

Abstract
Basic and clinical studies demonstrate that depression is associated with reduced size of brain regions that regulate mood and cognition, including the prefrontal cortex and the hippocampus, and decreased neuronal synapses in these areas. Antidepressants can block or reverse these neuronal deficits, although typical antidepressants have limited efficacy and delayed response times of weeks to months. A notable recent discovery shows that ketamine, a N-methyl-D-aspartate receptor antagonist, produces rapid (within hours) antidepressant responses in patients who are resistant to typical antidepressants. Basic studies show that ketamine rapidly induces synaptogenesis and reverses the synaptic deficits caused by chronic stress. These findings highlight the central importance of homeostatic control of mood circuit connections and form the basis of a synaptogenic hypothesis of depression and treatment response.
Despite extensive research, the neurobiology of major depressive disorder (MDD) remains poorly understood due to lack of biomarkers, relatively low rates of heritability, and heterogeneity of precipitating factors, including stress (1–3). Studies of antidepressant mechanisms and the development of more effective therapeutic agents have also progressed slowly. The widely prescribed serotonin selective reuptake inhibitors (SSRIs), derived from drugs developed more than 50 years ago, take weeks to months to produce a therapeutic response and are only moderately effective, leaving more than one-third of depressed individuals resistant to drug treatments (3).
However, basic and clinical studies have begun to shed light on this widespread, debilitating illness that is characterized by loss of pleasure (anhedonia); decreased cognition and memory; and disrupted sleeping, eating, ambulation, and sexual activity. There are consistent reports of decreased size of brain regions implicated in depression, as well as neuronal atrophy, including loss of synapses in MDD and rodent chronic stress models. Recent studies report what is arguably the most important discovery in half a century: the therapeutic agent ketamine that produces rapid (within hours) antidepressant actions in treatment-resistant depressed patients (4, 5). Notably, the rapid antidepressant actions of ketamine are associated with fast induction of synaptogenesis in rodents and reversal of the atrophy caused by chronic stress (6, 7).
We propose a hypothesis that depression is caused by disruption of homeostatic mechanisms that control synaptic plasticity, resulting in destabilization and loss of synaptic connections in mood and emotion circuitry. We compare and contrast the mechanisms underlying typical antidepressants and ketamine, particularly the induction of synaptogenesis. Together, these studies provide a framework for current and future studies of the neurobiology of depression and for the treatment and prevention of MDD and other stress-related illnesses.
Neuronal Atrophy and Synaptic Loss in Depression
Brain-imaging studies of depressed patients provide strong and consistent evidence of decreased volume of cortical and limbic brain regions— such as the prefrontal cortex (PFC) and the hippocampus—that control emotion, mood, and cognition, suggestive of neuronal atrophy that is related to length of illness and time of treatment (8, 9). Functional imaging studies report reductions of connectivity of the hippocampus and the PFC, as well as other brain regions, although there are also reports of increased connectivity of some regions, indicating a more complex disruption of brain circuits (10, 11), possibly due to dysregulation of reciprocal connections (9). Accordingly, it has been proposed that dysregulation rather than an overall increase or decrease of connectivity within PFC networks and their target limbic regions are responsible for the disturbances in emotional, cognitive, and autonomic regulation in mood disorders (9). Also, whereas imaging provides connectivity information on a particular brain region, it does not provide information on the integrity and efficiency of connections, which will require further analysis.
Postmortem studies of MDD report a reduction in the size but not number of pyramidal neurons in the dorsal lateral (dl) PFC (12). There is also evidence that GABAergic interneurons (GABA, γ-aminobutyric acid) are decreased in the dlPFC, along with consistent reports of decreased glia (12, 13). Similar reductions in neuronal cell body size and neuropil have been reported in the hippocampus of patients with MDD (14). Magnetic resonance spectroscopy demonstrates altered levels of GABA and glutamate cycling, indicating an imbalance of these major neurotransmitters in MDD patients (15).
