

Abstract
There are two known mRNA degradation pathways, 3′ to 5′ and 5′ to 3′. We identified likely pathogenic variants in two genes involved in these two pathways in individuals with intellectual disability. In a large family with multiple branches, we identified biallelic variants in DCPS in three affected individuals; a splice site variant (c.636+1G>A) that results in an in-frame insertion of 45 nucleotides and a missense variant (c.947C>T; p.Thr316Met). DCPS decaps the cap structure generated by 3′ to 5′ exonucleolytic degradation of mRNA. In vitro decapping assays showed an ablation of decapping function for both variants in DCPS. In another family, we identified a homozygous mutation (c.161T>C; p.Phe54Ser) in EDC3 in two affected children. EDC3 stimulates DCP2, which decaps mRNAs at the beginning of the 5′ to 3′ degradation pathway. In vitro decapping assays showed that altered EDC3 is unable to enhance DCP2 decapping at low concentrations and even inhibits DCP2 decapping at high concentration. We show that individuals with biallelic mutations in these genes of seemingly central functions are viable and that these possibly lead to impairment of neurological functions linking mRNA decapping to normal cognition. Our results further affirm an emerging theme linking aberrant mRNA metabolism to neurological defects.
Introduction
Gene expression levels in biological systems are highly regulated to ensure optimal cell function. Hence, RNA levels are specifically regulated by either transcription rates or mRNA stability and degradation. In particular, mRNA degradation provides an important mechanism for rapid response to cellular events (1–3). In mammalian cells, two main exonucleolytic mRNA decay pathways have been described. In the 3′ to 5′ decay pathway, the poly (A) tail is initially removed by deadenylation followed by degradation of the mRNA body from the 3′ end by the exosome exonuclease complex. The remaining monomethylguanosine mRNA cap (m7G cap) structure is subsequently hydrolyzed by the scavenger decapping enzyme [DCPS (MIM610534)] (4,5). The 5′ to 3′ decay pathway also starts with the deadenylation of the poly (A) tail, then the m7G cap is decapped by the decapping enzyme 2 [DCP2 (MIM 609844)] or the NUDT16 decapping enzyme (6), followed by 5′ to 3′ exoribonucleolytic decay of the remaining mRNA by XRN1 (MIM 607994) (7,8). Several proteins are required for efficient DCP2 activity including DCP1A [decapping enzyme 1 A (MIM 607010)], EDC4 [enhancer of mRNA decapping 4 (MIM 606030)], DDX6 [DEAD/H Box 6 (MIM 600326)] and EDC3 [enhancer of mRNA decapping 3 (MIM 609842)]. This decapping complex is conserved in all eukaryotes (9,10). Thus, a change in mRNA degradation rates or specificity is expected to have numerous effects on the expression levels of various pathways and disturb the cellular balance and function.
Here, we report on mutations in DCPS and EDC3 in two families (Family A and Family B, respectively), with autosomal recessive intellectual disability. In vitro biochemical assays support the reduced activities for both DCPS variants and for the EDC3 variant.
Results
In Family A, from the Azad Kashmir region of Pakistan, there are three affected individuals in three different branches (Fig. 1), all born to consanguineous parents. We examined the affected individuals at ages of 21 years (III-2), 17 years (IV-3) and 10 years (IV-7). The pregnancy and delivery were reported to be normal for III-2 and IV-3; however, IV-7 was born premature, in the 28th week of pregnancy. For all three affected persons, motor development milestones were achieved late (III-2 and IV-3) or not achieved at all (IV-7), also all have intellectual disability, ranging from mild to severe [III-2: mild to moderate (IQ: 45–55); IV-3: moderate (IQ: 35–45); IV-7: severe (IQ: <35)]. No clear facial dysmorphic features or abnormalities of skin, hair, joints, vision or hearing were found. A prominent upper jaw is noted in III-2 and IV-3; however, photographs were not available to determine whether this is a family trait. Head circumference is in the normal range for III-2 and IV-3 (56 and 55 cm, respectively) but below the third centile for IV-7 (48 cm) (11) and heights for III-2, IV-3 and IV-7 were 157.5, 147, and 119.5 cm, respectively. For IV-3 and IV-7, this is below the second centile for Pakistani children (12) (no data is available for the Pakistani adult population). Neurological investigation showed normal cranial nerves, pupillary response to light and eye movement. No history of other brain issue or epilepsy was reported by their parents. Individual IV-7 also has muscle hypotonia, motor functions are severely delayed and she is unable to walk. The affected individuals recognize their parents and homes and have a rather shy demeanor but are generally cooperative. III-2 and IV-3 sometimes show aggressive behavior. III-2 can speak in sentences, albeit slurred, and he shows some autistic features. The other two show little or no speech development. Magnetic resonance imaging of the brain of individual IV-3 at the age of 17 was unremarkable (Supplementary Material, Fig. S1).
Figure 1.

Pedigree of Pakistani Family A. Individuals indicated with black shaded symbols are affected with apparent NS-ARID and photographs of affected individuals III-2, IV-3 and IV-7.
