

Abstract
Mutations in the ABCA4 gene are a common cause of autosomal recessive retinal degeneration. All mouse models to date are based on knockouts of Abca4, even though the disease is often caused by missense mutations such as the complex allele L541P;A1038V (PV). We now show that the PV mutation causes severe human disease whereas the V mutation alone causes mild disease. Mutant ABCA4 proteins expressed heterologously in mammalian cells retained normal cellular localization. However, basal and all-trans-retinal-stimulated ATPase activities were reduced substantially for P and PV but only mildly for V. Electron microscopy revealed marked structural changes and misfolding for the P and PV mutants but few changes for the V mutant, consistent with the disease severity difference in patients. We generated Abca4PV/PV knock-in mice homozygous for the complex PV allele to investigate the effects of this misfolding mutation in vivo. Mutant ABCA4 RNA levels approximated WT ABCA4 RNA levels but, surprisingly, only trace amounts of mutant ABCA4 protein were noted in the retina. RNA sequencing of WT, Abca4−/− and Abca4PV/PV mice revealed mild gene expression alterations in the retina and RPE. Similar to Abca4−/− mice, Abca4PV/PV mice showed substantial A2E and lipofuscin accumulation in their RPE cells but no retinal degeneration up to 12 months of age. Thus, rapid degradation of this large misfolded mutant protein in mouse retina caused little detectable photoreceptor degeneration. These findings suggest likely differences in the unfolded protein response between murine and human photoreceptors and support development of therapies directed at increasing this capability in patients.
Introduction
Two parallel paths toward the design of effective treatments for human inherited retinal degeneration consist of understanding the detailed human phenotype resulting from specific gene mutations, and the use of animal models that replicate key aspects of this phenotype in proof-of-concept studies. For most recessively inherited diseases resulting from loss of gene function, the model of choice has been a knockout mouse wherein the gene of interest is completely inactivated. But, there also is emerging evidence that the severity of recessive diseases can be affected by a toxic gain-of-function (1). This type of pathogenesis results when the mutated protein acquires new properties that render it cytotoxic. Therefore, animal models with the exact genetic changes found in humans are needed to determine the effects of such mutations, as well as to investigate the pathological role of the mutated protein.
The photoreceptor cell–specific ATP-binding cassette transporter gene (ABCA4) is mutated in most patients with autosomal recessive Stargardt disease (STGD1) or fundus flavimaculatus and less frequently in those with cone-rod dystrophy and other inherited retinal diseases. ABCA4 is a critical enzyme of the visual (retinoid) cycle involved in flipping N-retinylidene-phosphatidylethanolamine from the extracellular to cytoplasmic side of internal disc membranes of both rod and cone photoreceptor cells (2). By catalyzing this reaction, ABCA4 provides a means of transport for the spent potentially toxic visual chromophore, all-trans-retinal, an aldehyde which otherwise becomes trapped within disk membranes owing to its reversible conjugation with phosphatidylethanolamine (PE) (Fig. 1). Once relocated to the cytoplasmic side of disc membranes, all-trans-retinal can reenter the visual cycle with the help of retinol dehydrogenases (3).
Figure 1.
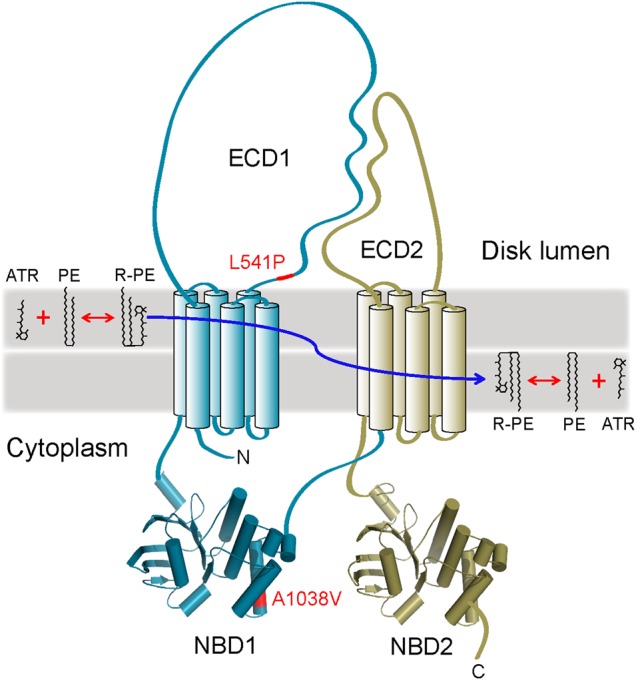
A topological model of ABCA4. This protein is organized in two symmetrical but non-identical parts (colored cyan and gold). Transmembrane helices are shown as cylinders. Homology models of the two nucleotide-binding domains, NBD1 and NBD2, are shown as cartoons. The positions of the L541P and A1038V mutations (abbreviated as P and V, respectively, in the manuscript) are indicated with a red color. The generally accepted role of ABCA4 is the transport of N-retinylidene-phosphatidylethanolamine (R-PE), a product of all-trans-retinal (ATR) reversible conjugation with PE, to the cytoplasmic side of the OS disk membrane, thereby preventing accumulation of R-PE inside the disk. ECD1, ECD2: exocytoplasmic domains 1 and 2, respectively.
The number of different mutations associated with ABCA4-associated retinal degeneration is extensive, with over 800 documented to date (4) involving alleles predicted to code for missense, premature truncation, frameshift or splicing defects. To a first approximation, two inherited alleles make additive contributions, which result in a wide spectrum of retinal disease expression in different patients (5). At one end of this spectrum are alleles associated with a mild retinopathy confined to the macula at the fifth or later decade of life. At the other extreme are alleles causing severe disease that expands across the retina well beyond the macula within the first decade of life. Establishing the relationship between the ABCA4 genotype and the resulting severity of the related retinal disease is critical for the appropriate selection of sub-populations of patients to test experimental treatments that could preserve, or potentially improve, their eyesight. A recent evaluation of patients over a decade revealed that some missense mutations cause a more severe phenotype than produced by early truncations in the ABCA4 genes. It was hypothesized that ABCA4 protein misfolding caused by some missense mutations could be toxic to the cellular machinery responsible for trafficking a protein from its site of biosynthesis to the outer segment (OS) membranes of rod and cone photoreceptors (5).
A relatively common founder mutation in patients of German origin (6) is the complex L541P;A1038V (PV) allele (two missense changes, P and V, on the same allele) of ABCA4. The PV mutation in humans dramatically accelerates the initiation of retinopathy compared with early truncations, suggesting the presence of an additional toxic gain-of-function component (5). Furthermore, it was shown that the PV mutant of ABCA4 is retained in the inner segments of photoreceptor cells in transgenic frogs (7). Thus, both loss-of-activity and misfolding components could contribute to the increased severity of disease caused by this complex mutation.
Here, we performed in-depth functional and structural studies to investigate the molecular basis of the retinopathy resulting from the PV allele and the potential role of protein misfolding in pathogenesis of this disease. We also produced Abca4 knock-in mice homozygous for the PV allele and compared their retinal morphology and function to those of Abca4−/− mice and WT controls.
Results
Understanding the contribution of different ABCA4 alleles to the resulting severity of human retinopathies
Common among all patients with ABCA4-associated retinal degeneration (ABCA4-RD) is a maculopathy involving the foveal region of the retina. This defect is exemplified by atrophy of the RPE and retina apparent upon lipofuscin imaging of patient 1 (P1) at 16 years of age (Fig. 2A, left). ABCA4-RD is progressive, with greater retinal involvement and a corresponding increase in visual disability. Such progression has at least two spatiotemporally distinct components (5). One is the centrifugal expansion of the maculopathy illustrated by the enlarged region of atrophy in P1 imaged at 29 years of age (Fig. 2A, right). The other component that homogeneously affects all cells across the retina (5) can be assessed by visual function testing. ABCA4-RD patients progress from having normal to abnormal peripheral retinal function as shown for P1 (Fig. 2B). Serial data obtained over a decade in a large population of ABCA4-RD patients suggest that such progression follows a delayed exponential to a first approximation (5). The delay and the rate are two parameters of such a model. Although it is plausible that the rate is impacted by different mutant proteins, the longitudinal data are well described with a simpler model holding the rate invariant (at 1.1 log/decade for rod-mediated retina-wide function), leaving the delay, or age of disease initiation (ADI) in the periphery as the only variable parameter.
Figure 2.