Studies of neuronal morphology in human postmortem brain tissue have been hampered by poor fixation and tissue condition, but we recently reported that the number of synapses, determined by electron microscopy, is decreased in the dlPFC of depressed patients (16). Evidence in support of a reduction of synapse number is provided by reports of decreased levels of synaptic signaling proteins in MDD patients, including decreased levels of glutamate receptor subtypes, presynaptic neurotransmitter vesicle-associated proteins, and postsynaptic structural and functional proteins in the dlPFC, the hippocampus, and other forebrain structures (16–19). In many cases, the molecular alterations associated with depression can be distinguished from the medication status of the patients, although studies are not always sufficiently powered to make this distinction.
Stress Causes Neuronal Atrophy and Decreases Synaptic Density
Rodent studies have provided detailed evidence of neuronal atrophy, reduced synaptic density, and cell loss in models of depression and stress (Fig. 1). Chronic unpredictable stress (CUS), a putative model of depression, results in behavioral abnormalities, including core symptoms of depression such as anhedonia (7). CUS exposure causes a reduction in the length and branching of apical dendrites and decreases the number and function {i.e., reduction in serotonin [5-hydroxytryptamine (5-HT)] and hypocretin-induced excitatory post-synaptic potentials} of spine synapses in layer V pyramidal neurons of the medial PFC (Fig. 2) (7). Other chronic stress paradigms (such as restraint stress) cause similar reductions in dendrite complexity and spine density in PFC neurons and CA3 pyramidal cells of the hippocampus (20–22). Chronic stress also decreases neurogenesis (23) in the adult hippocampus and the number of glia in the medial PFC (2, 24), consistent with reports in postmortem MDD (12).
Fig. 1.

Atrophy of cortical neurons is caused by chronic stress or a BDNF polymorphism. Representative confocal photomicrographs of labeled layer V pyramidal neurons in the medial PFC are shown. (Top) Effects of restraint stress (~30 min per day, 7 days) on dendrite length and branching. (Bottom left) Pyramidal neuron dendrites from wild-type (WT; Val66Val66) mice compared with mice with a knock-in of one or both Met alleles. (Bottom right) The Met allele decreases the transport of BDNF transcripts to dendrites and activity-dependent release of BDNF, which leads to atrophy of pyramidal neurons, including decreased number and length of dendrite branches. There is a gene dose effect, with Val66Met66 knock-in mice showing an intermediate effect and Met66Met66 mice a greater effect compared with WT Val66Val66 mice. Adapted from (20, 32).
Fig. 2.

Chronic stress decreases synaptic connections and produces depressive-like behavior: rapid reversal by ketamine. (A) Confocal photomicrographs of labeled layer V pyramidal neurons in the medial PFC, showing the effects of CUS (21 days) on spine-synapses in layer V pyramidal neurons and reversal by a single dose of ketamine 1 day later. (B) Effects of chronic stress ± ketamine administration on 5-HT– or hypocretin (Hcrt)– induced excitatory postsynaptic potentials (EPSPs). (C and D) Quantitative analysis of the effects of CUS ± ketamine on spine density and the corresponding regulation of spine synapse function, 5-HT– or Hcrt-induced EPSP frequency (percentage of control). (E and F) Influence of CUS ± ketamine on behavior in (E) the sucrose preference test (measured by percentage of preference for a sucrose solution) and (F) novelty suppressed feeding, measured by latency to feed in an open field (measured in seconds). These models provide measures of anhedonia and anxiety, respectively, and are rapidly (1 day) reversed by ketamine, compared with the requirement for long-term administration (3 weeks) of a typical antidepressant. Error bars indicate SEM; asterisks indicate significance from control (C to F) or between CUS and CUS+ket (E, Hcrt). Adapted from (7, 20).
Atrophy of PFC pyramidal neurons is observed after as little as 1 week of restraint stress (20 to 30 min per day), indicating that these cells are particularly sensitive (20). High levels of social, environmental, and work-related stressors are experienced routinely in everyday life and probably cause PFC neuronal atrophy in many individuals. Dendrite complexity and synaptic density can also be increased or the effects of stress reversed by antidepressants, enriched environment, and exercise (6, 7, 21). In contrast, chronic stress causes hypotrophy of neurons in the amygdala and nucleus accumbens (24, 25), effects that could contribute to disrupted emotion, motivation, and reward behaviors that are regulated by these brain regions, demonstrating the involvement of a broader neurocircuit in the pathophysiology of depression.
