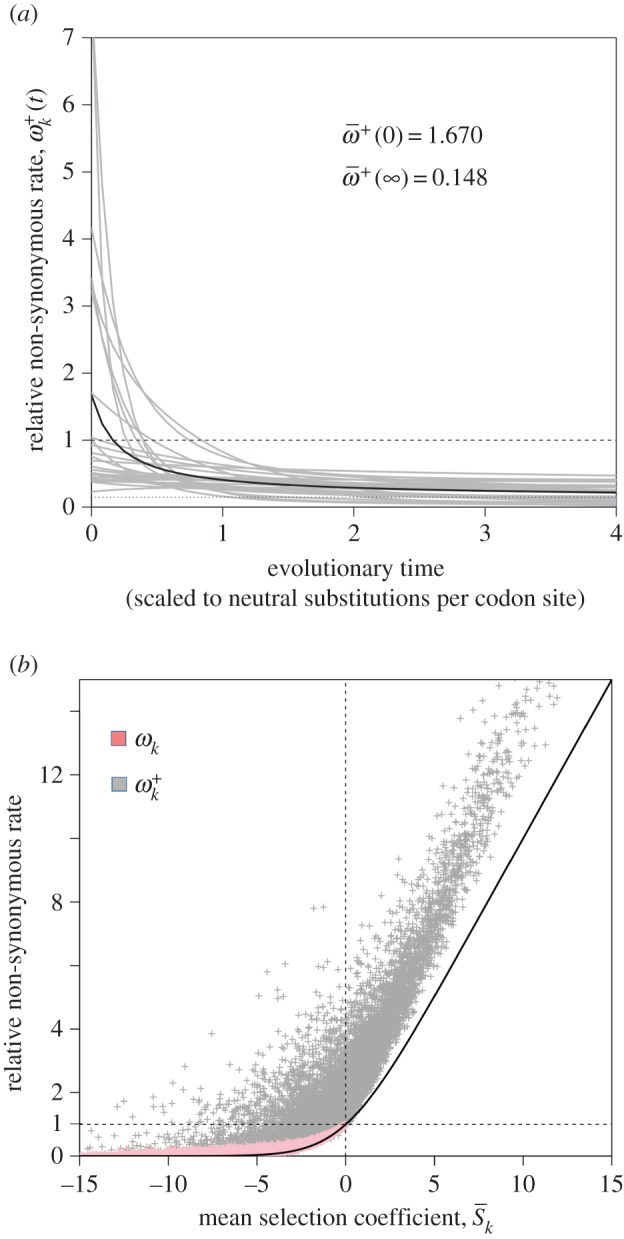
(a) Decay in the relative non-synonymous rate after a host shift for 25 adaptive sites (grey lines) in the pb2 gene of influenza. The solid line is the mean across the 25 sites, 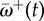 . As time passes,
. As time passes, 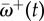 approaches the long-term mean
approaches the long-term mean 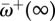 (dotted line). (b) The relative non-synonymous rate as a function of mean selection coefficient at sites. Pink dots: fitness values for 10 000 sites were sampled from normal distributions with mean 0 and σ = 0, … , 10. Then, equations (3.1) and (5.1) were used to calculate ωk and
(dotted line). (b) The relative non-synonymous rate as a function of mean selection coefficient at sites. Pink dots: fitness values for 10 000 sites were sampled from normal distributions with mean 0 and σ = 0, … , 10. Then, equations (3.1) and (5.1) were used to calculate ωk and 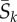 . Grey dots: another set of 10 000 fitness values were sampled as above, then equations (4.2) and (5.1) were used to calculate
. Grey dots: another set of 10 000 fitness values were sampled as above, then equations (4.2) and (5.1) were used to calculate 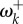 and
and 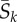 under the environment shift model. Solid line: S/(1 − exp(−S)).
under the environment shift model. Solid line: S/(1 − exp(−S)).
5. Conclusion
Previous authors have shown that the relationship between the non-synonymous rate and the selection coefficient is approximately ω = S/(1 − exp(−S)) [13,14], but the approximation relies either on the infinite-sites model or assumes that all mutant amino acids have the same fitness. Equations (3.1) and (4.1) provide more realistic approximations but are hard to visualize. Consider a site fixed for I. The probability that the next mutation will be J is 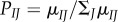 for I ≠ J. Over time, the proportion of I to J mutations at the site will be πI,kPIJ. Thus, the average selection coefficient on mutations at site k is
for I ≠ J. Over time, the proportion of I to J mutations at the site will be πI,kPIJ. Thus, the average selection coefficient on mutations at site k is
| 5.1 |
Figure 2b shows ωk as a function of 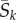 for simulated sites when fitnesses are constant or when they shift with the environment. Note that the approximation ω = S/(1 − exp(−S)) provides a reasonable lower bound on ωk. In general,
for simulated sites when fitnesses are constant or when they shift with the environment. Note that the approximation ω = S/(1 − exp(−S)) provides a reasonable lower bound on ωk. In general, 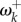 increases with
increases with 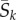 , but the relationship is not as simple as in the previous approximations [13,14].
