

Abstract
The depth-differentiation hypothesis proposes that the bathyal region is a source of genetic diversity and an area where there is a high rate of species formation. Genetic differentiation should thus occur over relatively small vertical distances, particularly along the upper continental slope (200–1000 m) where oceanography varies greatly over small differences in depth. To test whether genetic differentiation within deepwater octocorals is greater over vertical rather than geographical distances, Callogorgia delta was targeted. This species commonly occurs throughout the northern Gulf of Mexico at depths ranging from 400 to 900 m. We found significant genetic differentiation (FST = 0.042) across seven sites spanning 400 km of distance and 400 m of depth. A pattern of isolation by depth emerged, but geographical distance between sites may further limit gene flow. Water mass boundaries may serve to isolate populations across depth; however, adaptive divergence with depth is also a possible scenario. Microsatellite markers also revealed significant genetic differentiation (FST = 0.434) between C. delta and a closely related species, Callogorgia americana, demonstrating the utility of microsatellites in species delimitation of octocorals. Results provided support for the depth-differentiation hypothesis, strengthening the notion that factors covarying with depth serve as isolation mechanisms in deep-sea populations.
Keywords: deep sea, population genetics, connectivity, adaptive divergence, octocoral, Gulf of Mexico
1. Introduction
The depth-differentiation hypothesis proposes that the majority of genetic differentiation and biodiversity in the deep sea is generated across the relatively narrow continental slope [1]. Not only do water mass characteristics, pressure and food supply change rapidly with increasing depth across the continental slope, but this region can also exhibit topographic complexity (e.g. submarine canyons, bioherm formation, authigenic carbonates), experience rapid fluctuations in current patterns, be subjected to low levels of dissolved oxygen, and contain high levels of hydrocarbon seepage [2]. Strong environmental gradients with depth coupled with high habitat heterogeneity [3,4] in the bathyal region can create different selective regimes, thus potentially promoting adaptive divergence in deep-sea species.
Several studies have demonstrated high levels of genetic differentiation across relatively small depth ranges in the deep sea, particularly across depth horizons of 1000 and 3000 m [5–9]. Population divergence over relatively narrow bathymetric gradients may be due to either historical patterns in colonization or the environmental changes (e.g. pressure, temperature and dissolved oxygen) that occur with changes in depth [6,7,9]. Few studies, however, have addressed the depth-differentiation hypothesis on species inhabiting the upper continental slope (200–1000 m), yet this region is where environmental changes occur most rapidly. Furthermore, this bathymetric zone may be a source of genetic diversity to down slope areas [1,10] leading to the formation of deeper-occurring populations and species. Thus, it could be expected that isolation by depth and increased genetic diversity would occur over relatively small vertical distances. In contrast to population differentiation with depth, numerous examples have demonstrated high connectivity across expansive geographical distances (hundreds to thousands of kilometres) in the deep sea [11,12].
Deep-sea corals (including scleractinians, antipatharians and octocorals) occur along the continental slope in a diversity of marine habitats worldwide. These foundation species create habitat for numerous other species and are long-lived and slow-growing [13,14]. Most species reproduce sexually either by broadcast spawning or brooding [15]. Brooding of larvae can occur internally within the polyps or externally on the surface of the colonies [16,17]. Deep-sea corals are susceptible to anthropogenic disturbances, including fishing, hydrocarbon extraction and mining [18,19]. Their prevalence and importance in creating habitat on the continental slope necessitate a better understanding of the level of connectivity among populations occurring across depth and geographical boundaries.
Previous studies have examined population differentiation in mesophotic corals (50–200 m) on the continental shelf [20] and in deep-sea (more than 200 m) corals on the continental slope [21] and seamounts [22]. These studies have indicated different degrees of population connectivity, which can be related to differences in both the reproductive biology and the specific region and depth of sampling of the coral species. Populations of the scleractinian coral Lophelia pertusa, a broadcast spawner, were differentiated across regions (e.g. Gulf of Mexico (GoM), southeastern US and northeastern Atlantic), while populations were panmictic in similar depth ranges within each region [21]. In comparison, Oculina varicosa, although also a broadcast spawner, exhibited pronounced population divergence across the continental shelf off Florida at a depth of 50 m [20].
In the northern GoM, at least 162 species of octocorals contribute to habitat heterogeneity across the continental slope to depths of approximately 3000 m. In particular, species within the genus Callogorgia (Family Primnoidae) are the most abundant octocorals inhabiting the upper continental slope in this region [23]. Strong niche segregation with depth occurs within the genus, with three Callogorgia spp. [Callogorgia delta Cairns & Bayer 2002, Callogorgia americana Cairns & Bayer 2002, Callogorgia gracilis (Milne Edwards & Haime 1857)] occupying distinct depth zones. Thus depth (and the cofactors that vary with depth) significantly influences the evolution and ecology of these deep-sea octocorals [23].
Callogorgia delta commonly occurs in the northern GoM at depths of 400–900 m, providing an ideal system in which to examine the depth-differentiation hypothesis at a finer scale within a species inhabiting the bathyal region. If the bathyal region is a source of increased genetic differentiation in the deep sea, then it could be expected that genetic differentiation within C. delta exists on the upper continental slope and is greater over vertical rather than geographical distances. This study also provided the opportunity to examine the level of genetic differentiation between C. delta and C. americana; two species only recently elevated to species status [24]. Closely related octocorals can be difficult to identify visually, and inclusion of samples from several species in a population genetic dataset can lead to erroneous inferences of population connectivity [25]. We thus tested whether the here developed microsatellite markers could distinguish between C. delta and C. americana, yielding a molecular method for delimiting Callogorgia species.
2. Material and methods
(a). Field methods
In the GoM, 116 specimens of C. delta and 29 specimens of C. americana were collected across nine sites at depths between 340 and 848 m using remotely operated vehicles (i.e. ROVs Jason II, SeaEye Falcon) and the human-occupied vehicle (HOV) Johnson-Sea-Link during five cruises from 2008 to 2010 (figure 1). Surveyed sites were named in accordance with the GoM planning areas and lease blocks (managed by the Bureau of Ocean Energy Management) in which they occur: e.g. Viosca Knoll (VK) 826 and 862/906, Mississippi Canyon (MC) 751, and 885, Green Canyon (GC) 235, 246, 249, 338 and Garden Banks (GB) 299 (figure 1).
Figure 1.