Family B, from Syria, includes three healthy and two affected children, born to healthy first-degree cousins (Fig. 2). We examined the affected siblings at ages of 12 (IV-2) and 7 years (IV-4). Pregnancy, delivery and birth parameters of both children were reported to be normal by the parents and the midwife. Both children developed unremarkably in the first year of life. Then, a slower cognitive development in comparison to their healthy siblings was observed. Nevertheless, motor development was unremarkable. At the time of examination, both children present with mild intellectual disability with an estimated IQ of 50–70. Sleeping pattern is unremarkable, and neither child has epilepsy. Further, IV-4 has sensory hearing loss (right ear: 35–40 dB; left ear: not measurable). Wearing a hearing aid has not improved his language and pronunciation. He also has sectoral heterochromia iridum. Other than this, the affected children have no dysmorphological facial features (Fig. 2). At the time of examination, IV-2 had normal height (145 cm at age of 12 years, −0.7 SD) and a reduced head circumference (50.5 cm, −3 SD), and IV-4 had normal growth parameters and a reduced head circumference (48 cm, −3.6 SD). The healthy parents were in the lower normal spectrum regarding height and head circumferences (father 54 cm, mother 52 cm).
Figure 2.

Pedigree of Syrian family B. Individuals indicated with black shaded symbols are affected with apparent NS-ARID and pictures of the affected persons: There are no significant facial characteristics of the affected individuals, the affected boy has heterochromia iridum and wears a hearing aid. Both show mild ID.
For Family A, 500K SNP microarray genotyping (Affymetrix) was performed for the affected individuals III-2 and IV-3 and unaffected individual III-3. Pathogenic copy number variants were excluded using dChip (13). Positional mapping was conducted using dChip (http://www.hsph.harvard.edu/cli/complab/dchip/) and HomozygosityMapper (http://www.homozygositymapper.org) software (14,15) under the hypothesis of a founder mutation. Microsatellite markers were used to corroborate autozygosity, including in the right-hand branch of the pedigree (i.e. in IV-7). No common region of homozygosity could be identified for all affected individuals, but we identified a single candidate region of 11.3 MB on chromosome 11 that is shared by individuals III-2 and IV-3. Exome sequencing was performed using DNA from individual III-2. Details on sequencing procedure and bioinformatics processing are described elsewhere (16). Whole-exome sequencing (WES) resulted in an average of 91-fold read coverage. 93.75% of the target sequences were covered with a depth of at least 20×, and 97.97% were covered with a depth of at least 10×. Filtering revealed one homozygous candidate mutation in the decapping scavenger gene, DCPS (NM_014026.3): chr11:126 208 295G>A;c.636+1G>A. This variant is highly conserved, predicted to prevent correct splicing (17), and is not reported in the Exome Aggregation Consortium browser (ExAC; Cambridge, MA, USA; http://exac.broadinstitute.org; Dec 2014). A G to T at this position, however, is reported in 1 out of 126 460 alleles. Reverse transcription (RT)–PCR followed by Sanger sequencing revealed a shift to a cryptic splice site 45 bp downstream from the canonical splice donor with correct splicing to exon 5. This maintains the reading frame and results in the addition of 15 amino acids (5′ Val-Lys-Val-Ser-Gly-Trp-Asn-Val-Leu-Ile-Ser-Gly-His-Pro-Ala 3′) to the DCPS protein (Supplementary Material, Fig. S2). Affected individuals III-2 and IV-3 are homozygous for this variant, while individual IV-7 is heterozygous. We thus sequenced all coding exons of DCPS using Sanger sequencing and identified in individual IV-7 a second heterozygous candidate mutation in DCPS (NM_014026.3): chr11:126 215 441C>T; c.947C>T; p.Thr316Met. This variant was genotyped across the family, and only the mother (III-11) was identified with this allele (heterozygous). This variant (rs137941190) is conserved, is predicted to be deleterious (17–19) and is present in the ExAC browser at a low frequency of 0.001263.
For Family B, SNP microarrays (Affymetrix SNP Chip 6.0) were run for both affected individuals and their healthy siblings, and pathogenic copy number variants were excluded using Affymetrix Genotyping Console Software (Version 3.0.2). Positional mapping was performed using HomozygosityMapper (15) under the hypothesis of a founder mutation as described in detail in a previous study (20). Mapping for Family B revealed two candidate regions of a total length of 33 Mb on chromosomes 15 and 19. Exome sequencing was performed using DNA from individual IV-2, as described elsewhere (21). WES resulted in an average of 43.2-fold read coverage. 57.1% of the target sequences were covered with a depth of at least 20× and 67.4% were covered with a depth of at least 10×. The candidate mutation in EDC3 (NM_025083.3): chr15:74 967 305A>G;c.161T>C; p.Phe54Ser on chromosome 15q24.1 was the only variant located in a candidate region, is highly conserved is and predicted in silico to be damaging (17–19). It is not reported in the ExAC browser or in >700 in-house exomes. As the coverage was low, we repeated sequencing of the coding elements (CDS) in the candidate regions (496 genes) by a custom targeted sequencing using DNA from individual IV-4. We achieved a mean coverage of 299 reads, at least 20× coverage for 80.8% of the target sequence and at least 10× for 84.6%. Combining reads from exome and CDS custom targeted sequencing, 85.4% of the CDS target region was covered at least 10× in at least one of the siblings. No additional candidate variants were identified using this approach.