Estimated contributions of individual ABCA4 alleles to disease severity in patients with ABCA4-associated retinal degeneration. (A) Lipofuscin autofluorescence imaging in P1 shows a small region of retinal and RPE atrophy (dark) near the fovea surrounded by healthy tissue (brighter homogeneous regions). Thirteen years later, imaging of the same eye reveals a substantially enlarged central region of complete atrophy surrounded by heterogeneity implying a partial loss of RPE cells. The circular region around the optic nerve is relatively well preserved. (B) Gray scale mapping of retina-wide rod sensitivity loss (RSL) plotted at the same two ages shows near normal values at age 16 (left) and substantial losses at age 29 (right). (C) Mean peripheral RSL (symbols) as a function of age in P1. Lines show the delayed exponential fit to the data and the estimated age of retina-wide disease initiation (ADI) (dashed arrow). (D) Comparison of RSL in P5 at age 65 and in P4 at age 59. (E) Mean peripheral RSL as a function of age in P5 and P4 and estimated ADI values from an imputed fit to a delayed exponential. (F) Comparison of RSL in P2 at age 31 and in P3 at age 32. PV refers to the L541P;A1038V allele, and V refers to the A1038V allele without the L514P allele. (G) Mean peripheral RSL as a function of age in P3 and P2 and estimated ADI values from an imputed fit to a delayed exponential. Note that the RSL for P3 is within the normal range and, thus, the ADI estimate shown is a minimum. Inferior (I), superior (S), nasal (N) and temporal (T) visual fields.
The ADI provides a single quantitative measure that can be used to compare disease severity resulting from different combinations of ABCA4 alleles with the assumption of additive contributions from each allele (5). Earlier ADIs presage more severe disease, whereas ADIs at older ages imply milder disease. Indeed, some individuals in the latter group never develop peripheral disease. For example, P1 had an ADI of 20 years (Fig. 2C), which resulted from the sum of 10.6 years [a previously estimated offset (5)], with a −2.1 year delay owing to the IVS35+2 T > C allele, and a +11.7 year delay from the G863A allele. Based on these estimates, it can be concluded that IVS35 + 2 T>C is a much more severe disease-causing allele than G863A. When data are available for only a single time point, an ADI can also be imputed (5). P5 (homozygous for P1380L alleles) at age 65 manifested a severe retina-wide loss of visual function (Fig. 2D, left) with an imputed ADI of 25 years (Fig. 2E) placing the P1380L allele intermediate in severity between IVS35 + 2 T>C and G863A. P4, in contrast, revealed a less severe disease (Fig. 2D, right) with an ADI of 44 years (Fig. 2E) consistent with the compound heterozygosity for a severe allele (P1380L, 7.4 year delay) with a mild allele (G1961E, 26.7 year delay).
Comparing the human disease severity resulting from PV and V alleles in the ABCA4 gene
Using the above-mentioned ADI approach, we first compared the severity of retinopathy associated with the complex PV allele with that of the simple V allele in unrelated patients also carrying the P1380L allele. P2 at age 31 showed a substantial loss of retina-wide function (Fig. 2F, left) with an imputed ADI of 16 years (Fig. 2G), suggesting the PV contribution is a delay of −1.6 years, whereas P3 at age 32 showed nearly normal retina-wide function (Fig. 2F, right) suggesting that the V mutation alone caused a delay of at least +13 years. These results taken together with previous work (5) support the hypothesis that the complex PV allele of ABCA4 causes a more severe retina-wide disease at earlier ages than does the simple V allele.
Cellular localization and expression levels of P, V and PV mutant ABCA4 proteins
To investigate the effects of mutations on the activity and structure of ABCA4, we expressed the human WT protein as well as single P and V mutants and the double PV mutant in HEK293 mammalian cells. Here, we found that the P and V mutations as well as their combination did not affect the cellular localization of ABCA4, such that both the WT and mutated proteins localized mainly in the endoplasmic reticulum (ER) (Fig. 3A). This observation agrees with the results of several previous studies demonstrating ABCA4 localization to both the ER and intracellular vesicles (8–10). Although trapping in the ER could indicate misfolding of an expressed protein, it has been clearly shown that recombinant ABCA4 has normal ATPase activity that can be stimulated by its proposed transported substrates (2,8,9,11,12). Moreover, our recent study provided evidence that WT ABCA4 is fully folded despite being trapped in the ER (13). In this regard, when expressed in the photoreceptors of transgenic frogs, the P and PV mutants are retained in the inner segment, whereas the V variant successfully reaches the OS of the cell (7).
Figure 3.

Biochemical properties of wild-type (WT) and mutated human ABCA4 expressed in HEK293 cells. (A) Confocal microscopy did not reveal differences in cellular ABCA4 localization. Calreticulin was used as the ER marker. (B) WT ABCA4, the P and V single mutants and the PV complex mutant were expressed at comparable levels in HEK293 cells as determined by immunoblotting. Equal amounts of total membrane protein were loaded on the gel. MW indicates molecular weight. (C) An SDS–PAGE gel illustrating purification of WT and mutated ABCA4 from HEK293 cells by lectin affinity (Galanthus nivalis lectin, GNL) and immunoaffinity (Rim3F4 monoclonal antibody) chromatography. All proteins were purified to homogeneity. (D) Normalized basal (black bars) and all-trans-retinal-stimulated [100 μm all-trans-retinal (ATR)] ATPase activity of WT and mutant ABCA4. The basal ATPase activity level of WT ABCA4 was assigned the normalized value of 1. The P and V mutations severely reduced basal ATPase activity. Furthermore, unlike the V mutant, which demonstrated a near-native level of stimulation with ATR, the P and PV mutants did not respond to the addition of the transport all-trans-retinal substrate.
Given the lack of reliable tools for assessing the folding of ABCA4 molecules, the stability of this protein and effects of its mutations are commonly evaluated based on expression levels. It is expected that a destabilizing mutation would render a protein more susceptible to proteolytic degradation. For instance, expression levels of both the P and V variants of ABCA4 were found to be slightly decreased compared with that of the WT protein, whereas the PV mutant was not investigated (9). In contrast, we found that the expression levels of the WT and all three mutant proteins were equivalent as determined by western blotting (Fig. 3B). This observation suggests that the mutations did not accelerate the degradation of ABCA4 expressed in cultured mammalian cells. Discrepancies between our findings and previously published results could stem from the known variability in expression levels associated with the use of a transient expression system.
An efficient purification protocol is essential for obtaining pure protein samples and minimizing potential interference from contaminants. Recombinant ABCA4 is typically isolated in one step using immunoaffinity chromatography (2,9,12). Here, we introduced an additional step, namely lectin affinity chromatography that exploits the glycosylation present in the exocytoplasmic domains of ABCA4 (Fig. 3C). This improvement facilitated achieving a high purity for all the four variants under investigation. Moreover, all our ABCA4 variants were recognized by the Galanthus nivalis lectin, suggesting that the high-mannose nature of ABCA4 glycosylation was not grossly affected by these mutations. However, despite the similar expression levels of the four proteins, the purification yields were somewhat lower for the P and PV mutants (Fig. 3C). This could indicate that the efficiency of binding to the lectin affinity resin was reduced as a result of altered glycosylation. Partial misfolding could lead to a similar effect by influencing the accessibility of the glycans.
ATPase activity of mutant ABCA4 variants
Basal and all-trans-retinal-stimulated ATPase activities of WT and mutated ABCA4 were determined after reconstitution of these proteins into lipid vesicles. The WT protein demonstrated a typical 2-fold increase in ATP hydrolysis rate upon addition of 100 μm all-trans-retinal (Fig. 3D). Although the basal ATPase activity of the V mutant was somewhat reduced (∼70% of WT ABCA4), it was still stimulated by all-trans-retinal nearly 1.8-fold, suggesting that this mutant form of ABCA4 is capable of transporting the substrate. In contrast, both the P and PV ABCA4 variants exhibited a dramatically reduced basal ATPase activity (∼30% of WT ABCA4), which did not increase upon the addition of all-trans-retinal. Thus, ABCA4 variants carrying the P mutation were functionally impaired. The remarkably reduced ATPase activity of the P mutant was demonstrated previously by Sun et al. (9). According to that report, however, the activity of the human V variant was even lower and not stimulated by all-trans-retinal. We verified our results by repeating the assay many times with independent protein preparations.
Stability and structural changes induced by ABCA4 mutations
To shed some light on the effects of the mutations on the stability and structure of ABCA4, the WT and mutated proteins were imaged by negative-stain electron microscopic (EM) (Fig. 4A). This approach allowed direct visual examination of individual protein molecules and identification of potential misfolding. Separate particles with consistent shapes and sizes were present in micrographs prepared with WT ABCA4 and the V mutant, indicating structural homogeneity. In stark contrast, large heterogeneous particles with irregular shapes were evident in the images of both the P and PV proteins, implying nonspecific protein oligomerization. Smaller particles that could represent partially unfolded ABCA4 were also abundant.
Figure 4.

Evaluation of conformational homogeneity and structures of WT and mutated ABCA4. (A) Micrographs of WT ABCA4 and the P, V and PV mutants negatively stained with uranyl acetate. The WT protein and the V mutant are represented by homogeneous, monomeric particles, suggesting correct protein folding. In contrast, aggregation and structural heterogeneity are evident for the P and PV mutants. Homogeneous particles are enclosed in white squares; red arrows indicate misfolded/aggregated protein. (B) Structures of WT and mutated ABCA4 obtained by single-particle analysis (0°, 90°, 180° and 270° vertical angular views). The structure of the A1038V mutant closely resembles that of the WT protein, which, in turn, is equivalent to native ABCA4 protein purified from bovine rod photoreceptors (14). In contrast, the exocytoplasmic region that hosts the P mutation is absent in the structures of the P and PV mutants, suggesting misfolding and gross conformational impairment. ECDs, TMDs and CDs: exocytoplasmic, transmembrane and cytoplasmic domains, respectively. Lines indicate the putative position of the OS disc membrane.