Mechanisms Underlying Disruption of Synaptic Homeostasis and Behavior
The reason that neuronal atrophy in response to stress has evolved is unclear. One possibility is that the mechanisms underlying these effects are designed to be engaged only briefly during the fight-or-flight response and then rapidly turned off when the stress and/or danger has subsided. Acute stress responses are necessary for shunting energy and resources to the appropriate organs and tissues for maximal responses to danger situations. However, continued or long-term stress exposure and sustained activation of the accompanying physiological signals have negative effects on brain and many other organ systems. This hypothesis is supported by studies demonstrating that acute stress enhances glutamate transmission in the PFC and related cognitive function, but that chronic stress disrupts these processes (26).
One of the most frequently studied stress-activated systems is the hypothalamic-pituitary-adrenal (HPA) axis. Chronic exposure to adrenal glucocorticoids causes atrophy of neurons in the PFC and hippocampus, similar to the effects of chronic stress (20–22). Activation of the HPA axis and loss of negative feedback occur in more than 50% of depressed patients, highlighting the impact of this endocrine system in mood disorders.
Alterations of brain-derived neurotrophic factor (BDNF)
Stress and adrenal-glucocorticoids influence neuronal systems at many levels, but neurotrophic factors (particularly BDNF) have been of particular interest with respect to atrophy of neuronal connections (2, 24). BDNF is required for neuronal development early in life and for neuronal survival and function, including activity-dependent synaptic plasticity, in the adult brain (24). Stress or glucocorticoid exposures decrease the expression of BDNF in the hippocampus and the PFC, and levels of BDNF are decreased in postmortem brains of depressed patients (2, 24). Conversely, chronic administration of a typical anti-depressant increases BDNF expression in these brain regions, and the behavioral actions of antidepressants are blocked in BDNF deletion mutant mice (24). BDNF deletion mutants are also more vulnerable to stress (24, 27), and targeted deletion of BDNF in the hippocampus is sufficient to cause depressive behaviors (28). In contrast to the PFC and the hippocampus, BDNF activity in the mesolimbic dopamine system produces prodepressive effects, demonstrating the region-specific actions of this plasticity-related factor (2).
As might be expected for an activity-dependent trophic factor, BDNF substantially affects dendrite complexity and spine density (Fig. 1). BDNF heterozygous deletion mutant mice exhibit decreased dendrite length and branching in the hippocampus and occlusion of the effects of stress (29, 30). Mice with a knock-in of a human loss-of-function BDNF gene variant (Val66Met) also show reduced dendrite length and decreased spine-synapse density, maturity, and function in the hippocampus and/or the PFC (Fig. 1) (27, 29, 31, 32). The Met polymorphismimpairsthetransportofBDNFmRNA to subsynaptic sites, a requirement for activity-dependent local translation of proteins essential for synaptic formation and maturation (Fig. 1) (27, 29, 31, 32). Brain-imaging studies demonstrate that carriers of the Met polymorphism have decreased hippocampal volume and reduced executive function (8, 29) and, if exposed to early-life stress, are more vulnerable to depressive symptoms (33).
Disrupted synaptic signaling pathways
Stress and depression also disrupt BDNF–tropomyosinrelated kinase B (TrkB) receptor signaling, including reductions of the downstream extracellular signal–regulated kinase (ERK) and Akt pathways in the hippocampus and PFC (24, 34). These pathways positively influence synaptic maturation and stability via regulation of synaptic protein synthesis and glutamate receptor cycling (35, 36). Decreased Akt activity in the ventral tegmental dopamine neurons is associated with increased susceptibility to social defeat stress and is reversed by anti-depressant treatment in rodents and in human postmortem tissue (37). In addition, stress and depression increase levels of a negative regulator of ERK signaling, mitogen-activated protein kinase phosphatase 1, that is sufficient to cause depressive behaviors (34) and decrease a downstream target of BDNF, neuritin, that is required for normal antidepressant behavior (38). Chronic stress also inhibits glutamate signaling and transmission via ubiquitin-mediated degradation of glutamate receptors in the PFC that are correlated with cognitive function (26). In addition, the reduction of synaptic density and proteins in response to stress and in the PFC of depressed patients is associated with increased expression of a transcriptional repressor of synaptic proteins (16).