To gain further information on potential pathogenic effect of the identified variants, we performed molecular modeling using Modeler 9.9 (22). In the altered DCPS protein carrying a 15-residue (DCPSIns15) insertion, the loop connecting the amino acids lysine (p.Lys207) and tyrosine (p.Tyr217) that build the active decapping site (23) is extended (Supplementary Material, Fig. S3A). As a consequence, the active site becomes distorted and the long loop also interferes with the DCPS homodimer interface. Therefore, we expect the dimer to have reduced stability and enzymatic activity. Molecular modeling of the homodimeric DCPS also predicted that p.Thr316 forms stabilizing water-mediated interactions with the glutamine at position p.314 and with the glutamate at position p.303 (Supplementary Material, Fig. S3C). These polar interactions can no longer be formed in altered DCPS with a methionine at position p.316; thus, DCPS homodimer stability may be decreased. Molecular modeling of EDC3 with the amino acid serine at position p.54 instead of phenylalanine predicted that the substitution at p.54, which is located within the LSm domain, leads to a disruption of the domain structure due to a poorly packed hydrophobic core. Energetic calculations indicate a loss of 4–5 kcal/mol of stabilization energy for p.Ser54, suggesting that the domain might entirely unfold (Supplementary Material, Fig. S4). Since the LSm domain is crucial for the interaction between EDC3 and DCP2, we anticipate that this variant will significantly impair protein function.
Functional decapping assays were carried out to test whether the respective mutations adversely affect decapping. The scavenger decapping activity of recombinant proteins corresponding to DCPS wild-type (DCPSWT), DCPS with the 15 amino acid insertion (DCPSIns15) and DCPS with the p.Thr316Met substitution (DCPST316M) were tested. Decapping activity from the DCPSIns15 and DCPST316M variants was almost completely ablated relative to DCPSWT (Fig. 3A). Similarly, proteins extracted from lymphoblastoid cell lines from affected individuals homozygous for the splice site variant (III-2 and IV-3) were assayed and shown to have significantly reduced or no detectable mRNA decapping activity in comparison to their unaffected heterozygous siblings (III-3 and IV-2) (Fig. 3B). RT–PCR of total cellular RNA derived from the lymphoblastoid cells revealed the presence of mRNA corresponding to DCPSIns15 allele in the two homozygous cells and DCPSWT and DCPSIns15 alleles in the III-3 heterozygous cells (Fig. 3C). Western blot analysis of lymphoblastoid cell lysates confirmed the generation of DCPSIns15 protein in the homozygous individuals with the complete absence of DCPSWT protein and, most importantly, also revealed greatly reduced steady-state levels of DCPSIns15 in both homozygous and heterozygous cells (Fig. 3D).
Figure 3.

Functional analysis of DCPS variants. (A) DCPS in vitro decapping assay. mRNA decapping was assayed using recombinant full-length DCPS wild-type (DCPSWT) or altered [splice mutation/45 bp insertion (DCPSIns15) and p.Thr316Met(DCPST316M)] DCPS protein in pET28a (Novagen, EMD Biosciences) vector expressed and purified from Escherichia coli BL21(DE3) cells. Migration of the input cap structure (m7GpppG) or m7GMP product (m7Gp) is designated, with the red ‘p’ denoting the location of the 32P. The percent decapping is indicated at the bottom of the panel. (B) DCPS activity in lymphoblastoid cell lines. Lymphoblastoid cell lines from homozygous affected (III-2 and IV-3) and heterozygous unaffected (III-3 and IV-2) family members were used for decapping assays. Decapping products and labeling are as in (A). (C) RT–PCR analysis for DCPS. RT–PCR across the DCPS splice mutation (from exons 3 to 6) was performed using mRNA from patients (III-2 and IV-3) and unaffected family member (III-3), plus cartoon indicating exon usage. (D) Western Blot of DCPS. Wild-type and variant DCPS protein from the indicated lymphoblast cell lines were detected by western blotting to demonstrate migration of the variant DCPS protein in homozygous and heterozygous individuals. A wild-type lymphoblastoid cell line is shown to indicate the position of wild-type DCPS.
We also assayed the propensity of EDC3 wild-type (EDC3WT) and EDC3 with the p.Phe54Ser variant (EDC3F54S) to stimulate DCP2 decapping in vitro. As expected, EDC3WTenhanced DCP2 decapping activity by 2-fold while the same was not observed with EDC3F54S. EDC3F54S even demonstrated inhibitory activity at higher concentrations (Fig. 4). This suggests that DCP2 decapping is impaired in affected individuals.
Figure 4.