, but the relationship is not as simple as in the previous approximations [13,14].
In the site-wise mutation–selection model, one calculates the selection coefficients first, and hence one may know whether a site has been under positive selection without calculating ωk [8]. However, the model is over-parametrized, computationally expensive, and fitnesses can be well estimated only in large datasets [9]. Instead, the model should be of advantage in evolutionary reasoning and in model building. For example, the behaviour of ωk under more complex models (such as frequency-dependent selection [10], adaptation to gradual environment changes [10] or selection on codon usage [6]) can also be studied under the site-wise mutation–selection framework. This will be a worthwhile effort as it will shed light on our ability to detect adaptive evolution in molecular sequences.
Acknowledgements
I thank Ziheng Yang, Richard Goldstein and Asif Tamuri for valuable comments.
Data accessibility
The data accompanying with this study are available at Dryad doi:10.5061/dryad.3r3q4.
Funding statement
M.d.R. is supported by BBSRC (UK) grant no. BB/J009709/1 awarded to Ziheng Yang.
Conflicts of interest
I have no competing interests.
References
- 1.Halpern AL, Bruno WJ. 1998. Evolutionary distances for protein-coding sequences: modeling site-specific residue frequencies. Mol. Biol. Evol. 15, 910–917. ( 10.1093/oxfordjournals.molbev.a025995) [DOI] [PubMed] [Google Scholar]
- 2.Fisher R. 1930. The genetic theory of natural selection. Oxford, UK: Clarendon Press. [Google Scholar]
- 3.Wright S. 1931. Evolution in Mendelian populations. Genetics 16, 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holder MT, Zwickl DJ, Dessimoz C. 2008. Evaluating the robustness of phylogenetic methods to among-site variability in substitution processes. Phil. Trans. R. Soc. B 363, 4013–4021. ( 10.1098/rstb.2008.0162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spielman SJ, Wilke CO. 2015. The relationship between dN/dS and scaled selection coefficients. Mol. Biol. Evol. 32 ( 10.1093/molbev/msv003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Z, Nielsen R. 2008. Mutation–selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol. Biol. Evol. 25, 568–579. ( 10.1093/molbev/msm284) [DOI] [PubMed] [Google Scholar]
- 7.Rodrigue N, Philippe H, Lartillot N. 2010. Mutation–selection models of coding sequence evolution with site-heterogeneous amino acid fitness profiles. Proc. Natl Acad. Sci. USA 107, 4629–4634. ( 10.1073/pnas.0910915107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamuri AU, dos Reis M, Goldstein RA. 2012. Estimating the distribution of selection coefficients from phylogenetic data using sitewise mutation–selection models. Genetics 190, 1101–1115. ( 10.1534/genetics.111.136432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamuri AU, Goldman N, dos Reis M. 2014. A penalized-likelihood method to estimate the distribution of selection coefficients from phylogenetic data. Genetics 197, 257–271. ( 10.1534/genetics.114.162263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.dos Reis M. 2013. Population genetics and substitution models of adaptive evolution. (http://arxiv.org/abs/1311.6682).
- 11.Tamuri AU, dos Reis M, Hay AJ, Goldstein RA. 2009. Identifying changes in selective constraints: host shifts in influenza. PLoS Comput. Biol. 5, e1000564 ( 10.1371/journal.pcbi.1000564) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.dos Reis M, Hay AJ, Goldstein RA. 2009. Using non-homogeneous models of nucleotide substitution to identify host shift events: application to the origin of the 1918 ‘Spanish’ influenza pandemic virus. J. Mol. Evol. 69, 333–345. ( 10.1007/s00239-009-9282-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen R, Yang Z. 2003. Estimating the distribution of selection coefficients from phylogenetic data with applications to mitochondrial and viral DNA. Mol. Biol. Evol. 20, 1231–1239. ( 10.1093/molbev/msg147) [DOI] [PubMed] [Google Scholar]
- 14.Bustamante CD. 2005. Population genetics of molecular evolution. In Statistical methods in molecular evolution (ed. Nielsen R.), pp. 63–99. New York, NY: Springer. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data accompanying with this study are available at Dryad doi:10.5061/dryad.3r3q4.