Map of sampling locations of C. americana (GB299, VK862/906) and C. delta (all other sites). Colours denote clusters identified by STRUCTURE (figure 2). Number of samples (n) and depth range of collections are noted. (Online version in colour.)
Photographs were taken of each coral colony prior to sampling, and branches were subsampled from each colony using the ROV manipulator arm. On the research vessel, branches were divided into 2–3 cm sections and tissue subsamples were subsequently frozen at −80°C, preserved in 95% ETOH (stored at −20°C) and placed in a high-salt EDTA preservative (stored at −80°C). Voucher specimens of each individual colony were preserved in 95% ETOH or dried. Morphological characters and DNA barcodes were used to identify species [23]. A subset of representative voucher specimens were accessioned at the National Museum of Natural History of the Smithsonian Institution, Washington DC, USA (USNM1202708–1202713).
(b). Molecular methods
DNA was extracted using a Qiagen DNeasy kit. Genomic DNA from two individuals of C. delta was sequenced on a half plate on Roche-454 (Engencore, Colombia, SC, USA) following [26]. Roche 454 reads (electronic supplementary material, table S1) were input into the software program QDD v. 2 [27], enabling microsatellite selection and subsequent primer design (see the electronic supplementary material). Thirty-nine primer pairs predicted to produce products more than 120 bp were then tested for amplification across C. delta and C. americana collected from different sites. Of the 39 primer pairs tested, 10 amplified in both species across all sites in the GoM (the electronic supplementary material, table S2). PCR products were analysed on an ABI 3130XL Genetic Analyzer (University of Pennsylvania) with a Gene Scan 500LIZ size standard.
(c). Microsatellite analyses
The quality and applicability of microsatellite markers were assessed using a variety of analyses. First, fragments were sized using the microsatellite plug-in for Geneious v. 6 (created by Biomatters, http://www.geneious.com/) with Gene Scan 500LIZ size standards (Applied Biosystems, Inc.). MICROCHECKER v. 2.2 [28] was used to check for genotyping errors across all individuals. INEst v. 2 [29] was used to check for null alleles while taking into account the possibility of inbreeding within a population. INEst was run using both ‘nfb’ (accounting for null alleles, inbreeding and genotyping errors) and ‘nb ‘(null alleles and genotyping errors) models on the entire dataset and for each population (ngen = 500 000, burnin = 5000). The deviance information criterion (DIC) was used to determine which model performed better, and thus whether inbreeding was significant in the populations. Linkage disequilibrium was tested using Fisher's exact test (GENEPOP on the Web) [30] followed by a Bonferroni adjustment among all pairs of loci across all populations to determine whether the loci were non-randomly associated with one another. Departures from Hardy–Weinberg equilibrium and observed and expected heterozygosity at each locus and among populations were tested (GenALEx v. 6.5) [31]. The statistical power of the microsatellite data was assessed for each species using PowSim v. 4.1 [32]. Because of the low statistical power for C. americana (see the electronic supplementary material), this species was not used to test the depth-differentiation hypothesis.
We searched for duplicate genotypes among the samples (GenALEx). Duplicate multi-locus genotypes (MLGs) were subsequently removed from the dataset for further analyses (five total matching genotypes). Probability of identity (PI and PIsibs) was calculated to determine the probability of multi-locus genotypes matching at random with increasing numbers of loci (GenALEx).
We also searched for outlier loci potentially under selection in C. delta using two programs (LOSITAN [33,34] and BAYESCAN [35]), following [36]. LOSITAN implements an FST outlier test. Simulations were run under infinite allele and stepwise mutation models (SMMs) for 10 000 replications. The Bayesian program BAYESCAN, based on the multinomial Dirichlet model, calculates differences in allele frequencies in each subpopulation from a common migrant gene pool. Default parameters were used and acceptance rates were between 0.25 and 0.45.
STRUCTURE v. 2.3 [37], a Bayesian model-based clustering approach, was used to determine the number of populations (designated by K) both within C. delta and between C. delta and C. americana by assigning the probability of membership of individuals iteratively to each K. For C. delta, model priors included location information [38], an admixture model (i.e. individuals having mixed ancestry) and correlated allele frequencies [39]. The location prior does not bias detecting structure when no actual structure is present, but values of r ≤ 1 (electronic supplementary material, figure S1) signify that the locations are informative to population structure [38]. A total of 1 000 000 MCMC generations were run following a burn-in of 250 000 generations. Five independent chains were run to test each value of K (K = 1–8 for C. delta). The level of genetic admixture between C. delta and C. americana was examined using STRUCTURE with parameters as above without a location prior and K = 1–3. STRUCTURE Harvester v. 0.6 [40] was used to choose K with the delta K criterion [41]. For the chosen K, results from each of the five iterations were aligned using CLUMPP v. 1.1 [42] and plotted in DISTRUCT v. 1.1 [43].
To examine the amount of genetic differentiation among populations, FST [44] was calculated between sites and/or species (GENALEx). Based on the STRUCTURE results for C. delta, two sites (GC338 and GC249) located only approximately 5 km apart in similar depths were pooled for these analyses. An analysis of molecular variance (AMOVA) [45] was conducted to test for significance among pairwise FST values across all sites (GenALEx). Additionally, AMOVA was re-calculated after removing two sites (GC235 and GC246) that had small sample sizes. If AMOVA results were significant for C. delta, partial Mantel tests were used to test for significant correlations between FST [linearized to FST/(1 − FST)] values with vertical distance given geographical distance and with geographical distance given vertical distance (IBD on the Web v. 3.5, 1000 randomizations) [46]. Vertical and geographical distances were log transformed. Partial Mantel tests were also performed without GC235 and GC246. All analyses were repeated for both candidate neutral loci only and candidate selective loci.
BOTTLENECK v. 1.2 [47] was used to determine whether any C. delta populations experienced a recent reduction in population size at putative neutral loci. Bottleneck was calculated under three mutation models, including the infinite alleles model (IAM), two-phase model (TPM) and the SMM. A Wilcoxon sign-rank test and the relative distribution of allele frequencies (mode-shift indicator) were used to assess whether any of the populations experienced a recent bottleneck.