Using single-particle analysis, we first reconstructed a three-dimensional EM-map of WT ABCA4 (Fig. 4B). This structure was highly similar to that of native ABCA4 isolated from bovine photoreceptors (14), including the exocytoplasmic regions, transmembrane portion and cytoplasmic moiety. Remarkably, all these features were preserved in the structure of the V mutant, which closely resembled that of the WT protein (Fig. 4B). This observation strongly indicates a minimal effect of the V mutation on the conformation and stability of ABCA4, which is in line with its well-preserved ATPase activity. To obtain structures of the P and PV variants, we excluded all particles representing aggregated protein (Fig. 4A, red arrows) and assumed that the remaining small fraction of homogeneous particles (∼20% of total) was representative of the population of ABCA4 molecules that were relatively well folded. Whereas the resulting 3D maps revealed the cytoplasmic and transmembrane moieties in the P and PV mutants (Fig. 4B), the entire exocytoplasmic region harboring the P mutation was absent in both cases, indicating that it failed to form a stable conformation. This result suggests that the P mutation strongly destabilizes ABCA4, inducing partial misfolding and gross conformational changes.
Generation of homozygous dual knock-in mice with complex PV alleles
To evaluate the complex PV allele in vivo, we designed a gene-targeting strategy to produce homozygous dual knock-in (Abca4PV/PV) mice (Fig. 5A, see also Materials and Methods). Briefly, after two separate homologous recombination events, we introduced two point mutations in the Abca4 gene: L541P at exon 12 (abbreviated as P) and A1038V at exon 21 (abbreviated as V). The mouse also had two additional residual sequences from the recombination: a LoxP-derived insertion before exon 12 and a FRT/LoxP insertion after exon 22. The DNA sequence was confirmed for these two regions of this large Abca4 gene. Primers flanking the 141-bp FRT/LoxP residue were used for genotyping. A PCR product of larger size was amplified from homozygous Abca4PV/PV mice as compared with WT mice (boxed inset in Fig. 5A). Heterozygous Abca4WT/PV mice had both DNA fragments.
Figure 5.

Targeting strategy for generating Abca4PV/PV mice and confirmation of the intended mutations. (A) Targeting strategy. Top row: the targeting construct to introduce the first mutation in the Abca4 gene was designed such that the 5′ short homology arm (SA1) extended ∼2.8 kb to exon 12. The 3′ long homology arm (LA1) started after exon 12 and was ∼5.4 kb long. A LoxP-flanked Neo cassette was inserted on the 5′ side of exon 12. The T–C mutation in exon 12 (red P) was generated by PCR mutagenesis. Middle row: the targeting construct to introduce the second mutation in the Abca4 gene was designed such that the 5′ LA2 extended ∼6.5 kb to exon 21 and the ∼2.2 kb-long SA started after exon 22. The FRT-Neo-FRT/Lox cassette was inserted on the 3′ side of exon 22 and the C–T mutation in exon 21 (red V) was generated by PCR mutagenesis. Bottom row: the Neo cassette before exon 12 was eliminated by electroporation of a Cre-expressing vector into recombinant ES cells, whereas the other Neo cassette after exon 22 was removed by cross-breeding with FLP mice. As a result, Abca4PV/PV mice were generated with two point mutations, P and V at codons 541 and 1038, encoding the dual mutant L541P;A1038V ABCA4. These mice lacked Neo cassettes but had a LoxP residual sequence (P) in the intron between exon 11 and exon 12 as well as an FRT/LoxP residual sequence between exon 22 and exon 23, which was used for genotyping. The two knock-in loci were also cloned and sequenced to confirm their correct targeting. Inset: Genotyping. PCR primers were designed to amplify the intron with the 141-bp FRT/LoxP residual sequence. The WT allele produced a 412-bp PCR product, and the PV Abca4 allele produced a 583-bp PCR product. The negative control revealed no template contamination. (B) RNA-Seq reveals Abca4 gene expression changes in Abca4−/− and Abca4PV/PV compared with WT mouse retinas. Single-base resolution of the RNA-Seq run of Abca4−/− retina reveals ablation of the Abca4 transcript in exon 1 (top panel, indicated with red line) where there was a neomycin cassette. However, comparable transcripts were present in Abca4WT/WT and Abca4PV?PV retinas (middle and lower panel). (C) SNPs in exon 12 (blue) and 21 (red) of Abca4PV/PV retinal Abca4 transcripts revealed by RNA-seq.
Expression changes in the retina and RPE coding genes resulting from the PV mutation in Abca4PV/PV mice
To investigate which genes and gene regulation networks are affected in Abca4−/− and Abca4PV/PV mice, we performed RNA-Seq transcriptome analyses, comparing the global retinal transcriptome of 4-week-old Abca4−/−, Abca4PV/PV and control WT mice. First, we verified the absence of the Abca4 exon 1 fragments in the Abca4−/− transcript (Fig. 5B) and two single-nucleotide polymorphisms (SNPs) in exon 12 and 21 of the Abca4PV/PV transcripts (Fig. 5C). Here, we identified 474 differentially expressed genes between Abca4−/− and WT mice, 290 genes between Abca4PV/PV and WT mice and 323 genes between Abca4−/− and Abca4PV/PV mice in these retinal samples (Supplementary Material, Fig. S1). We also found 430 differentially expressed genes between Abca4−/− and WT mice, 354 genes between Abca4PV/PV and WT mice and 347 genes between Abca4−/− and Abca4PV/PV mice from the total number of RPE samples (Supplementary Material, Figure S1). Most of these genes were not enriched in the retinal and RPE transcriptome (FPKM < 10) and not highly differentially expressed (−1 < log 2 < 1) between the two compared genotypes. When we excluded those genes, 31 differentially expressed genes were obtained in Abca4−/− versus WT, 21 genes in Abca4PV/PV versus WT and 25 genes in Abca4−/− versus Abca4PV/PV retina transcriptomes; 28 differentially expressed genes in Abca4−/− versus WT, 18 genes in Abca4PV/PV versus WT and 6 genes in Abca4−/− versus Abca4PV/PV in the RPE transcriptomes were also identified (Supplementary Material, Figs S2 and S3; Supplementary Material, Tables S1 and S2). Among those genes, we found several lens-specific genes in both retina and RPE transcriptomes and photoreceptor-specific structure and phototransduction-related genes in RPE transcriptomes from contamination during eye tissue preparation. Genes related to sex distinction, hemoglobin and CD antigens were also included in this list, but they were likely caused by animal variations rather than ablation/mutation of Abca4 genes. The rest of these genes were either functionally uncharacterized or probably not linked to retinal/RPE dysfunction. Thus, we conclude that the observed gene expression differences in either Abca4−/− or Abca4PV/PV mice when compared with their WT counterparts were generally minor. Also, we failed to detect any upregulation in the unfolded protein response (UPR) genes. No notable changes in genes or gene regulation pathways were found between Abca4−/− and Abca4PV/PV mice.
Immunocytochemistry and retinal structure as a function of age
Real-time PCR and immunoblots were used to verify Abca4 gene and protein expression further in both WT and Abca4PV/PV retinas from 1-month-old mice with age-matched Abca4−/− retinas serving as negative controls. Abca4 RNA levels were found to be slightly decreased in retinas from Abca4PV/PV mice but showed no statistically significant difference compared with those from WT mice (Fig. 6A). We did not detect any Abca4 RNA by real-time PCR with primers spanning the exon 1 and 2 junction in the Abca4−/− retinas (Fig. 6A), a result consistent with the RNA-Seq findings. But, surprisingly, only trace amounts of mutant ABCA4 protein were detected in Abca4PV/PV retina with the monoclonal antibody TMR4 (Fig. 6B). Similar results were obtained with two other antibodies, TMR1 and RIM.
Figure 6.