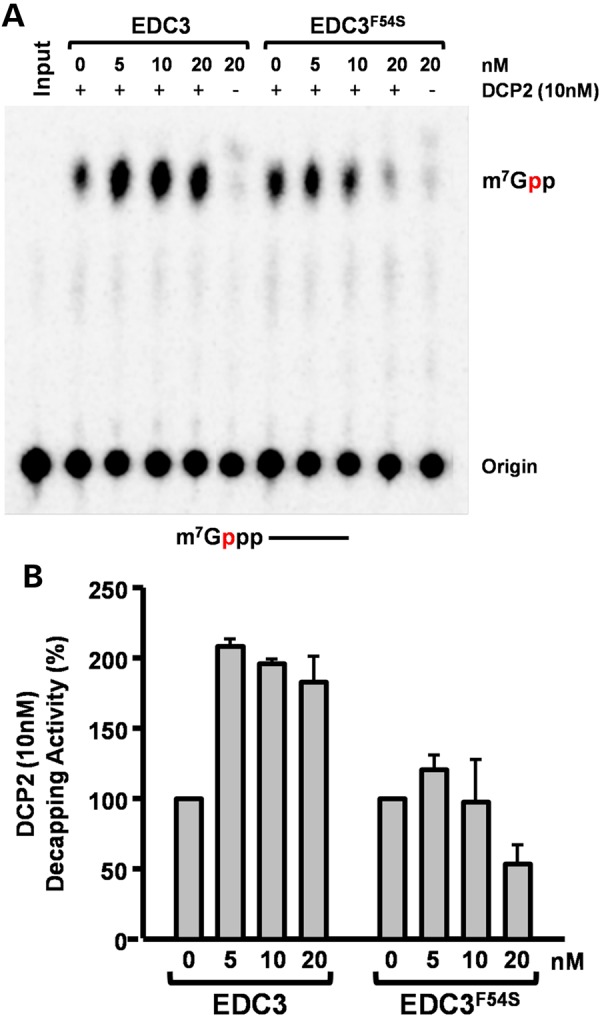
Functional analysis of EDC3 variant. EDC3-DCP2 decapping assay. Wild-type EDC3 enhances DCP2-mediated decapping in vitro, while a similar enhancement was not observed with the Phe54Ser variant (EDC3F54S), and inhibition of decapping was detected at the highest concentration tested. Decapping assays were carried out with 32P-cap-labeled RNA with 10 nm DCP2 and the indicated amounts of EDC3 proteins as previously described (24). (A) A representative decapping assay is shown and labeled as in the legend to Figure 3A. A schematic of the substrate with the m7Gppp denoting the cap and the line representing the RNA is shown at the bottom. (B) The average of three independent decapping assays in the presence of the different EDC3 proteins is shown with error bars representing ±standard deviation.
Discussion
Taken together, in two families with a non-specific form of intellectual disability, we identified biallelic variants in DCPS or EDC3. In silico parameters and molecular modeling suggested deleterious effects of all variants. Functional analyses validated this by revealing an impairment of protein function.
In cells, a disruption of DCPS decapping activity is expected to lead to the accumulation of m7GpppN cap structures, which can sequester the nuclear cap binding protein and decrease the efficiency of first intron splicing (25) and potentially alter cytoplasmic mRNA translation by sequestering the cytoplasmic cap-binding protein (26). In addition, at least in Saccharomyces cerevisiae and Caenorhabditis elegans, DCPS orthologs contribute to Xrn1-mediated exonucleolytic decay (27–29). Whether DCPS fulfills a similar role in mammalian cells remains to be determined. Genomic mRNA and proteomic analysis of the individuals expressing homozygous DCPSIns15 will enable us to uncover the underlying molecular defect attributed to expression of this variant protein.
In an effort to generate a Dcps null mouse, we found that Dcps is an essential gene and its deletion is embryonic lethal (M. Kiledjian, unpublished observations), demonstrating a role for Dcps protein in embryogenesis. CRISPR screens of human genes also indicate the requirement of DCPS for cellular viability (30), underlining the importance of this protein in fundamental cellular functions. The viability of individuals with a decapping-compromised DCPS suggests that the decapping activity of DCPS is only important for normal cognition, while an additional decapping-independent function of DCPS may also be required for normal development and survival. However, this additional function is yet to be determined. An alternative explanation is that, while low levels of mRNA decapping are important in all tissues, neural development and/or function may be more sensitive to disruption of decapping activity than other tissues. Further, we consider it unlikely that fully null homozygous mutations in DCPS will be identified in a clinical context and that bi-allelic mutations would always be hypomorphic or with restricted effect on specific isoforms or functions of the protein.
EDC3 interacts with and stimulates DCP2 (31). The identified alteration in EDC3 is situated in the interacting LSm domain and, thus, possibly disturbs this interaction. Our functional analyses showed that the altered EDC3 does not enhance DCP2 activity and at high concentration even inhibits DCP2 activity. Therefore, EDC3F54S is compromised in its ability to stimulate DCP2 decapping in the affected individuals, leading to aberrant accumulation of a subset of mRNAs. This may have adverse consequences on normal neural function, and this dysregulation of mRNA is a possible cause of intellectual disability in Family B.
The individuals examined in this report have intellectual disability as a common phenotype. Nevertheless, there are some significant differences in further details. Individual IV-7 of Family A with the compound heterozygote DCPS mutation also presents with a neuromuscular phenotype with extremely delayed motor development. Although this may be a result of the different genotype, it is also possible that prematurity (birth in the 28th pregnancy week) is a contributory factor for this phenotypic variability. It cannot be excluded that a variant in an intronic or regulatory region at DCPS, or an independent mutation in another gene may also play a role in this individual.
The individuals with EDC3 mutations in Family B present with a milder form of intellectual disability than individuals in Family A. Nevertheless, the phenotypes of the patients with variants in EDC3 and in DCPS are rather similar, although DCPS and EDC3 are involved in different pathways of RNA decapping and at different stages of RNA degradation.