3. Results
(a). Microsatellite marker data
Of 39 primer pairs that were tested for amplification across individuals, 10 loci were polymorphic and consistently amplified in either C. delta (n = 116) or C. americana (n = 29) (electronic supplementary material, tables S2 and S3). However, CA3 did not amplify well in C. americana and CA1 did not amplify well in C. delta (electronic supplementary material, tables S2 and S3). No loci were in linkage disequilibrium (all loci, p > 0.05, Fisher's exact test). Three of the same MLGs of C. delta were found at VK826 and two were found at GC249. Two of the same C. americana MLGs were found at GB299. Removing these individuals resulted in 113 C. delta and 28 C. americana for further analyses. With at least eight loci, the probability of identifying identical multi-locus genotypes at random decreased to 0–0.05% for both species at both probability calculations. Estimates of null allele frequencies ranged from 0 to 5% across all loci (INEST, ‘nb’ model best for each species; electronic supplementary material, table S4). Except, the null allele estimate was 25% for CA3; however, this locus did not amplify in all individuals. Both BAYESCAN and LOSITAN indicated that two loci (CA3 and CA7) were outliers and thus potentially under selection.
The number of alleles per locus ranged from 3 to 22, and the majority were private alleles within a species (electronic supplementary material, table S2). With the exception of CA3, CA7 and CA10 (14 to 22 alleles per locus), there were few alleles at each of seven loci (three to seven alleles per locus). Including all loci, the mean number of alleles per locus ranged from 5.2 ± 0.84 s.e. in C. americana to 7.50 ± 1.85 s.e. in C. delta. Without the three loci with the higher number of alleles, the mean number of alleles per locus ranged from 3.86 ± 0.55 s.e. in C. americana to 4.14 ± 0.40 s.e. in C. delta.
Several loci showed negative fixation (F) indices, suggesting heterozygote excess; however, departures from HWE were not significant in most cases (electronic supplementary material, table S3). Three loci showed significant departures from HWE within a particular site for C. delta: CA3 at MC751 and MC885 (χ2, p < 0.005), CA5 at GC235, MC751 and MC885 (χ2, p < 0.005), and CA9 at MC751 for C. delta (χ2, p < 0.005). CA5 and CA9 were in heterozygote excess at the specific sites. Two loci showed significant departures from HWE for C. americana: CA2 and CA7 at GB299 (χ2, p < 0.005); however, CA3 only amplified in five individuals collected from GB299 and was thus not used in the analyses.
Power analysis indicated that the probability of detecting genetic differentiation in C. delta when FST ≥ 0.0250 was 100%. The probability of identifying significant genetic structure when the true FST = 0 was less than 6% in all simulations (electronic supplementary material, table S5).
(b). Genetic differentiation between Callogorgia delta and Callogorgia americana
STRUCTURE indicated little to no genetic admixture between C. delta and C. americana at eight loci (CA2, CA4–10). Two populations (K = 2) corresponded to the two species (figure 2). In addition, there was high genetic differentiation between the two species (FST = 0.434, p = 0.001; AMOVA; electronic supplementary material, table S6) and no evidence of hybridization. The proportion of membership to one of the species clusters for each individual was more than 99%.
Figure 2.
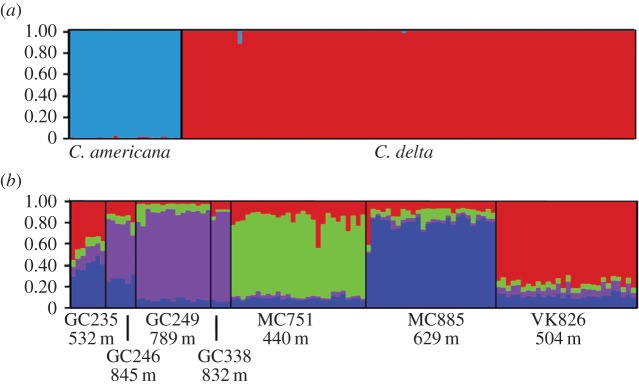
Average probability of membership graphs (STRUCTURE) for (a) Callogorgia spp. (n = 141, K = 2) and (b) C. delta (n = 113, K = 4). Mean depth of collections for each site is included. (Online version in colour.)
(c). Genetic differentiation within Callogorgia delta
Genetic differentiation was apparent within C. delta sampled from seven sites across the northern GoM using nine loci (CA2–10) (figure 2). The FST value among populations was significant (FST = 0.042, p = 0.001) (AMOVA; electronic supplementary material, table S6). Removing the two sites that had few individuals (GC235 and GC246) from AMOVA analyses still yielded significant overall FST (FST = 0.042, p = 0.001). The overall inbreeding coefficient was low (AvgFi = 0.0065) and not a significant component of the model (DIC = 3493 for the ‘nfb’ model, DIC = 3491 for the ‘nb’ model). Inbreeding coefficients were also low for each population (AvgFi = 0.011–0.041) and not a significant component of the model for any population based on DIC (INEst; electronic supplementary material, table S4).
Bayesian clustering analysis (STRUCTURE) with a location prior converged well. Delta K (STRUCTURE Harvester) indicated that the most likely number of population clusters (K) present in the dataset was four (electronic supplementary material, figure S2). Membership in each of these four clusters corresponded well to most of the sites from which individuals were collected: GC249 and GC338, MC751, MC885 and VK826 (electronic supplementary material, table S7). However, at some sites, multiple lineages were represented. A high proportion of individuals from GC235 were assigned to both the VK826 cluster and the MC885 cluster and a high proportion of individuals from GC246 were assigned to both the GC249/338 cluster as well as the MC885 cluster.
Pairwise FST values among sites harbouring C. delta ranged between 0.021 and 0.078 with p-values ranging from 0.002 to 0.126 (AMOVA, table 1). After a Bonferroni correction, FST values between only a few pairs of sites remained significant (p < 0.003, AMOVA, table 1). The strongest differences were observed between MC751 with VK826, MC885 and GC249/338 (FST = 0.033–0.053, p < 0.003). Relatively high FST values were also found between VK826 and MC885 (FST = 0.040, p = 0.004) and VK826 and GC249/338 (FST = 0.052, p = 0.007); however, these results did not remain significant after the Bonferroni adjustment. Greater genetic differentiation was evident at increasing differences in depth, given geographical distance (partial Mantel test, r = 0.61, p = 0.002, figure 3). By contrast, no significant correlation was found with FST and geographical distance given vertical distance (r = 0.21, p = 0.22). Partial Mantel tests were also conducted following the removal of the two sites with low sample sizes (GC235 and GC246). In this analysis, a relatively high, but non-significant (r = 0.68, p = 0.08) correlation was found with FST and vertical distance given geographical distance. However, a significant correlation in FST with geographical distance given vertical distance was also evident (r = 0.78, p = 0.03).