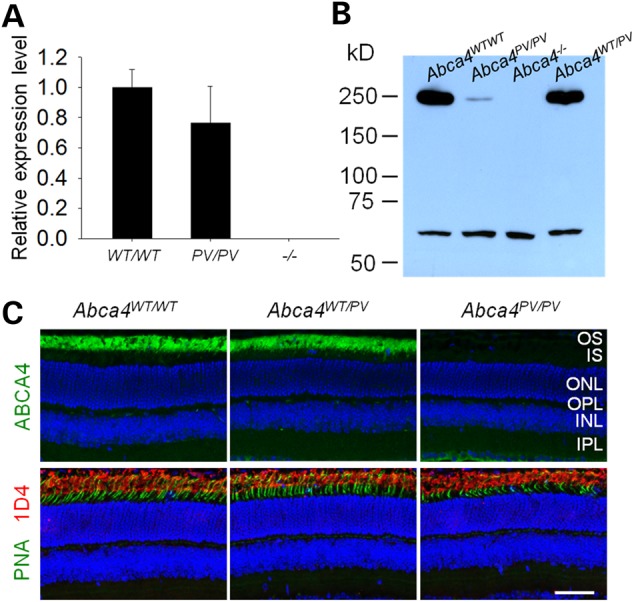
ABCA4 expression in 1-month-old mouse retinas. (A) Real-time PCR revealed that Abca4 mRNA levels normalized to Gapdh mRNA were comparable in retinas from WT and Abca4PV/PV mice but undetectable in retinas from Abca4−/− mice. (B) An immunoblot with TMR4 antibody featured a dramatic decrease in mutant ABCA4 (∼250 kDa) protein level in the retina of a 1-month-old Abca4PV/PV mouse. Retinas from Abca4WT/WT, Abca4PV/PV and Abca4−/− mice were used for comparison. The cross-reacting antigen (∼60 kDa) served as the loading control. (C) Immunohistochemistry of retinal cryosections from 1-month-old mice. ABCA4 protein (green) was undetectable in Abca4PV/PV retinal cross-sections (upper panels), but rods (red) and cones (green) were apparently normal when probed with 1D4 antibody and PNA lectin (lower panels). Nuclei were counter-stained with DAPI (blue). Scale bar: 50 µm.
Immunohistochemistry was performed with retinal cryosections from 1-month-old mice. ABCA4 protein (green) was localized normally at the photoreceptor OS both in WT retinas and retinas from Abca4WT/PV mice but was undetectable in retinas from Abca4PV/PV mice (Fig. 6C, upper panel). Anti-rhodopsin 1D4 antibody was used to label rod outer segments (red) and peanut agglutinin (PNA) to label cone sheaths (green), and no apparent rod/cone degeneration was noted in retinas from age-matched Abca4PV/PV mice (Fig. 6C, lower panels). We then used optical coherence tomography (OCT) to image retinal cross-sectional structures in vivo at high resolution (2.0 μm). Again, there was no evidence of progressive photoreceptor degeneration in Abca4PV/PV mice or Abca4−/− mice up to 12 months of age (Fig. 7A and B).
Figure 7.

Retinal degeneration was not evident in either Abca4PV/PV or Abca4−/− mice. (A) Representative OCT images of retinas from 6-month-old mice. (B) Thicknesses of the outer nuclear layer (ONL) measured 500 μm from the optic nerve head in nasal OCT images are shown from mice at various ages (n > 3). (C) Plastic sections of retinas from 14-month-old WT, Abca4PV/PV and Abca4−/− mice stained with toluidine blue. All retinal layers appear virtually normal in all three genotypes. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer. Scale bar: 50 µm. (D) Electron microscopy reveals excessive lipofuscin accumulation in Abca4PV/PV, and Abca4−/− mouse RPE at the age of 14 months. White arrows indicate irregularly shaped lipofuscin pigment granules that are distinct from oval melanosomes. Scale bar: 2.0 μm.
Stained with toluidine blue, plastic sections of eyecups from 14-month-old mice were viewed under a light microscope. Both Abca4PV/PV and Abca4−/− sections appeared virtually normal at the level of the retina and the RPE (Fig. 7C). More detailed EM studies of the RPE revealed dramatically increased irregular-shaped dark granules considered to be lipofuscin deposits accumulated in older (14-month-old) Abca4−/− and Abca4PV/PV mice (Fig. 7D, arrows).
To evaluate whether retinas of Abca4−/− and Abca4PV/PV mice were undergoing significant stress, we performed immunohistochemistry to detect glial fibrillary acidic protein (GFAP), an indicator for Müller glia activation (15). We reasoned that minor micro-environmental changes in the retina could potentially activate Müller cells, the resident glial cells maintaining surveillance of the retina, even without evidence of severe retinal degeneration. However, in both young and older (1- and 6-month-old) mice, GFAP staining was not increased compared with that in WT mouse retina (Supplementary Material, Fig. S4). In contrast, a retinitis pigmentosa rhodopsin E150K knock-in mouse model which we had characterized as a positive control (16) exhibited pronounced Müller cell activation.
Photoreceptor dark adaptation in Abca4−/− and Abca4PV/PV mice
The light sensitivity of rods appeared unaffected in Abca4−/− and Abca4PV/PV mice based on ERG responses similar to their WT counterparts at the light intensity of −0.286 (log cd·s/m2) used to measure rod dark adaptation in vivo (WT, 243.0 ± 22.8 µV; Abca4PV/PV, 301.7 ± 23.7 µV; Abca4−/−, 247.6 ± 11.4 µV). The effect of the dual PV mutation in Abca4 on rod dark adaptation in vivo was evaluated by measuring the recovery of ERG a-wave amplitudes after >90% bleaching of rod visual pigment (Supplementary Material, Fig. S5). Rod photoresponse recovery was similar in both Abca4PV/PV and Abca4−/− mice compared with WT controls at 1 month of age. Moreover, no difference in rod dark adaptation was observed between Abca4PV/PV mice and Abca4−/− animals. Findings were similar in 8-month-old mice. These observations indicate that recycling of 11-cis-retinal through the RPE visual cycle was not compromised in ABCA4 mutant mice.
To test the specific function of M/L-cones in mutant mice, we performed transretinal ERG recordings to obtain cone photoresponses across the whole isolated retina from 3-month-old Abca4PV/PVGnat1−/− mice and Gnat1−/− controls. The absence of rod transducin α-subunits in these mice eliminated interference from rod signaling (17). Recordings were carried out in the presence of synaptic inhibitors to block post-photoreceptor components of the photoresponse and isolate pure cone-driven signals. Cones from Abca4PV/PVGnat1−/− retinas produced robust responses (Fig. 8A and B). Interestingly, the Abca4PV/PVGnat1−/− cone responses appeared somewhat faster than those of control Gnat1−/− cones. Analysis of the dim flash response kinetics confirmed a significant (P < 0.01) acceleration in both the time to peak [from 89 ± 3 ms (n = 5) in Gnat1−/− controls to 65 ± 4 ms (n = 6) in Abca4PV/PVGnat1−/− cones] and the integration time [from 133 ± 15 ms (n = 5) in Gnat1−/− controls to 73 ± 8 ms (n = 6) in Abca4PV/PVGnat1−/− cones]. A similar response acceleration was observed in the cones of Abca4−/− mice (data not shown). Comparison of the averaged intensity-response functions indicated that the photosensitivity of dark-adapted M/L cones (determined as their half-saturated light intensity) was comparable in Abca4PV/PVGnat1−/− and control Gnat1−/− mice (Fig. 8C and D), whereas the maximum cone response amplitude in Abca4PV/PVGnat1−/− mice was slightly (∼20%) lower than that in Gnat1−/− controls (Fig. 8C). The robust cone signaling preserved in mutant mice at 3 months of age is consistent with the lack of apparent cone degeneration in Abca4PV/PVGnat1−/− mice (Fig. 6C). This observation allowed us next to investigate whether the mutant ABCA4 protein affects dark adaptation of mouse cones.
Figure 8.
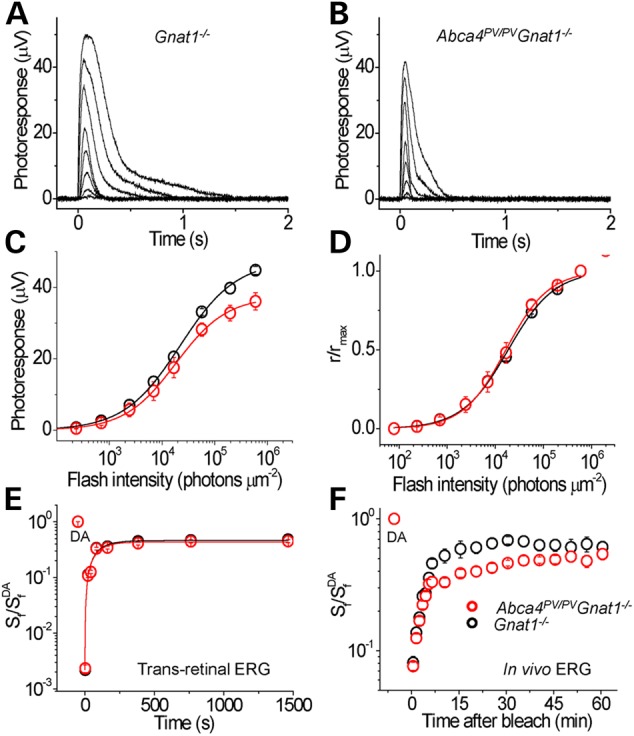
Cone photoresponses and recovery kinetics after photo bleaching recorded from isolated intact retinas of anesthetized control Gnat1−/− and Abca4PV/PVGnat1−/− mice. Representative responses to 20-ms 505-nm light test flashes with intensities increased in 0.5 log unit steps for Gnat1−/− (A) and Abca4PV/PVGnat1−/− (B) retinas. Absolute (C) and normalized (D) whole retina response amplitudes of Gnat1−/− (n = 5) and Abca4PV/PVGnat1−/− (n = 6) mice are shown as functions of flash strength. Points were fitted with Naka-Rushton hyperbolic functions. (E) Recovery of M/L-cone a-wave flash sensitivity (Sf) after a >90% bleach of cone visual pigment was identical in isolated retina ERG recordings the two mouse lines. (F) Recovery kinetics of M/L-cone ERG b-wave flash sensitivity (Sf) after a >90% bleach of cone visual pigment in live mice. Cone photosensitivity recovery was substantially delayed in Abca4 mutant mice during the period of 8–40 min after bleaching. Data are means ± SEM.