Another signaling molecule that is regulated by BDNF-Akt signaling and is implicated in synaptic homeostasis is glygocen synthase kinase 3 (GSK3). GSK3 is involved in synaptic deconsolidation or pruning—the opposite of synaptogenesis—via regulation of glutamate receptor cycling (35). GSK3 is inhibited by Akt and is activated by protein phosphatase 1 (PP1). An excess of GSK3-deconsolidation could contribute to the reduction in spine synapses in response to stress and depression (Fig. 3) (39). Postmortem studies report increased levels of activated GSK3 in MDD or bipolar depression, in support of this hypothesis (39). Levels of GSK3 activity in the nucleus accumbens are also associated with social defeat stress, and blockade of GSK3 in this brain region or in response to systemic administration of a GSK3 inhibitor produces antidepressant effects in behavioral models of depression (39, 40).
Fig. 3.
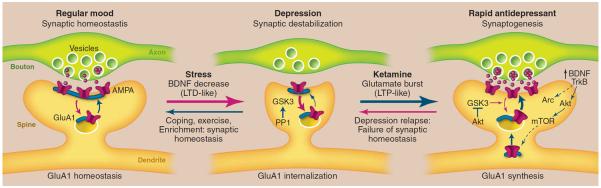
Model depicting the synaptogenic basis of depression and treatment response. (Left) Regular mood (synaptic homeostasis). Under normal conditions, levels of synapse number and function are maintained by homeostatic mechanisms that contribute to regular mood. This includes cycling of glutamate A1 (GluA1) receptors to and from the synapse. (Middle) Depression (synaptic destabilization). Chronic stress exposure decreases synaptic density, similar to the destabilization of spine synapses that occurs under conditions that cause long-term depression (LTD). Neuronal atrophy and synaptic loss are reversible, possibly more rapidly in resilient individuals with coping, exercise, or enriched environment. Stress also decreases BDNF, and BDNF deletion mutant mice exhibit similar synaptic deficits, suggesting that this neurotrophic factor may contribute to the loss of synapses and neuronal atrophy. GSK3, which is increased in depression, can be activated by PP1 and may also contribute to synaptic destabilization by promoting the internalization of GluA1. (Right) Rapid antidepressants (synaptogenesis). Ketamine rapidly increases glutamate transmission and synaptogenesis, similar to LTP. Ketamine-induction of synaptogenesis requires BDNF/TrkB-activation of Akt and mTOR signaling, resulting in increased translation of synaptic proteins, including GluA1, as well as Arc, which is required for the expansion and stabilization of spines. The actions of ketamine are also dependent on inhibition of GSK3, which could occur via stimulation of Akt or by blockade of NMDA receptors and PP1 (not shown in the figure). Depressed patients treated with ketamine relapse after 7 to 10 days, possibly due to failure of synaptic homeostasis, which could result from genetic mutations or environmental factors such as sustained stress.
Antidepressant Regulation of Synaptogenesis and Behavior
In contrast to the effects of stress and depression, typical antidepressants, with chronic treatment, produce a slow increase in neurotrophic factor expression and enhance synaptic plasticity. Moreover, the rapid-acting agent ketamine has fast and dramatic effects on synaptic function and spine density, providing further evidence for the relevance of synaptic alterations in depression and treatment response. Here, we provide a brief overview of typical antidepressants and synaptic plasticity and compare and contrast this with the actions of the rapidly acting drug ketamine.
Typical antidepressant drugs increase neuroplasticity
Evidence that typical antidepressants, such as the highly prescribed SSRIs, could influence synaptic plasticity is provided by electrophysiological studies demonstrating that chronic administration of fluoxetine enhances synaptic transmission and/or long-term potentiation (LTP), a cellular model of learning (41). Recent studies using elegant behavioral models of plasticity also provide evidence that chronic administration of fluoxetine reinstates ocular dominance plasticity in the adult mouse visual cortex and enhances the extinction of conditioned fear, an active learning process (42, 43). BDNF is required for the actions of fluoxetine on synaptic transmission, LTP, ocular dominance plasticity, and extinction training plasticity (41–43). There is also evidence that chronic antidepressant administration can increase spine density (44) or block the atrophy of dendrites and spines caused by chronic stress exposure (45, 46), although the role of BDNF in these effects has not been examined. The behavioral response to antidepressants is blocked in BDNF deletion or BDNF Met knock-in mice (24, 27, 29).