Table 1.
Pairwise FST values for C. delta among sites at all loci. FST values in bold are significant (AMOVA, Bonferroni adjustment, p ≤ 0.003). P-values are indicated above diagonal. n = number of unique multi-locus genotypes used in analyses. Depth range of collected specimens is noted.
| GC235 532–533 m n = 7 | GC246 844–848 m n = 6 | GC249 777–790 m n = 15 | GC338 830–838 m n = 4 | GC249/338 777–838 m n = 19 | MC751 437–445 m n = 27 | MC885 623–637 m n = 26 | VK826 454–545 m n = 28 | |
|---|---|---|---|---|---|---|---|---|
| GC235 | — | 0.109 | 0.058 | 0.123 | 0.081 | 0.044 | 0.029 | 0.126 |
| GC246 | 0.039 | — | 0.048 | 0.257 | 0.071 | 0.010 | 0.073 | 0.029 |
| GC249 | 0.038 | 0.048 | — | 0.239 | — | 0.004 | 0.021 | 0.001 |
| GC338 | 0.051 | 0.026 | 0.020 | — | — | 0.016 | 0.043 | 0.056 |
| GC249/338 | 0.035 | 0.038 | — | — | — | 0.002 | 0.007 | 0.007 |
| MC751 | 0.038 | 0.078 | 0.048 | 0.078 | 0.053 | — | 0.002 | 0.002 |
| MC885 | 0.041 | 0.032 | 0.029 | 0.057 | 0.034 | 0.033 | — | 0.006 |
| VK826 | 0.021 | 0.054 | 0.056 | 0.056 | 0.052 | 0.044 | 0.040 | — |
Figure 3.

Scatterplots of pairwise FST for C. delta with respect to vertical and geographical distance at (a,b) all nine loci; (c,d) two loci candidates for selection and (e,f) seven putative neutral loci.
Because both BAYESCAN and LOSITAN indicated that two loci were outliers and thus potentially under selection, we re-analysed the FST data using either the two candidate loci under selection or the seven putative neutral loci. Pairwise FST values at the two candidate loci were much higher (FST = 0.012–0.166) compared with the putative neutral loci (FST = 0.000–0.086) (electronic supplementary material, table S8). After a Bonferroni correction, numerous FST values between pairs of sites at the two candidate loci were significant (p < 0.003, AMOVA), whereas none were significant at the neutral loci (p > 0.003, AMOVA). Greater genetic differentiation with increasing vertical distance given geographical distance was evident at the two candidate loci (partial Mantel test, r = 0.56, p = 0.008, figure 3). No genetic differentiation with geographical distance given vertical distance was evident at these loci (r = 0.25, p = 0.24). Removing the two sites with small sample sizes revealed a significant correlation of FST with both vertical distance (r = 0.97, p = 0.04) and geographical distance (r = 0.93, p = 0.04). In comparison, no significant genetic divergence with vertical distance given geographical distance (r = 0.33, p = 0.08) or geographical distance given vertical distance (r = −0.05, p = 0.57) was evident at the seven neutral loci. Removing the two sites with small sample sizes resulted in no significant correlations of FST with vertical distance (r = 0.33, p = 0.27) or geographical distance (r = −0.11, p = 0.72).
BOTTLENECK results indicated that there was a shifted mode distribution of allele frequencies for C. delta at GC235 and GC249/338. In addition, there was significant heterozygote excess (Wilcoxon sign-rank test, p < 0.05) at both sites calculated under the IAM, TPM and SMM models.
4. Discussion
The hypothesis that there is no genetic differentiation within C. delta across the northern GoM can be rejected. Rather, results indicated that there is weak, but significant genetic differentiation across the sites surveyed. Different evolutionary processes could lead to the genetic differentiation observed within C. delta, including genetic drift due to a past reduction in population size, limited gene flow among sites and adaptive divergence in the presence of gene flow across a gradient of depth. Regardless of the precise mechanism, depth is an important factor influencing the population structure of C. delta across the slope in the northern GoM. A higher degree of genetic differentiation over vertical rather than geographical distance within C. delta re-enforces the importance of the environmental factors associated with depth as important abiotic gradients influencing the evolution of deep-sea populations and species.
(a). Utility of microsatellite markers in species delimitation
Determining species boundaries within octocorals has been problematic due to morphological gradations [48], phenotypic plasticity [48], the slow evolutionary rate of mitochondrial genomes [49] and the lack of phylogenetically informative, single copy nuclear loci that can be amplified across the clade [50]. Our microsatellite analyses indicated the utility of these loci in resolving species boundaries of octocorals that have been separated for millions of years. Callogorgia delta and C. americana were only recently elevated from sub-species to species status [24], with an estimated time since divergence of approximately 19 Ma [23]. Yet, the majority of microsatellite markers consistently amplified across C. delta and C. americana, resulting in a high FST value (0.434, p = 0.001) between species. Previous studies have also indicated the utility in using similar numbers of microsatellite loci to delimit species of scleractinian corals [51,52].
(b). Genetic differentiation across the Gulf of Mexico continental slope
The results from the STRUCTURE analyses suggested that genetic differentiation occurs among populations of C. delta across 400 m of depth and 400 km of distance in the northern GoM, with population clusters apparent at MC751, VK826, MC885 and GC249/338 (in order from shallowest to deepest). The remaining two sites, GC246 and GC235, contained individuals that were admixed across a few other populations, but this could be indicative of the low sample sizes (n < 7) collected from these two sites. This population structure pattern was further supported by FST results, as numerous pairwise comparisons showed significant differentiation between sites.
Our results also indicated that C. delta is more strongly isolated by depth rather than by geographical distance in the GoM. FST values were significantly correlated with larger differences in vertical rather than horizontal distances. This is further supported by the STRUCTURE analyses and the low and non-significant, FST value (0.021, p = 0.126) between two sites (VK826 and GC235) that were located 400 km apart yet in similar depth ranges. In comparison, MC751 (440 m) and MC885 (629 m) were significantly divergent (FST = 0.033, p = 0.002), despite the fact that these two sites were spatially only separated by 15 km. When the sites with small sample sizes were excluded from the analysis, high correlations with both depth and geographical distance were found, suggesting that geographical distance also has a role in isolating populations. These results, however, are confounded by the fact that the deeper site, GC249/338, was also the furthest from VK and MC areas.