M/L-cone dark adaptation was first tested in isolated Abca4PV/PVGnat1−/− retinas by tracing the recovery of cone ERG a-wave flash sensitivity (Sf) after applying 3 s of green LED light. This light bleached >90% of cone visual pigment which instantly reduced cone sensitivity by almost three log units. We found that the kinetics of postbleach cone photosensitivity recovery in the dark was identical in the two mouse lines and could be characterized with a single exponential function with a time constant of ∼1.5 min (Fig. 8E). Thus, cone pigment regeneration through the retina's visual cycle was unaltered in our mutant mice. Next, we conducted a similar experiment in live 3-month-old Abca4PV/PVGnat1−/− mice in which both the retina and adjacent RPE were in their native physiological environment. Here, the cone bipolar cell-driven ERG b-wave was used to assess cone function owing to a barely detectable photopic a-wave in vivo. The lack of rod signaling allowed us to obtain cone-driven ERG responses without the use of rod-suppressing background light. Cone b-wave responses from the Abca4PV/PVGnat1−/− mice appeared normal, with a maximal amplitude of 259 ± 12 µV (n = 16) compared with 266 ± 31 µV (n = 8) in Gnat1−/− controls. Monitoring the recovery of M/L-cone ERG b-wave flash sensitivity (Sf) after the >90% cone pigment bleach with 30 s green LED light revealed no difference in the initial rates (up to ∼6 min postbleach) of cone b-wave sensitivity recovery between Abca4PV/PVGnat1−/− and Gnat1−/− mice (Fig. 8F). This result is consistent with the finding obtained with isolated retinas and indicates that the early phase of cone recovery, which is likely driven by the retina's visual cycle in vivo (18), was not affected by the lack of functional ABCA4. However, the later phase of cone dark adaptation in vivo was substantially delayed in PV mutant mice indicating slower supply of chromophore recycled through the RPE visual cycle to cones. The final level of cone sensitivity recovery during the 60-min postbleach period was similar in both strains (Fig. 8F). Thus, these observations demonstrate that the overall kinetics of mouse M/L-cone pigment regeneration was compromised by the absence of functional ABCA4 protein in adult Abca4PV/PVGnat1−/− mice.
Retinoid analysis in Abca4PV/PV mice
As ABCA4 is a visual cycle protein which plays an important role in the clearance of all-trans-retinal and regeneration of 11-cis-retinal after light exposure, the kinetics of all-trans-retinal reduction (Fig. 9A, left panel) and 11-cis-retinal formation (Fig. 9A, right panel) were examined after light exposure that bleached ∼90% of rod visual pigment. Mice were kept in darkness after light exposure until examined. No difference in the levels of all-trans-retinal and 11-cis-retinal were observed between Abca4PV/PV, Abca4WT/PV, Abca4−/− and WT mice at any time after this bleach in 6-week-old mice.
Figure 9.

Retinoid analyses in ocular tissues of Abca4PV/PV, Abca4WT/PV, Abca4−/− and WT mice. (A) Kinetics of ocular retinoids after light exposure in 6-week-old Abca4PV/PV, Abca4WT/PV, Abca4−/− and WT mice. The kinetics of all-trans-retinal reduction (left panel) and 11-cis-retinal formation (right panel) were examined after light exposure that bleached ∼90% of the visual pigment. Retinoids were quantified by normal-phase HPLC in samples collected from dark-adapted mice and at different time points after illumination. Symbols indicate means; error bars indicate standard deviations (n = 5). All data points obtained after bleaching were fitted by an exponential decay for all-trans-retinal and by an exponential rise to maximum for 11-cis-retinal. Rates of all-trans-retinal clearance were 4.92 ± 0.47, 4.03 ± 0.34, 4.18 ± 0.43 and 4.34 ± 0.40 s−1 for Abca4PV/PV, AbcaWT/PV, Abca4−/− and WT mice, respectively. Rates of 11-cis-retinal accumulation were 0.65 ± 0.26, 0.74 ± 0.22, 0.61 ± 0.26 and 0.83 ± 0.21 s−1 for Abca4PV/PV, AbcaWT/PV, Abca4−/− and WT mice, respectively. No significant changes in the mutant compared with WT control mice were observed. DA: dark-adapted. (B) Marked accumulation of A2E in the eyes of Abca4PV/PV and Abca4−/− mice. Amounts of A2E in the eyes of Abca4PV/PV, Abca4−/− and WT mice were quantified by reverse-phase HPLC. Mice were raised under a regular 12 h (∼10 lux) light/12 h dark cycle and A2E in whole eyes was measured at the ages of 1, 3, 6 and 15 months. Bars indicate standard errors of the means (n = 5). The symbols * and # indicate statistically significant changes (P < 0.005 compared with WT animals). No significant differences were found between Abca4PV/PV and Abca4−/− animals.
Amounts of A2E in the eyes of Abca4PV/PV, Abca4−/− and WT mice at the ages of 1, 3, 6, 12 and 15 months were quantified by reverse-phase high-performance liquid chromatography (HPLC) (Fig. 9B). Mice were raised under a regular 12-h light (∼10 lux)/12-h dark cycle. Age-dependent A2E accumulation was noted in all genotypes, with Abca4PV/PV and Abca4−/− mice accumulating about 5-fold more A2E than WT mice. No statistically significant differences in A2E accumulation were found between Abca4PV/PV and Abca4−/− animals.
Scanning laser ophthalmoscopy and two-photon excitation microscopy of RPE fluorophores
Confocal scanning laser ophthalmoscopy (SLO) was used to detect lipofuscin autofluorescence in vivo with a 488-nm laser excitation. A moderate increase in autofluorescence was detected in the fundus of Abca4PV/PV mice (1.60 ± 0.24-fold) as well as in Abca4−/− mice (1.37 ± 0.13-fold) as compared with the fundus of WT mice, whereas heterozygous Abca4WT/PV animals (1.08 ± 0.03-fold) did not exhibit any apparent fundus autofluorescence alterations (Fig. 10A and B). We had previously shown that all-trans-retinal condensation products over-accumulate in the eyes of Abca4−/−Rdh8−/− mice and that this increase in retinoid condensation products can be assessed by two-photon excitation microscopy (TPM) at shorter (∼730 nm) and longer (∼900 nm) excitation wavelengths (19–21). The ratio of fluorescence excited with longer wavelengths to that excited at shorter wavelengths increased with age in WT mice, and this increase was substantially enhanced in Abca4−/−Rdh8−/− mice. Here, we used 730- and 850-nm excitation light to compare the accumulation of retinoids in the RPE of WT, Abca4PV/PV and Abca4−/− mice (Fig. 10C). By comparing images of RPE obtained with these two excitation wavelengths, we found that the ratio of RPE fluorescence excited with 850 nm to that excited with 730 nm was significantly different between WT mice and mice with the ABCA4 PV dual mutation (P < 0.001). Additionally, we noted that this ratio tended to be higher in Abca4PV/PV mice (Fig. 10D). This result is consistent with the A2E measurements presented in Figure 9B and indicates that increased amounts of A2E accumulate in the RPE of Abca4−/− and Abca4PV/PV mice. Notably, A2E, SLO autofluorescence and TPM ratios were higher for Abca4PV/PV mice compared with Abca4−/− mice, even though differences were not statistically significant.
Figure 10.

Characterization of retinal and RPE autofluorescence in Abca4PV/PV, Abca4−/− and WT mice. Low magnification images of fundus autofluorescence captured by confocal scanning laser ophthalmoscopy (cSLO) in mice with indicated genotypes at the age of 3 months are shown in A. High magnification characterization of RPE fluorescence by TPM is presented in C. (A) Representative cSLO fundus images from mice of different genotypes. (B) Quantification of average intensities from circular fundus images of these mice. P-values versus WT (two-tailed Student's t test) are provided followed by the numbers of each mouse group in parentheses. (C) Images of the RPE in the eyes of Abca4PV/PV, Abca4−/− and WT mice. Images obtained with 850-nm excitation are shown in the upper row, and those obtained with 730-nm excitation are featured in the bottom row. Scale bars indicate 25 µm. (D) Ratios of fluorescence excited with 850 to that excited with 730-nm light. Ratios were normalized to the average 850/730 ratio for Abca4PV/PV mice. Bars indicate standard deviations (n = 5); P values are indicated in the figure.