Despite these positive neuroplasticity effects, SSRIs and other currently prescribed antidepressants have substantial limitations, including slow rates of response and modest therapeutic efficacy. These limitations could be due to genetic or environmental factors that increase the severity of depression and/or resistance to treatment in certain individuals. However, these agents target only monoamines, which are modulatory transmitters and would not be expected to produce a robust impact on synaptic transmission, activity-dependent release of BDNF, and synaptogenesis. Agents such as ketamine that produce more profound effects on fast excitatory glutamate transmission and increase BDNF release and synaptogenesis may be required to treat certain types or more severe forms of depression.
Rapid-Acting Antidepressants Increase Synaptogenesis
The recent discovery that ketamine, a N-methyl-D-aspartate (NMDA) receptor antagonist, produces rapid antidepressant responses in treatment-resistant depressed patients addresses the major limitations of available agents (4, 5, 47). A single dose of ketamine alleviates depressive symptoms within hours in patients who have failed to respond to two or more conventional antidepressants, and the effects are sustained for 7 to 10 days. Ketamine is also effective for bipolar depression and decreases suicide ideation (48). The fast, efficacious actions of ketamine by a different mechanism, NMDA receptor blockade, make this an exciting advance for depression therapeutics. The discovery that ketamine rapidly increases the number and function of synaptic connections has focused attention on synaptogenesis as a fundamental process for the treatment of depressive symptoms and also suggests that disruption of synaptogenesis and loss of connections underlies the pathophysiology of depression (Fig. 3).
Ketamine also produces fast antidepressant behavioral responses in rodent models, and these effects are correlated with rapid induction of synaptic proteins (within hours) and the number and function of spine synapses in layer V pyramidal neurons of the PFC (6). Ketamine rapidly reverses the deficit in spine density, as well as the anhedonia and anxiety caused by exposure to chronic stress (3 weeks) (Fig. 2) (7). These effects are preceded by activation of the mammalian target of rapamycin (mTOR) signaling pathway and are blocked by infusions of the selective mTOR inhibitor rapamycin (Fig. 3) (6, 7). The mTOR pathway, which is localized to dendrites and cell bodies and regulates translation, has been linked to activity-dependent, long-term synaptogenesis reliant on protein synthesis (36).
Ketamine rapidly and transiently increases glutamate transmission in the PFC, possibly via inhibition of tonically active GABAergic inter-neurons, and the resulting burst of glutamate is thought to underlie ketamine induction of synapto-genesis (6, 7). In addition, the synaptogenic actions of ketamine are blocked in mice with a knock-in of the BDNF Met allele that impairs BDNF mRNA transport to dendrites, as well as in conditional BDNF mutant mice (32, 49). These findings are consistent with cell culture studies demonstrating that glutamate receptor stimulation of synaptogenesis requires the release of BDNF to stimulate TrkB-mTOR signaling and synaptic protein synthesis (50). Activity-dependent release of BDNF could also explain why ketamine is more rapid and efficacious than conventional antidepressants, which increase the expression but not the release of BDNF. A requirement for the synthesis of BDNF, as well as other synaptic proteins, is supported by evidence that inhibition of elon gation factor 2 kinase and desuppression of translation are required for the actions of ketamine (49).
Ketamine also rapidly increases activity-regulated cytoskeleton-associated protein (Arc) (6), which is required for actin polymerization and stable expansion of dendritic spines (Fig. 3) (51). The actions of ketamine also require inhibition of GSK3 (52). As discussed above, GSK3 could contribute to synaptic deconsolidation and is increased in brains of MDD patients (39). The actions of ketamine may include blockade of NMDA receptors that lead to activation of GSK3 via PP1, thereby causing spine destabilization (35).
The rapid antidepressant actions of ketamine could involve a number of cortical and limbic brain regions, many of which are reported to show alterations of glutamate transmission and synaptic activity. We have found that infusion of rapamycin into the medial PFC, but not the dorsal striatum, blocks the effects of systemic ketamine administration (6). Additional blocking studies, as well as local infusions of ketamine, are needed to further define the brain regions and circuits underlying the rapid actions of ketamine. Also, because the interpretation of preclinical studies is limited by the animal models available, additional humanimaging studies will be needed to confirm the circuitry underlying the clinical response to ketamine.