We acknowledge that few C. delta individuals were sampled in the GC area. Obtaining samples from multiple localities in the deep sea is more difficult than in shallow-water environments [53], particularly as depth of sampling increases. We stress that more samples are needed to corroborate the pattern of isolation by depth in this study. However, if these results hold in the presence of additional samples, then the observed patterns of isolation by depth would strongly indicate that depth is a significant factor shaping populations in the deep sea. Recent studies [8,12] using microsatellite markers to distinguish populations of Lamellibrachia tubeworms in the deep (300–2600 m) northern GoM yielded similar results to those presented in this study. Depth-dependent gene flow was evident in Lamellibrachia spp., whereas continuous gene flow was apparent across 650 km in the GoM.
Gene flow in C. delta may be limited across the GoM through a number of different mechanisms. If long-distance dispersal via horizontal transport occurs, suitable substrate may not be available within a given depth range. Lack of habitat availability would result in a lack of successful recruitment. Sub-optimal habitat would lead to increased mortality, thus limiting gene flow. Alternatively, shorter larval lifespans along with slow current flow or mesoscale circulations (e.g. eddies) may result in local retention [54], thereby limiting connectivity. Although reproduction has not been studied in the genus Callogorgia, species within the family Primnoidae either broadcast spawn (e.g. Primnoa spp. [55]) or internally brood their larvae (e.g. Thouarella, Fannyella, [17,56]). Most planula larvae of brooding octocorals appear to settle shortly after release [16,56]. Thus, the dispersal distance of brooding species could be reduced if larvae settle close to the colonies, ultimately leading to limited gene flow between sites. If Callogorgia also brood larvae, this could contribute to limited larval dispersal among sites. However, if this were the case, we would suspect that either inbreeding would be significant at some sites or that isolation by distance would be evident using the putative neutral loci.
Limited gene flow could also occur across depth in the GoM due to the existence of water mass boundaries. In the GoM, four water masses dominate the continental slope. Sargasso Sea Water is predominant from 200 to 400 m; Tropical Atlantic Water (TAW) dominates depths ranging from 400 to 600 m; Antarctic Intermediate Water (AAIW) dominates depths ranging from 600 to 1000 m; and a mixture of North Atlantic Deep Water and Caribbean Water occurs below 1000 m [57]. These water mass boundaries (particularly across TAW to AAIW) could create a barrier to gene flow within Callogorgia by entraining larvae and inhibiting the relatively simplistic planula larvae from physically settling out of the water column. In addition, dispersing larvae may not be able to physiologically tolerate environmental parameters (pressure, temperature and dissolved oxygen) that change at water mass boundaries. Physiological intolerances of echinoderm larvae to temperature and/or pressure have been shown to limit distribution of bathyal species into either shallower or deeper depths depending upon their adult depth ranges [58,59].
Larvae that pass through water mass barriers and successfully form adult colonies may have a different range of physiological tolerances to environmental conditions than their source population. This could lead to selection, and thus adaptive divergence among populations occupying different depths [60]. Pre-reproductive selection could be common in sessile animals, as sessile species are not able to move away from environmental pressures once settled [60]. For Callogorgia occupying this area of the continental slope, variability in temperature and dissolved oxygen may be major factors influencing either larval or post-settlement survival. Where C. delta occurred, temperatures ranged from 5.0 to 10.0°C and dissolved oxygen from 1.5 to 3.5 ml l−1 [23]. Changes in only a few degrees of temperature could lead to adaptive protein changes and thus could influence species distributions [61]. The potential presence of microsatellite markers under selection (or linked to loci under selection) supports the idea that these processes may be at work here. Therefore, adaptive divergence in the presence of gene flow could lead to the weak, but significant genetic differentiation with depth observed in C. delta across the continental slope of the northern GoM.
Callogorgia delta may also have undergone a recent population bottleneck, as indicated by the low allelic diversity and heterozygote excess at several loci, specifically at the GC sites. Allelic diversity can often be reduced faster than heterozygosity, particularly if heterozygotes have a selective advantage [47,62]. It is possible that there was a decrease in the effective population size at sites in the GC region in the past. This region experienced fluctuations in the amount of freshwater discharge and water depth during glacial and inter-glacial periods over at least the past 16 000 years, which could have led to habitat fragmentation in the region through anoxic events or temperature and salinity fluctuations [63,64]. Alternative to the population bottleneck scenario, it is possible that the significant results are spurious due to poor sampling of individuals at the GC sites as statistical power increases with more loci and more individuals.
Disentangling the evolutionary mechanisms (e.g. selection versus limited gene flow) causing genetic differentiation across the GoM in C. delta will likely require additional data, as any of the scenarios described above could result in the pattern observed. Determining whether species in the genus Callogorgia are brooders or broadcast spawners would provide insight into their effective dispersal distances and whether reproductive mode could limit gene flow. However, our results indicated that the most likely scenario for the genetic differentiation in C. delta across depth is adaptive divergence in the presence of gene flow. First, there was no evidence for significant inbreeding within any site. Second, FST values calculated using only the candidate selective loci revealed much higher genetic differentiation between sites, whereas no genetic differentiation was evident at the seven neutral loci. In a similar study that examined population differentiation in the deep-sea fish Coryphaenoides rupestris, one microsatellite locus (out of 16 total) was found to be under selection with genetic differentiation greater at this locus across a depth boundary of 1200 m [36]. In our study, two loci, CA3 and CA7, were designated as outliers and thus potential candidates for selection; however, it is noted that these two loci had much higher allelic diversities and larger motif sizes than the putative neutral loci. Including more loci spread across the genome would help resolve whether CA3 and CA7 are in fact under selection or linked to loci under selection. Increasing the number of loci used would yield a more robust estimate of the average genetic differentiation among loci, and thus allow for more confidence when designating outliers. Nevertheless, our results provide evidence that environmental conditions are shaping the pattern of genetic differentiation in C. delta across a gradient of depth.