Discussion
Stargardt disease is an inherited retinal degeneration that starts in the macula and often expands to the rest of the retina with progressive vision loss and ultimate blindness. Most cases of this recessive disease are associated with mutations in the all-trans-retinal transporter, ABCA4 (5,22). Although causative mutations correlated with Stargardt disease have been identified, lack of understanding their pathophysiology has resulted in a paucity of clinical treatment options. Located in the rims of outer segment discs in photoreceptor cells, ABCA4 is a large glycoprotein that translocates all-trans-retinal from the disk lumen to the cytoplasmic leaflet. This translocation facilitates reduction of this toxic aldehyde, allowing retinoids to reenter the visual (retinoid) cycle. Mouse models generated to date are based on knockout of Abca4. Although Abca4−/− mice exhibit some features of human retinopathies, including accumulation of A2E and other retinal condensation products, they do not develop the most important feature of Stargardt disease, namely retinal degeneration at a young age (23,24). Moreover, only a small number of patients have two mutations that disable the expression of the ABCA4 gene; most alleles are either point mutations, frame shifts or splicing defects (4). This fact motivated us to focus on the complex PV double mutation with other ABCA4 mutants serving as controls. The PV mutation had been extensively characterized in patients by noninvasive methods and exhibited a more rapid progressive retinal degeneration than truncation mutations (5). The present work provides a more detailed analysis of the effects of this mutation on the structural, biochemical and genetic properties of ABCA4, as well as its physiological function.
All mutant ABCA4 proteins evaluated in this study demonstrated reduced ATPase activity. This reduction, however, was relatively modest for the V mutant protein, which showed a substantial level of basal ATPase activity along with a robust stimulation by its retinoid substrate, all-trans-retinal. In contrast, the basal ATPase activity of the P and PV mutants was severely diminished and unresponsive to this retinoid, suggesting that the P mutation is probably the main contributor to the severe phenotype observed in human carriers of the PV genotype. From the activity data alone, however, it was unclear whether the impairment imposed by the mutations is limited to the functional activity of ABCA4 or has more global effects on the protein.
Our structural analyses revealed the extent of changes in the conformation and stability of ABCA4 resulting from P and V mutations introduced separately or in combination. In line with only a slight decrease in ATPase activity, the V mutant in the first nucleotide-binding domain (NBD1, Fig. 1) retained the major structural features of the native protein, with only minimal amounts of misfolded and aggregated protein revealed by electron microscopy. This finding agrees well with the conservative nature of this mutation (Ala to Val residue). In contrast, introduction of a Pro residue in place of Leu at position 541 within the first exocytoplasmic domain (ECD1, Fig. 1) led to apparent misfolding and structural disorganization of the whole exocytoplasmic region of ABCA4, which comprises nearly half of the total amino acid sequence (Fig. 4B). Quite predictably, these global changes were accompanied with a strong tendency of the P and PV mutants to form heterogeneous oligomers. Probably, the diminished activity of these two forms results from their severely affected conformations. Moreover, changes of this scope could theoretically trigger cellular stress from the unfolded protein.
We suspected that a gain-of-function of the mutated protein, such as excessive basal ATPase activity leading to depletion of ATP in photoreceptors, could contribute to the more severe form of Stargardt disease. But, surprisingly, despite the similar amount of mutant Abca4 mRNA in knock-in mouse retina, the amount of mutant protein was dramatically reduced compared with the native form in WT mice. Hence, even if the gain-of function hypothesis might be true for human patients, it does not apply to the mouse model because the protein itself is degraded. Additionally, the disappearance of the mutated protein did not trigger a major retinal degeneration as happens with many rhodopsin mutants (16,25,26). Thus, both P23H and E150K rhodopsin mutant mice exhibit early onset rod photoreceptor death owing to the activation of ER-associated protein quality control and degradation machinery (16,25,26). We investigated the retinal morphology of our knock-in mice over a year using both an in vivo OCT technique and histology combined with light microscopy but failed to detect notable retinal degeneration. Both rod and cone cells appeared normal, and no active gliosis of Müller cells or early signs of retinal stress were detected in either Abca4PV/PV or Abca4−/− mouse retinas. Using electron microscopy, we did find many more electron dense granules considered to be lipofuscin deposits in the ABCA4-deficient mouse RPE cells (Fig. 7D). But, other than that, the retina appeared intact in these mutant mice.
Accumulation of lipofuscin is one of the most characteristic features of aging observed in RPE cells. However, we observed a greater excess of lipofuscin granules in the RPE cells from the mutant mice than expected from a normal aging process. Considering that lipofuscin in RPE cells is composed of different fluorescent compounds together with lipids and proteins, we used multiple approaches including SLO, two-photon microscopy (TMP) and HPLC to identify and quantify some of these components. SLO employed to visualize mouse fundus with an excitation wavelength of 488 nm revealed a bright fundus in the ABCA4 mutant mouse eye. The brightness was evenly and continuously distributed throughout the fundus, unlike the pathology seen in many retinal degenerative mouse models that presents as bright scattered spots (activated microglias/macrophages). This finding is consistent with previous findings in Abca4−/− mice (27), as well as with the homogeneously increased autofluorescence measured at early disease stages before retinal degeneration in human patients with ABCA4-RD (28). Two-photon microscopy has been successfully used to detect various chromophores in ocular tissues. Retinoid condensation products have an excitation peak around 850 nm that is distinct from the 730-nm excitation peak of retinyl esters stored in retinosomes (20,21). We used the ratio of fluorescence excited at 850–730 nm to quantify retinoid condensation products and found that this ratio was increased in both Abca4−/− as well as Abca4PV/PV mice. This increase was considerably higher than that detected by the SLO method. This result could be related to the differing imaging directions, i.e. SLO was measured from the front of the eye through the cornea, lens and vitreous body, whereas two-photon images were obtained from the sclera at the back of the eye. In addition, the excitation wavelengths do not match exactly for these two methodologies so that different populations of chromophores could have been imaged. A2E, one of the major lipofuscin fluorophores (29), was quantified with HPLC. Though age-dependent accumulation of A2E was found in all genotypes, accumulation was far greater in Abca4−/− and Abca4PV/PV mice than in WT mice (Fig. 9B).
RNA-Seq is a recently developed approach to transcriptome profiling that provides a more precise measurement of levels of transcripts and their isoforms than other methods (30). RNA-Seq analysis was performed with both retinal and RPE samples, which accurately verified their identities as derived from knockout and dual knock-in mice. However, we did not find any notable alterations in gene expression levels among all three genotypes. Thus, the Abca4PV/PV mouse serves as a model comparable with the knockout mouse and only partially recapitulates the phenotypes observed in Stargardt patients.
Correlated with the largely normal retinal morphology, visual function of the retina was preserved as revealed by normal rod ERG responses indicating that the mutant mice could sense light well under normal scotopic conditions (Supplementary Material, Fig. S5). In addition, the kinetics of rod response recovery from bleaching most of rhodopsin by bright light (which mainly depends on the rate of 11-cis-retinal regeneration in the RPE during rod dark adaptation) was virtually unaffected in ABCA4 mutant animals as measured by in vivo ERG. Indeed, no differences were found in postbleach levels of both all-trans- and 11-cis-retinal between WT and mutant mice (Fig. 9A). This result differs somewhat from the original finding in Abca4−/− mice (31). The reason for this discrepancy is unclear, but it could be due to the different light bleaching paradigms employed. Thus, the previous studies indicate that clearance of all-trans-retinal is more effective when higher amounts of all-trans-retinal are produced. Abca4−/−Rdh8−/− and Rdh8−/− mice show no difference in all-trans-retinal clearance, even though an apparent clearance delay in Abca4−/− mice is observed as compared with WT animals when 250 pmol/eye of all-trans-retinal is produced after light exposure (23). Furthermore, a more prominent delay in all-trans-retinal clearance was measured when 70 pmol/eye of all-trans-retinal was produced after light exposure (31). These observations suggest that all-trans-retinal dehydrogenases play more dominant roles than ABCA4 for clearing all-trans-retinal from photoreceptors in the presence of large amounts of all-trans-retinal. In contrast to experiments aimed at examining retinoid metabolism, after exposure to intensive light, A2E quantification was conducted in mice kept under room lighting conditions. Therefore, the significant difference in accumulated A2E between the genotypes in this study could have occurred because ABCA4 deficiency contributed more to all-trans-retinal clearance under mild lighting conditions. A contribution of 11-cis-retinal to A2E production should also be considered (32).