New rapid-acting therapeutic targets
The psychotomimetic effects and abuse potential limit the development and use of ketamine for the treatment of depression. However, characterization of the mechanisms underlying the actions of ketamine has led to new potential targets for drug envelopment. Basic and clinical studies show that selective NMDA receptor subtype 2B (NR2B) antagonists produce antidepressant behavioral and synaptogenic actions (6, 24). However, the clinical response to NR2B selective agents does not appear to be as rapid as the response to ketamine, and further studies are needed to address this issue, as well as the efficacy of these agents in treatment-resistant patients. Because the actions of ketamine require a burst of glutamate transmission, agents that either increase synaptic levels of glutamate or act at postsynaptic sites to enhance glutamate receptor signaling have been investigated. Blockade of mGluR2/3 receptors, which are located presynaptically and regulate the release of glutamate, produces rapid behavioral responses that require mTOR signaling (53, 54). Positive AMPA receptor potentiating agents produce antidepressant behavioral responses in rodents and stimulate mTOR signaling in cultured cells (15, 50), but further studies are required to determine if the behavioral effects are dependent on mTOR signaling and synaptogenesis.
Inhibition of GSK3, which is required for the actions of ketamine in rodent models (52), could also produce rapid ketamine-like effects or enhance the response to ketamine. Other neurotransmitter systems that indirectly increase glutamate via regulation of tonic firing of GABAergic interneurons may also produce ketamine-like effects. For example, the roles of glutamate, synaptogenesis, and mTOR signaling in the rapid antidepressant actions of scopolamine, a muscarinic receptor antagonist (55), are currently under investigation.
Conclusions
Evidence exists for a central role of homeostatic control of synaptic connections in key mood-related circuits in the etiology and treatment of depression. Further studies are required to determine the effects of stress and antidepressants on synaptogenesis in other circuits and populations of neurons (e.g., anterior cingulate cortex, nucleus accumbens, amygdala) and to uncover the functional importance of synaptic alterations on behavior in models of depression and anxiety, as well as cognition, learning, and memory. These studies will further define the neuronal and synaptic alterations underlying depression and will contribute to the development of safer, more efficacious antidepressants that last longer and act faster.
References
- 1.Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Arch. Gen. Psychiatry. 2005;62:617. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krishnan V, Nestler EJ. Nature. 2008;455:894. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trivedi M, et al. Am. J. Psychol. 2006;163:28. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 4.Berman RM, et al. Biol. Psychiatry. 2000;47:351. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 5.Zarate CA, Jr, et al. Arch. Gen. Psychiatry. 2006;63:856. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 6.Li N, et al. Science. 2010;329:959. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li N, et al. Biol. Psychol. 2011;69:754. [Google Scholar]
- 8.MacQueen G, Frodl T. Mol. Psychiatr. 2011;16:252. doi: 10.1038/mp.2010.80. [DOI] [PubMed] [Google Scholar]
- 9.Price JL, Drevets WC. Neuropsychopharmacology. 2010;35:192. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perlman G, et al. J. Affect. Disord. 2012;139:75. doi: 10.1016/j.jad.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeng LL, et al. Brain. 2012;135:1498. doi: 10.1093/brain/aws059. [DOI] [PubMed] [Google Scholar]
- 12.Rajkowska G, et al. Biol. Psychiatry. 1999;45:1085. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 13.Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. Neuropsychopharmacology. 2007;32:471. doi: 10.1038/sj.npp.1301234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stockmeier CA, et al. Biol. Psychiatry. 2004;56:640. doi: 10.1016/j.biopsych.2004.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanacora G, Zarate CA, Krystal JH, Manji HK. Nat. Rev. Drug Discov. 2008;7:426. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang HJ, et al. Nat. Med. 2012;18:1413. doi: 10.1038/nm.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duric V, et al. Int. J. Neuropsychopharmacol. 2012;1 [Google Scholar]