The role of both genes in mRNA degradation and thus regulation of gene expression suggests DCPS and EDC3 as excellent candidate genes for a neurodevelopmental phenotype. Regulation of gene expression is crucial for neuronal development and function. As an example, this has been shown in mutations in the transcription factor TAF2 (32). Further regulators of mRNA stability have been implicated in cognitive function. Mutations in VCX3A(MIM 300533), a gene associated with the inhibition of mRNA decapping (33), were reported to segregate with X-linked non-syndromic intellectual disability (34). Although a more recent study of X-linked ichthyosis [XLI (MIM 308100)] and intellectual disability suggests that deletion of VCX3A alone is insufficient to result in intellectual disability and may involve a dosage compensation of VCX paralogs (35), VCX3A is involved in neuritogenesis and is an inhibitor of DCP2 decapping (33). Consistent with a role for decapping regulation in neuronal function, enhanced mRNA decapping induced by silencing of VCX3A results in decreased primary and secondary neurite projections, while exogenous expression of VCX3A in rat primary hippocampal neurons promotes neurite arborization (33). A role for proper mRNA turnover in cognitive function is further evident in individuals with mutations in the non-sense-mediated mRNA decay pathway gene, UPF3B (MIM 300298), a protein involved in mRNA quality control (36).
Although at present we only have identified disruptions in either DCPS or EDC3 in two families, the presence of compound heterozygous variants in DCPS in an affected person in Family A supports a role for DCPS as the causative agent for intellectual disability in these individuals. Moreover, the identification of a second family, of Jordanian origin, with intellectual disability caused by a homozygous mutation in DCPS, leading to truncation of a major isoform and missense change in a minor isoform, reinforces the significance of DCPS for neuronal function (C. Ng et al., this issue). The presence of obvious physical anomalies and neuromuscular phenotype in the Jordanian family suggests that a relatively wide clinical spectrum can be anticipated for different types and locations of mutations in DCPS. Future identification of additional individuals with intellectual disability and disruptions in DCPS, EDC3 or other components of the decapping machinery would further substantiate a role for these genes in cognitive ability.
In conclusion, we report the investigation of two families with apparent non-specific autosomal recessive intellectual disability, leading to the identification of mutations in DCPS and EDC3. Both genes are involved in RNA metabolism and function in either mRNA scavenger decapping or mRNA decapping. At present, the molecular consequence of defects in these genes is not known, yet they share a physiological consequence of adverse overall cognition in individuals harboring the mutant alleles. Future identification of the downstream target mRNAs or proteins altered in the cells of affected individuals will provide insights into the contribution of DCPS and EDC3 in gene expression and cognitive function.
Materials and Methods
Patient ascertainment and clinical information
This study was approved by the Ethics Committees of the Universities of Bonn and Erlangen-Nürnberg in Germany, the Centre for Addiction and Mental Health in Toronto, Canada, and the National University of Science and Technology, Islamabad, Pakistan. Informed written consent was obtained from all examined persons before obtaining blood samples. Blood was drawn for genetic studies, and genomic DNA was extracted by standard methods. Neurological and medical assessment, as well as assessment of the level of ID, was performed on affected members by a team of experienced clinicians. For Family A, this involved assessment by a clinical psychologist experienced in assessment of intellectual disability in this culture (T.N.) using the Vineland Adaptive Behaviour Scale (second edition) (37). Photographs of family members were assessed by an experienced clinical geneticist for dysmorphic features.
Homozygosity-by-descent mapping
For Family A, SNP microarrays (Affymetrix 500K NspI, Santa Clara, CA, USA) were run for affected individuals III-2 and IV-3, and an unaffected individual III-3. Microarray genotyping was performed as a service by the London Regional Genome Centre (www.lrgc.ca). Genotypes were analyzed for HBD regions using dCHIP software (14) and HomozygosityMapper (15). Microsatellite markers were used to corroborate autozygosity, and the 11.3 Mb region on 11q24.1-q25 was the sole locus not excluded using this approach.
For Family B, SNP microarrays (Affymetrix SNP Chip 6.0, Santa Clara, CA, USA) were run for all affected and their healthy siblings. HomozygosityMapper (15) was used for homozygosity mapping.
WES and targeted sequencing
For Family A, WES was performed using the SOLiD 4 (Life Technologies, Carlsbad, CA, USA) platform and SureSelect Human All Exon 50 Mb (Agilent technologies, Santa Clara, CA, USA) DNA capture array, according to the manufacturers' recommended protocols. Details on the sequencing procedures and bioinformatics processing are described elsewhere (16). Sanger sequencing was used to confirm a homozygous variant identified as a potential splice site mutation in DCPS and its segregation in the family. Both III-2 and IV-3 were homozygous for the variant. No other homozygotes were identified in the family. Individual IV-7 was compound heterozygous for this variant and the Thr316Met variant.
For Family B, exome sequencing was performed using DNA from individual IV-2. DNA was enriched using the SureSelect Human All Exon 50Mb Kit (Agilent technologies) and paired-end sequenced on a SOLiD 4 instrument (Life Technologies). Custom targeted enrichment was performed using a custom targeted capture kit (Agilent technologies) for all coding elements (CDS) in the candidate regions (496 genes) using DNA from individual IV-4. The library was then sequenced on a SOLiD 4 platform. Details on sequencing procedure and bioinformatics processing are described elsewhere (21). PCR and Sanger sequencing were done according to standard protocols to exclude technical artifacts and to test for segregation.
mRNA analysis for DCPS
Whole blood mRNA from members of Family A was extracted from blood collected in Tempus™ RNA tubes (Life Technologies) using TRIzol. After first-strand cDNA synthesis using SuperScript III Reverse Transcriptase (RT) (Invitrogen, Carlsbad, CA, USA) and random hexamers (Fermentas, Waltham, MA, USA), the DCPS gene was amplified by RT–PCR. Primers from exon 2 to 5 were used: 5′-CACTTGTTCCCTCCAAGACAA-3′ (2F), 5′-TAAGGTCGCGTAGGGATCTG-3′ (5R), to generate a 352 bp amplicon. Sanger sequencing was used to confirm a change in splice donor usage at exon 4/intron 4 for the splice variant allele.