Cone responses were investigated in mice having a Gnat1−/− background with abolished rod phototransduction and responses. Responses of dark-adapted M/L-cones, measured both by ERGs in vivo and by transretinal recordings ex vivo, were comparable in 3-month-old ABCA4 dual knock-in and control mice indicating normal photopic function at this age. Perhaps surprisingly, the cone response kinetics of Abca4PV/PV mice were somewhat accelerated compared with these from controls. One possible explanation for this result is that ABCA4 plays a structural role in the cone outer segment such that its alteration indirectly affects cone response kinetics. Alternatively, the lack of a functional ABCA4 in cones could affect the lipid composition of their plasma membrane, influencing the interactions of membrane-bound phototransduction proteins. Finally, ABCA4 could directly modulate cone phototransduction by interacting with a component of the cone phototransduction cascade. The initial kinetics of cone dark adaptation were unaltered in isolated retinas of Abca4PV/PV mice. One of several possible explanations is that the chromophore could be provided to cones from the Muller cells (33). However, the second RPE-driven component (34) of cone sensitivity recovery was substantially delayed in vivo (Fig. 8F). This result is in stark contrast to our findings in rods and argues that ABCA4 plays an important role in cone pigment regeneration through the RPE visual cycle required to complete dark adaptation of cones following exposure to bright light (18).
The largest visual phenotypic difference between human and mice carrying the PV mutation is the difference in retinal degeneration. Initially, it was suggested that retinal disease was not present in young mice (31). A subsequent study verified this observation, and it is now accepted that well documented changes in retinal structure and function do occur in mice older than 1 year (23). The similarity is that accumulation of all-trans-retinal condensation products including A2E occurs throughout the life of these animals. Recently, the idea has evolved that condensation products of all-trans-retinal, though certainly toxic to some extent, appear to play a less important role in the pathology associated with Stargardt disease and AMD (19,35,36). Here, we have proposed that, rather than these condensation products, all-trans-retinal exerts a direct toxic effect on retinal cells resulting in degeneration (19,24,35,36). Retinol red/ox potentials and NADH/NADPH production could be the most likely reason for the differences between human and mouse photoreceptor cells. Notably, the flux of retinoids (rate of visual pigment regeneration) is substantially faster in humans than in mice, especially after strong bleaching (37). When this regenerative flux was lowered in Abca4−/−Rdh8−/− mice, their retinas became highly susceptible to degeneration. Finally, the higher levels of retinoids in the retina of humans, compared with those in mice, combined with a fully reversible all-trans-retinol ↔ all-trans-retinal reaction could result in higher levels of truly toxic all-trans-retinal. Thus, specific therapies geared toward lowering the effective concentration of all-trans-retinal should be critical for effective treatment (24).
Several published studies have assessed the stability of a number of ABCA4 mutants based on their expression levels in mammalian cell cultures (8,9). In the present work, we found that expression levels of the P and V mutant proteins as well as the PV double mutant were comparable with that of WT ABCA4. This contrasts with the results obtained in the mouse model, where the level of ABCA4 carrying the two mutations was drastically diminished early in life. Therefore, even though in the absence of more precise approaches the recombinant expression level is often used as an indicator of protein stability, results can be misleading when sufficient differences exist between the native and recombinant expression systems with respect to protein synthesis, transport and maintenance.
In conclusion, we generated a new mouse model of Stargardt disease with a complex mutation in the Abca4 gene. In contrast to previous Abca4−/− mice, Abca4PV/PV mice genetically resemble the human condition with a rapidly progressive disease phenotype. Structural instability of the mutant protein causes its rapid degradation and clearance from photoreceptor cells, suggesting that murine photoreceptor cells have a high capacity to degrade misfolded proteins, as is documented for even more highly expressed rhodopsin mutants (25,26,38). Whether impairing the UPR in Abca4PV/PV mice will result in more substantial retinal degeneration remains to be determined.
Materials and Methods
Human subjects
This study population involved five human subjects, each with two known ABCA4 alleles. Informed consent was obtained from all five individuals, and all procedures followed the guidelines of the Declaration of Helsinki and had institutional review board approval. A complete eye examination was performed for all subjects including best-corrected visual acuity and Goldmann kinetic perimetry examinations. Sensitivities were measured with a modified automated perimeter across the full visual field at 71 loci placed on a 12° square grid (5). Chromatic (500 nm, blue, and 650 nm, red) stimuli (1.7° diameter, 200 ms duration) were used under dark-adapted conditions to estimate rod-mediated vision. The preferred locus of fixation was documented by fundus imaging (39), and psychophysical tests were adjusted to each individual's preferred fixation locus. Sensitivity losses were calculated for each locus by comparison with locus-specific mean normal values. Mean retina-wide loss of sensitivity was calculated by including loci at ≥30° eccentricity from the fovea (5). In two individuals, macular imaging was performed with a confocal scanning laser ophthalmoscope (HRA2 or Spectralis, Heidelberg Engineering, Dossenheim, Germany) as previously described (40).
Animals
Abca4PV/PV mice were generated by inGenious Targeting Laboratories (Ronkonkoma, NY, USA), whereas Abca4−/− mice were bred as described previously (23). Gnat1–/– mice (41) were crossbred with Abca4PV/PV mice for cone recordings. Albino Abca4PV/PV mice were bred for TPM experiments by backcrossing with 129P3/J or 129X1/SvJ mice (The Jackson Laboratory, Bar Harbor, ME, USA). The lack of rd/rd and rd8 mutations in these animals was verified by published methods (42,43). All mice had a RPE65 Leu450 genetic background. Animals were routinely maintained in a 12-h light (<10 lux)/12-h dark cycle with ad libitum access to food and water. Anesthesia and euthanasia were performed in compliance with the American Veterinary Medical Association Guidelines on Euthanasia after approval by the Case Western Reserve University Institutional Animal Care and Use Committee.
Generation of Abca4PV/PV mice
The first targeting construct containing the L541P point mutation and a LoxP-flanked neomycin cassette before exon 12 of the mouse Abca4 gene was transfected by electroporation into C57BL/6 × 129/SvEv hybrid embryonic stem (ES) cells. A selected recombinant ES clone was then transiently transfected with a Cre recombinase expression vector to remove the Neo cassette. The resulting ES cells were further transfected with a second targeting construct containing the A1038V point mutation and an FRT-flanked Neo-cassette after exon 22. Targeted ES cells then were microinjected into C57BL/6 blastocysts. The resulting chimeras were mated with WT C57BL/6N mice to generate F1 heterozygous offspring. These heterozygous mice were crossed with 129S4/SvJ FLP+/+ mice (The Jackson Laboratory, stock #003946) to obtain F2 heterozygous knock-in FLP+/− mice with a deleted neomycin cassette. Such mice were further bred with 129S6/SvEvTac mice (Taconic, Hudson, NY, USA) to obtain F3 Neo-deleted heterozygous knock-in mice without the FLP transgene. F4 homozygous knock-in mice were obtained by cross-breeding F3 heterozygous mice. The final resulting Abca4PV/PV mice carried two point mutations in the Abca4 gene: L541P in exon 12 and A1038V in exon 21. These mice also had two additional residual sequences: a LoxP-derived insertion before exon 12 and an FRT/LoxP insertion after exon 22 (Fig. 5A).
Genotyping the Abca4PV/PV mouse
PCR primers for genotyping were designed to amplify the intron with a residual 141-bp FRT/LoxP insert to distinguish the knock-in allele from the WT allele (NDU2: 5′ GTG GCC TAT GGC TGA AAC AGT C 3′; NDD2: 5′ GAG GGA TAG ACA AGT AAA GGG C 3′).
RNA-Seq analyses
Mice were euthanized by cervical dislocation, their eyes were enucleated, and retinas and RPE/choroids were carefully dissected out and immediately placed in RNALater stabilization reagent (Qiagen, Germantown, MD, USA) between 3 :00 and 5:00 PM. Retinas and RPE/choroids were immediately homogenized and passed through a QIAShredder column (Qiagen) to amplify the homogenization step. Total RNA was then purified by using the RNeasy Mini Kit (Qiagen) with on-column DNase treatment (Qiagen) as per the manufacturer's directions. For transcriptome analyses, RNA-Seq libraries were prepared with the Illumina TruSeq Stranded total RNA kit with Ribo-zero Gold by following the protocol provided. Samples were pooled, denatured and diluted according to Illumina's best practices and loaded onto the HiSeq 2500 instrument using a Rapid Run Paired-end flow cell with a run type of 100 bp paired-end in the Case Western Reserve University Genomics Core Facility. Sequencing reads generated from the Illumina platform were trimmed for quality and adapter sequences. Reads were then aligned to the University of California, Santa Cruz mouse genome assembly and transcript notation (mm9). Reads were mapped with tophat software (44), which implements the bowtie2 (45) algorithm for sequence alignment and analyzes the mapped reads to identify splice junctions between exons. Tophat alignments were then analyzed with cufflinks (46). The RNA-Seq analysis package recorded the fragments per kilobase of exon per million fragments mapped (FPKM) values. FPKM values were then used to identify differentially expressed genes employing a significance cutoff for the adjusted P-value (q-value) of <0.05 as a statistically significant different expression. From significantly different expressed genes, we selected those with a minimum mean expression level of 10 FPKM and a fold change of either >2 or <0.5 (Log2 > 1 or <−1) for further investigation.