The pattern of genetic differentiation with depth has emerged as an important feature of coral population structure. Population differentiation across depth has been indicated in several species of shallow and mesophotic corals that exhibit different reproductive modes. This includes three species of broadcast spawning corals: O. varicosa across the Florida continental shelf (less than 2 to 80 m) [20], Eunicea flexuosa across the Caribbean basin (less than 5 to 25 m) [60] and Montastraea cavernosa off Florida (less than 10 to 25 m) [26]. Similarly, populations of the octocoral Paramuricea clavata, a brooding species, were differentiated over both markedly short vertical (10–40 m) and horizontal (less than 400 km) distances in the Mediterranean [65]. In comparing these examples with this study, a stronger pattern of isolation by depth compared with distance further suggests that adaptive divergence in the presence of gene flow is likely leading to depth differentiation in Callogorgia across the upper continental slope of the GoM.
(c). Further considerations
Adaptive divergence with depth is becoming increasingly recognized as an important process shaping population and species evolution in the deep sea [7–9]. Our data and results from recent studies [7–9] reveal the importance of considering different environmental conditions associated with depth that could lead to population isolation in the deep sea. These depth-related patterns necessitate additional research to disentangle the mechanisms responsible for population divergence in the deep-sea environment. Furthermore, our results have significant implications for conservation efforts. With the increasing potential for anthropogenic impacts to deep-sea communities [18–19], future design of protected areas in the deep sea could beneficially incorporate a variety of depth ranges and habitat types in order to capture the diversity within and among vulnerable marine ecosystems.
Data accessibility
Microsatellite allele calls are deposited in Dryad (doi:10.5061/dryad.fq7d1).
Acknowledgements
Particular thanks to C. Fisher, J. Brooks and TDI-Brooks for support and the US Geological Survey for cruise participation. The crews of the ROV Jason, ROV Seaview and HOV Alvin provided offshore support. We particularly thank W. Cho, J. Lunden, S. Herrera and D. Ruiz for help at sea. C. Doughty and R. Falco aided with DNA extractions. M. Devlin-Durante helped with initial microsatellite marker screening. J. Rosado operated the ABI at U Penn. S. Cairns provided taxonomic expertise. Finally, we thank R. Kulathinal, M. Springmann, A. Thaler and A. Sethuraman for helpful discussions.
Funding statement
Funding was provided by BOEM and NOAA-OER (BOEM contract no. M08PC20038) for the Lophelia II project led by TDI-Brooks International. A.M.Q. was funded by the Dr Nancy Foster Scholarship programme, Temple University Dissertation Completion Grant and the Lerner-Gray grant for marine research.
Author contributions
A.M.Q. and E.E.C. conceived and designed the study. A.M.Q. performed the research, analysed the data and wrote the article with contributions from E.E.C. I.B.B. helped with microsatellite marker design. I.B.B., T.M.S. and C.L.M. helped with population genetic analyses and edited the manuscript. All authors gave final approval for publication.
Conflict of interests
We have no competing interests. Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the US Government.
References
- 1.Rex MA, Etter RJ. 2010. Deep-sea biodiversity: pattern and scale. Cambridge, MA: Harvard University Press. [Google Scholar]
- 2.Levin LA, Etter RJ, Rex MA, Gooday AJ, Smith CR, Pineda J, Stuart CT, Hessler RR, Pawson D. 2001. Environmental influences on regional deep-sea species diversity. Annu. Rev. Ecol. Syst. 132, 51–93 (doi:10.1146/annurev.ecolsys.32.081501.114002) [Google Scholar]
- 3.Levin LA, Sibuet M. 2012. Understanding continental margin biodiversity: a new imperative. Annu. Rev. Mar. Sci. 4, 79–112 (doi:10.1146/annurev-marine-120709-142714) [DOI] [PubMed] [Google Scholar]
- 4.Cordes EE, Cunha MM, Galeron J, Mora C, Olu-Le Roy K, Sibuet M, Van Gaever S, Vanreusel A, Levin L. 2010. The influence of geological, geochemical, and biogenic habitat heterogeneity on seep biodiversity. Mar. Ecol. 31, 51–65 (doi:10.1111/j.1439-0485.2009.00334.x) [Google Scholar]
- 5.Etter RJ, Rex MA, Chase M, Quattro J. 2005. Population differentiation decreases with depth in deep-sea bivalves. Evolution 59, 1479–1491 (doi:10.1111/j.0014-3820.2005.tb01797.x) [PubMed] [Google Scholar]
- 6.Zardus JD, Etter RJ, Chase MR, Rex MA, Boyle EE. 2006. Bathymetric and geographic population structure in the pan-Atlantic deep-sea bivalve Deminucula atacellana (SCHENCK 1939). Mol. Ecol. 15, 639–651 (doi:10.1111/j.1365-294X.2005.02832.x) [DOI] [PubMed] [Google Scholar]
- 7.Jennings RM, Etter RJ, Ficarra L. 2013. Population differentiation and species formation in the deep sea: the potential role of environmental gradients and depth. PLoS ONE 8, e77594 (doi:10.1371/journal.pone.0077594) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cowart DA, Halanych KM, Schaeffer SW, Fisher CR. 2014. Depth-dependent gene flow in Gulf of Mexico cold seep Lamellibrachia tubeworms (Annelida, Siboglinidae). Hydrobiologia 736, 139–154 (doi:10.1007/s10750-014-1900-y) [Google Scholar]
- 9.Glazier AE, Etter RJ. 2014. Cryptic speciation along a bathymetric gradient. Biol. J. Linn Soc. 113, 897–913 (doi:10.1111/bij.12389) [Google Scholar]
- 10.Jablonski D, Bottjer DJ. 1990. Onshore–offshore trends in marine invertebrate evolution. In Causes of evolution: a paleontological perspective (eds RM Ross, WD Allmon), pp. 21–75 Chicago, IL: University of Chicago Press. [Google Scholar]