Molecular modeling for the mutations in DCPS and EDC3
Modeling of the mutations was based on the Dcps crystal structure (38). The p.T316M mutation was generated with SwissModel (39), and the p.Q212_L213Ins15 mutation was modeled with ModLoop (40). RasMol (41) was used for structure analysis and visualization. ClustalW2 comparison of DCPS Thr316 across species shows conservation through most vertebrates, also to archaeo-chordate Branchiostoma floridae (lancelet), also fruit fly and nematode (see Supplementary Material). Molecular modeling using GeneSilico fold recognition meta-server (42) and Modeler9.9 (22) using the closest related hydrolase (PDB code: 3LP5) as template highlighted the detrimental effect of the exchange at position 54 on the structure of EDC3 (Fig. 3). Phenylalanine 54 is located within the LSm domain and is highly conserved across evolution. ClustalW2 comparison of EDC3 54Phe across species shows it to be present across vertebrates all the way to fish, but not including lancelet (see Supplementary Material). The amino acid substitution at position p.54 leads to a disruption of the domain structure due to a poorly packed hydrophobic core. Energetic calculations indicate a loss of 4–5 kcal/mol of stabilization energy, suggesting that the domain might entirely unfold upon Phe54Ser mutation. Since the LSm domain is crucial for the interaction between EDC3 and DCP2, we anticipate that this variant will significantly impair protein function.
DCPS mRNA decapping assay
The DCPS gene was amplified from affected and unaffected individuals from Family A by RT–PCR. PCR products were purified after gel electrophoresis followed by elution from the gel using the QIAquick Gel Extraction Kit (Qiagen, Venlo, Netherlands). The full-length wild-type (DCPSWT), and mutant (splice mutation/45 bp insertion: DCPSIns15) and Thr316Met (DCPST316M) DCPS cDNAs were initially cloned into the pDRIVE vector (Qiagen) using primers 5′-ACCACAACGGGGCCAAAGGCAGTAAC-3′ (forward) and 5′-ACTCCTCCCCCAATCCCACACATCTG-3′ (reverse). PCR amplification of the full-length inserts with primers with BamHI and XhoI restriction sites was performed prior to subcloning into the corresponding restriction enzyme sites within vector pET28a (Novagen, EMD Biosciences, Danvers MA, USA) and confirmed by sequencing. Recombinant proteins expressed from the pET28a constructs were generated in Escherichia coli BL21(DE3) cells induced with 0.5 mm IPTG and purified according to the manufacturer (Novagen, EMD Biosciences), except that 300 mm urea and 0.5% TritonX-100 was included in the binding buffer. The nickel column was then washed twice using wash buffer with 800 mm urea and 0.5% TritonX-100 and once using wash buffer without urea and TritonX-100. Protein eluted from the nickel column was dialyzed against PBS and concentrated by Centricon centrifugal filter columns (Amicon, EMD Biosciences, Danvers MA) and stored at −80°C in 10% glycerol. Protein concentrations were determined by Bradford assay. Generation of labeled RNA and cap structures: unlabeled, uncapped RNA corresponding to the pcDNA3 polylinker spanning the SP6 and T7 promoters (pcP) was generated from SP6 RNA polymerase transcribed RNA, 32P-cap labeled and cap structure isolated as previously described (24). DCPS wild-type and mutant proteins at the indicated concentrations were incubated with 10 fmol of 32P-labeled methylated-capped RNA (32P m7Gp*ppG) substrate and decapping products resolved by polyethyleneimine (PEI) cellulose thin-layer chromatography (TLC) (Sigma-Aldrich, St Louis, MO, USA), as described previously (24). This buffer condition separated the input cap structure from the resulting m7GMP generated by the DCPS decapping activity.
DCPS in vitro decapping assays
Lymphoblast cell lines were established from whole blood from homozygous affected (III-2 and IV-3) and heterozygous unaffected (III-3 and IV-2) family members by Epstein-Barr virus transformation. Decapping assays were carried out with 5 µg total cell extract derived from family member lymphoblasts. Proteins or extract were incubated with the labeled cap structures in decapping buffer (10 mm Tris–HCl at pH 7.5, 100 mm KCl, 2 mm MgCl2, 2 mm DTT, 0.5 mm MnCl2). Decapping reactions were carried out for 5 min (for recombinant proteins) or 10, 20, 30 min at 37°C and stopped by extracting once with phenol : chloroform (1:1). Decapping products were resolved by PEI-cellulose TLC plates (Sigma-Aldrich) and developed with 0.45 m (NH4)2SO4 in a TLC chamber at room temperature. The TLC plates were air-dried and exposed to Storm PhosphorImager (Molecular Dynamics, GE Healthcare Life Sciences, Little Chalfont, UK) for quantitation.