- 18.Feyissa A, Chandran A, Stockmeier CA, Karolewicz B. Prog. Neuro-Psychopharmcol. Biol. Psychiatry. 2009;33:70. doi: 10.1016/j.pnpbp.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J, et al. J. Affect. Disord. 2012;138:494. doi: 10.1016/j.jad.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 20.Liu R-J, Aghajanian GK. Proc. Natl. Acad. Sci. U.S.A. 2008;105:359. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McEwen BS, Eiland L, Hunter RG, Miller MM. Neuropharmacology. 2012;62:3. doi: 10.1016/j.neuropharm.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison JH, Baxter MG. Nat. Rev. Neurosci. 2012;13:240. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisch AJ, Petrik D. Science. 2012;338:72. doi: 10.1126/science.1222941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duman RS, Voleti B. Trends Neurosci. 2012;35:47. doi: 10.1016/j.tins.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christoffel DJ, et al. J. Neurosci. 2011;31:314. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuen EY, et al. Neuron. 2012;73:962. doi: 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu H, et al. J. Neurosci. 2012;32:4092. doi: 10.1523/JNEUROSCI.5048-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taliaz D, Stall N, Dar DE, Zangen A. Mol. Psychiatr. 2010;15:80. doi: 10.1038/mp.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z-Y, et al. Science. 2006;314:140. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magariños AM, et al. Hippocampus. 2011;21:253. doi: 10.1002/hipo.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiaruttini C, et al. Proc. Natl. Acad. Sci. U.S.A. 2009;106:16481. doi: 10.1073/pnas.0902833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu R, et al. Biol. Psychol. 2012;71:996. [Google Scholar]
- 33.Gatt J, et al. Mol. Psychiatr. 2009;14:681. [Google Scholar]
- 34.Duric V, et al. Nat. Med. 2010;16:1328. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collinridge GL, Peineau S, Howland JG, Wang YT. Nat. Rev. Neurosci. 2010;11:459. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- 36.Hoeffer CA, Klann E. Trends Neurosci. 2010;33:67. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krishnan V, et al. Biol. Psychol. 2008;64:691. [Google Scholar]
- 38.Son H, et al. Proc. Natl. Acad. Sci. U.S.A. 2012;109:11378. [Google Scholar]
- 39.Li X, Jope RS. Neuropsychopharmacology. 2010;35:2143. doi: 10.1038/npp.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkinson MB, et al. J. Neurosci. 2011;31:9084. doi: 10.1523/JNEUROSCI.0039-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bath KG, et al. Neuropsychopharmacology. 2012;37:1297. doi: 10.1038/npp.2011.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karpova NN, et al. Science. 2011;334:1731. doi: 10.1126/science.1214592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maya Vetencourt JF, et al. Science. 2008;320:385. doi: 10.1126/science.1150516. [DOI] [PubMed] [Google Scholar]
- 44.Ampuero E, et al. Neuroscience. 2010;169:98. doi: 10.1016/j.neuroscience.2010.04.035. [DOI] [PubMed] [Google Scholar]
- 45.Bessa J, et al. Mol. Psychiatr. 2009;14:739. doi: 10.1038/mp.2008.119. [DOI] [PubMed] [Google Scholar]
- 46.Magariños AM, McEwen BS. Neuroscience. 1995;69:89. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- 47.Machado-Vieira R, Ibrahim L, Henter ID, Zarate CA., Jr Pharmacol. Biochem. Behav. 2012;100:678. doi: 10.1016/j.pbb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Machado-Vieira R, Ibrahim L, Henter ID, Zarate CA., Jr Pharmacol. Biochem. Behav. 2012;100:678. doi: 10.1016/j.pbb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Autry AE, et al. Nature. 2011;475:91. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jourdi H, et al. J. Neurosci. 2009;29:8688. doi: 10.1523/JNEUROSCI.6078-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bramham CR. Curr. Opin. Neurobiol. 2008b;18:524. doi: 10.1016/j.conb.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 52.Beurel E, Song L, Jope RS. Mol. Psych. 2011;16:1068. doi: 10.1038/mp.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dwyer JM, Lepack AE, Duman RS. Int. J. Neuropsychopharmacol. 2012;15:429. doi: 10.1017/S1461145711001702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koike H, Iijima M, Chaki S. Neuropharmacology. 2011;61:1419. doi: 10.1016/j.neuropharm.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 55.Drevets W, Furey ML. Biol. Psychol. 2010;67:432. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]