- 11.Thaler AD, Zelnio K, Saleu W, Schultz TF, Carlsson J, Cunningham C, Vrijenhoek R, Van Dover CL. 2011. The effects of spatial scale on the population dynamics of Ifremeria nautilei, a hydrothermal vent endemic gastropod from the southwest Pacific. BMC Evol. Biol. 11, 372 (doi:10.1186/1471-2148-11-372) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cowart DA, Huang C, Arnaud-Haond S, Carney SL, Fisher CR, Schaeffer SW. 2013. Restriction to largescale gene flow versus regional panmixia among cold seep Escarpia spp. (Polychaeta, Siboglinidae). Mol. Ecol. 22, 4147–4162 (doi:10.1111/mec.12379) [DOI] [PubMed] [Google Scholar]
- 13.Andrews AH, Cordes EE, Mahoney MM, Munk K, Coale KH, Cailliet GM, Heifetz J. 2002. Age, growth and radiometric age validation of a deep-sea, habitat-forming gorgonian (Primnoa resedaeformis) from the Gulf of Alaska. Hydrobiologia 471, 101–110 (doi:10.1023/A:1016501320206) [Google Scholar]
- 14.Roark EB, Guilderson TP, Dunbar RB, Ingram B. 2006. Radiocarbon-based ages and growth rates of Hawaiian deep-sea corals. Mar. Ecol. Prog. Ser. 327, 1–14 (doi:10.3354/meps327001) [Google Scholar]
- 15.Kahng SE, Benayahu Y, Lasker HR. 2011. Sexual reproduction in octocorals. Mar. Ecol. Prog. Ser. 443, 265–283 (doi:10.3354/meps09414) [Google Scholar]
- 16.Cordes EE, Nybakken JW, VanDykhuizen G. 2001. Reproduction and growth of Anthomastus ritteri (Octocoraillia: Alcyonacea) from Monterey Bay, California, USA. Mar. Biol. 138, 491–501 (doi:10.1007/s002270000470) [Google Scholar]
- 17.Orejas C, López P, Gili JM, Teixidó N, Gutt J, Arntz W. 2002. Distribution and reproductive ecology of the Antarctic octocoral Ainigmaptilon antarcticum in the Weddell Sea. Mar. Ecol. Prog. Ser. 231, 101–114 (doi:10.3354/meps231101) [Google Scholar]
- 18.Ramirez-Llodra E, et al. 2011. Man and the last great wilderness: human impact on the deep sea. PLoS ONE 6, e22588 (doi:10.1371/journal.pone.0022588) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White HK, et al. 2012. Impact of the Deepwater Horizon oil spill on a deep-water coral community in the Gulf of Mexico. Proc. Natl Acad. Sci. USA 109, 20 303–20 308 (doi:10.1073/pnas.1118029109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eytan RI, Hayes M, Arbour-Reily P, Miller M, Hellberg ME. 2009. Nuclear sequences reveal mid-range isolation of an imperilled deep-water coral population. Mol. Ecol. 18, 2375–2389 (doi:10.1111/j.1365-294X.2009.04202.x) [DOI] [PubMed] [Google Scholar]
- 21.Morrison CL, Ross SW, Nizinski MS, Brooke S, Järnegren J, Waller RG, Johnson RL, King TL. 2011. Genetic discontinuity among regional populations of Lophelia pertusa in the North Atlantic Ocean. Conserv. Genet. 12, 713–729 (doi:10.1007/s10592-010-0178-5) [Google Scholar]
- 22.Baco AR, Shank TM. 2005. Population genetic structure of the Hawaiian precious coral Corallium lauuense (Octocorallia: Coralliidae) using microsatellites. In Cold-water corals and ecosystems (eds Freiwald A, Roberts JM.), pp. 663–678 Berlin, Germany: Springer. [Google Scholar]
- 23.Quattrini AM, Georgian SE, Byrnes L, Stevens A, Falco R, Cordes EE. 2013. Niche divergence by deep-sea octocorals in the genus Callogorgia across the continental slope of the Gulf of Mexico. Mol. Ecol. 22, 4123–4140 (doi:10.1111/mec.12370) [DOI] [PubMed] [Google Scholar]
- 24.Bayer FM, Cairns SD, Cordeiro RTS, Pérez CD. 2014. New records of the genus Callogorgia (Anthozoa: Octocorallia) in the western Atlantic, including the description of a new species. J. Mar. Biol. Assoc. UK, 1–7 (doi:10.1017/S0025315414001957) [Google Scholar]
- 25.Pante E, et al. 2015. Species are hypotheses: avoid connectivity assessments based on pillars of sand. Mol. Ecol. 24, 525–544 (doi:10.1111/mec.13048) [DOI] [PubMed] [Google Scholar]
- 26.Serrano X, Baums IB, O'Reilly K, Smith TB, Jones RJ, Shearer TL, Nunes FLD, Baker AC. 2014. Geographic differences in vertical connectivity in the Caribbean coral Montastraea cavernosa despite high levels of horizontal connectivity at shallow depths. Mol. Ecol. 23, 4226–4240 (doi:10.1111/mec.12861) [DOI] [PubMed] [Google Scholar]
- 27.Meglécz E, Costedoat C, Dubut V, Gilles A, Malausa T, Pech N, Martin JF. 2010. QDD: a user-friendly program to select microsatellite markers and design primers from large sequencing projects. Bioinformatics 26, 403–404 (doi:10.1093/bioinformatics/btp670) [DOI] [PubMed] [Google Scholar]
- 28.Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P. 2004. MICROCHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538 (doi:10.1111/j.1471-8286.2004.00684.x) [Google Scholar]
- 29.Chybicki IJ, Burczyk J. 2009. Simultaneous estimation of null alleles and inbreeding coefficients. J. Heredity 100, 106–113 (doi:10.1093/jhered/esn088) [DOI] [PubMed] [Google Scholar]
- 30.Raymond M, Rousset F. 1995. GENEPOP (v. 1.2): population genetics software for exact tests and ecumenicism. J. Heredity 86, 248–249. [Google Scholar]
- 31.Peakall ROD, Smouse PE. 2006. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryman N, Palm S. 2006. PowSim: a computer program for assessing statistical power when testing for genetic differentiation. Mol. Ecol. Notes 6, 600–602 (doi:10.1111/j.1471-8286.2006.01378.x) [DOI] [PubMed] [Google Scholar]
- 33.Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G. 2008. LOSITAN: a workbench to detect molecular adaptation based on a FST-outlier method. BMC Bioinformatics 9, 323 (doi:10.1186/1471-2105-9-323) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beaumont MA, Nichols RA. 1996. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. B 263, 1619–1626 (doi:10.1098/rspb.1996.0237) [Google Scholar]
- 35.Foll M, Gaggiotti OE. 2008. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (doi:10.1534/genetics.108.092221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White TA, Stamford J, Rus Hoelzel A. 2010. Local selection and population structure in a deep-sea fish, the roundnose grenadier (Coryphaenoides rupestris). Mol. Ecol. 19, 216–226 (doi:10.1111/j.1365-294X.2009.04446.x) [DOI] [PubMed] [Google Scholar]