Western blotting of DCPS from protein extracted from lymphoblast cell lines from Family A
Western blot analysis was carried out with 120 µg cell extract resolved on 12.5% sodium dodecyl sulphate–polyacrylamide gel electrophoresis. The DCPS proteins were detected with affinity purified rabbit antibodies directed to DCPS (1:200) (43) and visualized using secondary antibodies coupled to horse radish peroxidase (Jackson Immuno Research) and ECL chemiluminescence (GE Healthcare Life Sciences).
EDC3-DCP2 in vitro decapping assays
Full-length EDC3 was amplified from wild-type cDNA using RT–PCR and cloned into a TOPO-PCR2.1 vector (Life technologies). Subcloning into a pET28a vector (Novagen) was conducted using the restriction enzymes HindIII and SacI, and correct cloning was confirmed using Sanger sequencing. Recombinant proteins were expressed and purified as indicated above, and decapping assays were carried out with32P-cap-labeled RNA with 10 nm DCP2 and the indicated amounts of EDC3 proteins as previously described (24).
Supplementary Material
Funding
This research was supported by a grant from the Canadian Institutes of Health Research (#MOP-102758) to J.B.V., grants from the Deutsche Forschungsgemeinschaft (DFG, AB393/2-2) and Erlanger Leistungsbezogene Anschubsfinanzierung und Nachwuchsförderung (ELAN; 13-05-10-1-Abou Jamra) to R.A.J., and NIH grant GM067005 to M.K.
Supplementary Material
Acknowledgements
We wish to thank the family members for their willing participation and cooperation with this study. We thank Arif Ekici for support with the SNP arrays and NGS; Farah Radwan for excellent technical assistance. We also thank Dr Dan Doherty and Dr Gisele Ishak for assistance interpreting MRI data.
Conflict of Interest statement. None declared.
References
- 1.Adjibade P., Mazroui R. (2014) Control of mRNA turnover: implication of cytoplasmic RNA granules. Semin. Cell Dev. Biol., 34, 15–23. [DOI] [PubMed] [Google Scholar]
- 2.Garneau N.L., Wilusz J., Wilusz C.J. (2007) The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol., 8, 113–126. [DOI] [PubMed] [Google Scholar]
- 3.Wu X., Brewer G. (2012) The regulation of mRNA stability in mammalian cells: 2.0. Gene, 500, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu H., Kiledjian M. (2006) Decapping the message: a beginning or an end. Biochem. Soc. Trans., 34, 35–38. [DOI] [PubMed] [Google Scholar]
- 5.Milac A.L., Bojarska E., Wypijewska del Nogal A. (2014) Decapping Scavenger (DcpS) enzyme: advances in its structure, activity and roles in the cap-dependent mRNA metabolism. Biochim. Biophys. Acta, 1839, 452–462. [DOI] [PubMed] [Google Scholar]
- 6.Song M.G., Li Y., Kiledjian M. (2010) Multiple mRNA decapping enzymes in mammalian cells. Mol. Cell, 40, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arribas-Layton M., Wu D., Lykke-Andersen J., Song H. (2013) Structural and functional control of the eukaryotic mRNA decapping machinery. Biochim. Biophys. Acta, 1829, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y., Kiledjian M. (2010) Regulation of mRNA decapping. Wiley Interdiscip. Rev. RNA, 1, 253–265. [DOI] [PubMed] [Google Scholar]
- 9.Coller J., Parker R. (2005) General translational repression by activators of mRNA decapping. Cell, 122, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fenger-Gron M., Fillman C., Norrild B., Lykke-Andersen J. (2005) Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol. Cell, 20, 905–915. [DOI] [PubMed] [Google Scholar]
- 11.Tanner J.M. (1978) In Forfar J.O., Arneil G.C. (eds), Textbook of Pediatrics. Churchill-Livingstone, Edinburgh, UK, pp. 253–303. [Google Scholar]
- 12.Kelly A.M., Shaw N.J., Thomas A.M., Pynsent P.B., Baker D.J. (1997) Growth of Pakistani children in relation to the 1990 growth standards. Arch. Dis. Child., 77, 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X., Li C., Paez J.G., Chin K., Janne P.A., Chen T.H., Girard L., Minna J., Christiani D., Leo C., et al. (2004) An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res., 64, 3060–3071. [DOI] [PubMed] [Google Scholar]
- 14.Lin M., Wei L.J., Sellers W.R., Lieberfarb M., Wong W.H., Li C. (2004) dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics, 20, 1233–1240. [DOI] [PubMed] [Google Scholar]
- 15.Seelow D., Schuelke M., Hildebrandt F., Nurnberg P. (2009) HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic Acids Res., 37, W593–W599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mir A., Sritharan K., Mittal K., Vasli N., Araujo C., Jamil T., Rafiq M.A., Anwar Z., Mikhailov A., Rauf S., et al. (2014) Truncation of the E3 ubiquitin ligase component FBXO31 causes non-syndromic autosomal recessive intellectual disability in a Pakistani family. Hum. Genet., 133, 975–984. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz J.M., Cooper D.N., Schuelke M., Seelow D. (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods, 11, 361–362. [DOI] [PubMed] [Google Scholar]