- 37.Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hubisz M, Falush D, Stephens M, Pritchard J. 2009. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Res. 9, 1322–1332 (doi:10.1111/j.1755-0998.2009.02591.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falush D, Stephens M, Pritchard JK. 2003. Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Earl DA, vonHoldt BM. 2012. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Res. 4, 359–361 (doi:10.1007/s12686-011-9548-7) [Google Scholar]
- 41.Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (doi:10.1111/j.1365-294X.2005.02553.x) [DOI] [PubMed] [Google Scholar]
- 42.Jakobsson M, Rosenberg NA. 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (doi:10.1093/bioinformatics/btm233) [DOI] [PubMed] [Google Scholar]
- 43.Rosenberg NA. 2004. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138 (doi:10.1046/j.1471-8286.2003.00566.x) [Google Scholar]
- 44.Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (doi:10.2307/2408641) [DOI] [PubMed] [Google Scholar]
- 45.Excoffier L, Smouse PE, Quattro JM. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jensen JL, Bohonak AJ, Kelley ST. 2005. Isolation by distance, web service. BMC Genetics 6, 13 (doi:10.1186/1471-2156-6-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piry S, Luikart G, Cornuet JM. 1999. BOTTLENECK: a program for detecting recent effective population size reductions from allele data frequencies. Montpellier, France: CBGP. See http://www1.montpellier.inra.fr/CBGP/software/Bottleneck/pub.html. [Google Scholar]
- 48.Sánchez JA, Aguilar C, Dorado D, Manrique N. 2007. Phenotypic plasticity and morphological integration in a marine modular invertebrate. BMC Evol. Biol. 7, 122 (doi:10.1186/1471-2148-7-122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shearer TL, Van Oppen MJH, Romano SL, Wörheide G. 2002. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol. Ecol. 11, 2475–2487 (doi:10.1046/j.1365-294X.2002.01652.x) [DOI] [PubMed] [Google Scholar]
- 50.McFadden CS, Benayahu Y, Pante E, Thoma JN, Nevarez PA, France SC. 2011. Limitations of mitochondrial gene barcoding in Octocorallia. Mol. Ecol. Res. 11, 19–31 (doi:10.1111/j.1755-0998.2010.02875.x) [DOI] [PubMed] [Google Scholar]
- 51.Baums IB, Johnson ME, Devlin-Durante MK, Miller MW. 2010. Host population genetic structure and zooxanthellae diversity of two reef-building coral species along the Florida Reef Tract and wider Caribbean. Coral Reefs 29, 835–842 (doi:10.1007/s00338-010-0645-y) [Google Scholar]
- 52.Boulay JN, Hellberg ME, Cortés J, Baums IB. 2014. Unrecognized coral species diversity masks differences in functional ecology. Proc. R. Soc. B 281, 20131580 (doi:10.1098/rspb.2013.1580) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gage JD, Tyler PA. 1991. Deep-sea biology: a natural history of organisms at the deep-sea floor. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 54.Cowen RK, Lwiza KM, Sponaugle S, Paris CB, Olson DB. 2000. Connectivity of marine populations: open or closed? Science 287, 857–859 (doi:10.1126/science.287.5454.857) [DOI] [PubMed] [Google Scholar]
- 55.Feehan KA, Waller RG. 2015. Notes on the reproduction of eight species of eastern Pacific cold-water octocorals. J. Mar. Biol. Assoc. UK, 1–6 (doi.:10.1017/S0025315415000053) [Google Scholar]
- 56.Brito TAS, Tyler PA, Clarke A. 1997. Reproductive biology of the Antarctic octocoral Thouarella variabilis Wright and Studer 1889. In Proc. 6th Int. Conf. Coelenterate Biology, The Leeuwenhorst, Noordwijkerhout, The Netherlands, 16–21 July 1995, pp. 63–69 Leiden, The Netherlands: Natural History Museum of Leiden. [Google Scholar]
- 57.Rivas D, Badan A, Ochoa J. 2005. The ventilation of the deep Gulf of Mexico. J. Phys. Oceanogr. 35, 1763–1781 (doi:10.1175/JPO2786.1) [Google Scholar]
- 58.Young CM, Tyler PA, Gage JD. 1996. Vertical distribution correlates with pressure tolerances of early embryos in the deep-sea asteroid Plutonaster bifrons. J. Mar. Biol. Assoc. UK 76, 749–757 (doi:10.1017/S002531540003143X) [Google Scholar]
- 59.Tyler PA, Young CM. 1998. Temperature and pressure tolerances in dispersal stages of the genus Echinus (Echinodermata: Echinoidea): prerequisites for deep-sea invasion and speciation. Deep Sea Res. II 45, 253–277 (doi:10.1016/S0967-0645(97)00091-X) [Google Scholar]
- 60.Prada C, Hellberg ME. 2013. Long prereproductive selection and divergence by depth in a Caribbean candelabrum coral. Proc. Natl Acad. Sci. USA 110, 3961–3966 (doi:10.1073/pnas.1208931110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Somero GN. 1995. Proteins and temperature. Annu. Rev. Phys. 57, 43–68 (doi:10.1146/annurev.ph.57.030195.000355) [DOI] [PubMed] [Google Scholar]
- 62.Cornuet JM, Luikart G. 1996. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leventer A, Williams DF, Kennett JP. 1983. Relationships between anoxia, glacial meltwater and microfossil preservation in the Orca Basin, Gulf of Mexico. Mar. Geol. 53, 23–40 (doi:10.1016/0025-3227(83)90032-4) [Google Scholar]
- 64.Poore RZ, Dowsett HJ, Verardo S, Quinn TM. 2003. Millennial to century-scale variability in Gulf of Mexico Holocene climate records. Paleoceanography 18, 1048 (doi:10.1029/2002PA000868) [Google Scholar]
- 65.Mokhtar-Jamaï K, Pascual M, Ledoux JB, Coma R, Féral JP, Garrabou J, Aurelle D. 2011. From global to local genetic structuring in the red gorgonian Paramuricea clavata: the interplay between oceanographic conditions and limited larval dispersal. Mol. Ecol. 20, 3291–3305 (doi:10.1111/j.1365-294X.2011.05176.x) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Microsatellite allele calls are deposited in Dryad (doi:10.5061/dryad.fq7d1).