- 18.Ng P.C., Henikoff S. (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abou Jamra R., Wohlfart S., Zweier M., Uebe S., Priebe L., Ekici A., Giesebrecht S., Abboud A., Al Khateeb M.A., Fakher M., et al. (2011) Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur. J. Hum. Genet., 19, 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murakami Y., Tawamie H., Maeda Y., Buttner C., Buchert R., Radwan F., Schaffer S., Sticht H., Aigner M., Reis A., et al. (2014) Null mutation in PGAP1 impairing Gpi-anchor maturation in patients with intellectual disability and encephalopathy. PLoS Genet., 10, e1004320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez R., Sali A. (2000) Comparative protein structure modeling. Introduction and practical examples with modeller. Methods Mol. Biol., 143, 97–129. [DOI] [PubMed] [Google Scholar]
- 23.Gu M., Fabrega C., Liu S.W., Liu H., Kiledjian M., Lima C.D. (2004) Insights into the structure, mechanism, and regulation of scavenger mRNA decapping activity. Mol. Cell, 14, 67–80. [DOI] [PubMed] [Google Scholar]
- 24.Liu S.W., Jiao X., Welch S., Kiledjian M. (2008) Analysis of mRNA decapping. Methods Enzymol., 448, 3–21. [DOI] [PubMed] [Google Scholar]
- 25.Shen V., Liu H., Liu S.W., Jiao X., Kiledjian M. (2008) DcpS scavenger decapping enzyme can modulate pre-mRNA splicing. RNA, 14, 1132–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bail S., Kiledjian M. (2008) DcpS, a general modulator of cap-binding protein-dependent processes? RNA Biol., 5, 216–219. [DOI] [PubMed] [Google Scholar]
- 27.Bosse G.D., Ruegger S., Ow M.C., Vasquez-Rifo A., Rondeau E.L., Ambros V.R., Grosshans H., Simard M.J. (2013) The decapping scavenger enzyme DCS-1 controls microRNA levels in Caenorhabditis elegans. Mol. Cell, 50, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H., Kiledjian M. (2005) Scavenger decapping activity facilitates 5′ to 3′ mRNA decay. Mol. Cell. Biol., 25, 9764–9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinturel F., Brechemier-Baey D., Kiledjian M., Condon C., Benard L. (2012) Activation of 5′-3′ exoribonuclease Xrn1 by cofactor Dcs1 is essential for mitochondrial function in yeast. Proc. Natl Acad. Sci. USA, 109, 8264–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G., et al. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Decker C.J., Teixeira D., Parker R. (2007) Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J. Cell Biol., 179, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hellman-Aharony S., Smirin-Yosef P., Halevy A., Pasmanik-Chor M., Yeheskel A., Har-Zahav A., Maya I., Straussberg R., Dahary D., Haviv A., et al. (2013) Microcephaly thin corpus callosum intellectual disability syndrome caused by mutated TAF2. Pediatr. Neurol., 49, 411–416 e411. [DOI] [PubMed] [Google Scholar]
- 33.Jiao X., Chen H., Chen J., Herrup K., Firestein B.L., Kiledjian M. (2009) Modulation of neuritogenesis by a protein implicated in X-linked mental retardation. J. Neurosci., 29, 12419–12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukami M., Kirsch S., Schiller S., Richter A., Benes V., Franco B., Muroya K., Rao E., Merker S., Niesler B., et al. (2000) A member of a gene family on Xp22.3, VCX-A, is deleted in patients with X-linked nonspecific mental retardation. Am. J. Hum. Genet., 67, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Esch H., Hollanders K., Badisco L., Melotte C., Van Hummelen P., Vermeesch J.R., Devriendt K., Fryns J.P., Marynen P., Froyen G. (2005) Deletion of VCX-A due to NAHR plays a major role in the occurrence of mental retardation in patients with X-linked ichthyosis. Hum. Mol. Genet., 14, 1795–1803. [DOI] [PubMed] [Google Scholar]
- 36.Tarpey P.S., Raymond F.L., Nguyen L.S., Rodriguez J., Hackett A., Vandeleur L., Smith R., Shoubridge C., Edkins S., Stevens C., et al. (2007) Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet., 39, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sparrow S.S., Cicchetti D.V., Balla D.A. (2008) Vineland Adaptive Behavior Scales - Second Edition - Expanded Interview Form - Vineland-II. Pearson Assessments, Livonia, MN. [Google Scholar]
- 38.Chen N., Walsh M.A., Liu Y., Parker R., Song H. (2005) Crystal structures of human DcpS in ligand-free and m7GDP-bound forms suggest a dynamic mechanism for scavenger mRNA decapping. J. Mol. Biol., 347, 707–718. [DOI] [PubMed] [Google Scholar]
- 39.Guex N., Peitsch M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 40.Fiser A., Sali A. (2003) ModLoop: automated modeling of loops in protein structures. Bioinformatics, 19, 2500–2501. [DOI] [PubMed] [Google Scholar]
- 41.Sayle R.A., Milner-White E.J. (1995) RASMOL: biomolecular graphics for all. Trends Biochem. Sci., 20, 374. [DOI] [PubMed] [Google Scholar]
- 42.Kozlowski L.P., Bujnicki J.M. (2012) MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinformatics, 13, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu H., Rodgers N.D., Jiao X., Kiledjian M. (2002) The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J., 21, 4699–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.