Real-time PCR
Total RNA (∼1 μg) from mouse retinas and RPE/choroids was treated with DNase (Qiagen) and reverse-transcribed with a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) in a total volume of 20 μl. Next, 0.5% of this reaction mixture was used as a template for an RT–PCR reaction with Power SYBR Green PCR Master Mix (Applied Biosystems) following the manufacturer's instructions. Primers used to amplify Abca4 were as follows: forward, 5′ GGG CTT CCT CAG ACA GAT ACA G 3′; reverse, 5′ GAG TGG GTT GGC ATT CCT CA 3′. Mouse Gapdh served as a control with the primers: forward, 5′ GTG AAG GTC GGT GTG AAC GG 3′; reverse, 5′ GCC GTT GAA TTT GCC GTG AG 3′. All real-time experiments were carried out with an Applied BioSystems Step-One Plus RT–PCR machine.
Ultra-high-resolution SD-OCT and SLO imaging
For all in vivo imaging experiments, mice were anesthetized by intraperitoneal (IP) injection of a cocktail (10 μl/g body weight) containing ketamine (10 mg/ml) and xylazine (0.4 mg/ml) in phosphate-buffered saline (PBS) (HyClone Laboratories, South Logan, UT, USA). Pupils were dilated with 1% tropicamide (Bausch and Lomb, Rochester, NY, USA). Ultra-high-resolution SD-OCT (Bioptigen, Morrisville, NC, USA) was employed for imaging (47). Four pictures acquired in the B-scan mode were used to construct each final averaged 2D SD-OCT image. Mouse fundus imaging was performed with a confocal scanning laser ophthalmoscope (cSLO; SpectralisHRA2, Heidelberg Engineering, Heidelberg, Germany) with a 55° lens. A near infrared reflectance image (IR mode, 820-nm laser) was used to align the fundus camera and the pupil to acquire an evenly illuminated fundus image. The FA mode (488-nm laser) was employed for detecting autofluorescence.
Generation of monoclonal antibodies
Anti-ABCA4 monoclonal antibodies (TMR1 and TMR4) were raised against full-length ABCA4 isolated from bovine retinas following a published procedure (14). The purified protein was emulsified with an adjuvant system (Sigma–Aldrich, St. Louis, MO, USA) according to the manufacturer's protocol, and the antigen (50 μg/mouse) was injected intraperitoneally into 4-week-old BALB/c mice (The Jackson Laboratory). This immunization procedure was repeated three times every 10–12 days. After the third immunization, mouse serum titers were checked by immunoblotting with native bovine ABCA4. Purified native bovine ABCA4 (30 μg/mouse) in PBS was used for the final immunization performed 48 h before fusion. Hybridoma cell lines were prepared by fusion of SP2 mouse myeloma cells (American Type Culture Collection, Manassas, VA, USA) (7.7 × 106) with splenocytes (2.3 × 107) from an immunized mouse by using polyethylene glycol 1500 (Roche Applied Science, Indianapolis, IN, USA). Culture supernatants from the resulting hybridomas were screened with purified ABCA4 and ABCA4 domains expressed in bacteria (13), and bovine rod outer segment proteins were detected by enzyme-linked immunosorbent assays and immunoblotting. Positive hybridomas were subcloned three times by the method of limiting dilution in microtiter plates. IgG isotypes were examined with a mouse monoclonal isotyping test kit (Roche Applied Science).
Other antibodies
1D4 and Rim3F4 mouse monoclonal antibodies (mAbs) were kind gifts from Dr R. Molday (University of British Columbia) (48,49). Rabbit anti-GFAP polyclonal antibody (pAb) was from Dako (Carpinteria, CA, USA).
Immunohistochemistry
Cryosections prepared as described in a previous study (16) were incubated with primary antibodies: 1D4 (5 mg/ml; 1 : 10 000), Rim3F4 (1 mg/ml, 1 : 50) or anti-GFAP antibody (1 : 400) and biotinylated PNA (1 : 400, Vector Laboratories, Burlingame, CA, USA) and then stained with Alexa Fluor 488- or Cy3-conjugated goat anti-mouse IgGs (1 : 500, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) or Alexa Fluor 488-conjugated streptavidin (1 : 500, Invitrogen, Carlsbad, CA, USA). Fluorescence was detected with a Leica TCS SP5 upright confocal microscope.
Immunoblotting
Retinas from both eyes of a mouse were homogenized in 100 μl of ice-cold RIPA buffer (150 mm NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 50 mm Tris, pH 8.0) with 2% protease inhibitors (Sigma–Aldrich). Ten microliters of each protein sample was loaded on an SDS–PAGE gel and transferred to PVDF membranes. Monoclonal primary antibody TMR4 (1 : 400) used to probe for ABCA4 protein, and the combination was detected by incubation with goat anti-mouse secondary antibodies conjugated with horseradish peroxidase (1 : 1000). After incubation with a chemiluminescent substrate (SuperSignal West Pico Substrate, Thermo Scientific, Rockford, IL, USA), signals were detected by exposure to X-ray film (CL-XPosure, Thermo Scientific) and developed with an X-ray film processor (Mini-medical 90, ImageWorks, Elmsford, NY, USA).
Histology
Light and transmission EM histological experiments were performed in accordance with published protocols (16). Bright-field images were captured by a Leica CTR6000 microscope attached to a CCD camera (Micropublisher 5.0 RTV, Qimaging Surray BC, Canada). EM imaging was accomplished with a Tecnai T12 electron microscope (FEI).
Electroretinography (ERG)
Mice were dark-adapted overnight and anesthetized with an IP injection of a mixture of ketamine (100 mg/kg) and xylazine (20 mg/kg). Pupils were dilated with a drop of 1% atropine sulfate. Mouse body temperature was maintained at 37°C with a heating pad or circulating water bath. Full-field ERG responses were recorded from both eyes with a UTAS E3000 MF or BigShot system (LKC Technologies, Gaithersburg, MD, USA). For rod dark adaptation experiments, 1-month-old mice were exposed for 2 min or 8-month-old mice were exposed for 20 s to a 500 lux Ganzfeld background white light to bleach >90% of rhodopsin. After the bleach, recovery of the ERG response was recorded in the dark. For cone dark adaptation experiments, mice with a Gnat1−/− background were exposed to bright light delivered by a pair of 520-nm LEDs focused on the surface of mouse eye cornea for 30 s to bleach >90% of the M/L-cone pigments. The bleached fraction was estimated with the following equation:
where F is the fraction of bleached pigment, t is the duration of exposure to light (s), I is the bleaching light intensity of unattenuated 520-nm LED light (∼1.3 × 108 photons µm−2 s−1) and P is the photosensitivity of the mouse cone at the wavelength of peak absorbance (7.5 × 10−9 µm2), adopted from Nikonov et al. (50). After the bleach, the recovery of cone ERG b-wave flash sensitivity (Sf) was followed in the dark.
Transretinal ERG recordings
After dark-adaption overnight, mice were euthanized by CO2 asphyxiation, and their retinas were removed under infrared illumination. A whole single retina was placed in the recording chamber with the photoreceptor side up between two electrodes connected to a differential amplifier. The retina was perfused with Locke's solution (112.5 mm NaCl, 3.6 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 10 mm HEPES, pH 7.4, 20 mm NaHCO3, 3 mm Na succinate, 0.5 mm Na glutamate, 0.02 mm EDTA and 10 mm glucose). The perfusion solution was supplemented with 1–2 mm L-glutamate and 10–40 µm DL-2-amino-4-phosphonobutyric acid (DL-AP4) to block synaptic components of the photoresponse (51), 20–70 µm BaCl2 to suppress the slow glial PIII component (52) and MEM vitamin and amino acid solutions (Sigma–Aldrich) to retain retinal viability. The solution was continuously bubbled with a 95% O2/5% CO2 mixture and maintained at 36–37°C.
Light stimulation was applied in 20-ms test flashes of calibrated 505-nm LED light. For light uniformity, a glass optical diffuser was placed between the LED and the retina. The stimulating light intensity was controlled by a computer in 0.5 log unit steps. Photoresponses were amplified by a differential amplifier (DP-311; Warner Instruments, Hamden, CT, USA), low-pass filtered at 300 Hz (8-pole Bessel), digitized at 1 kHz and stored in a computer for further analysis. The intensity–response relationships were fitted with Naka-Rushton hyperbolic functions, as follows:
for fitting absolute data, or
for fitting normalized data, where R is the transient peak response amplitude, Rmax is the maximal response amplitude, I is the flash intensity, n is the Hill coefficient and I1/2 is the half-saturating light intensity. Time to peak was measured from the dim flash response as the time between the flash onset to the transient peak of the response; integration time was measured from dim flash responses as the area of the response normalized by its amplitude.
Retinoid analyses
Retinoid extraction, derivatization and separation by HPLC were performed on samples extracted from eyes. Mice were euthanized by cervical dislocation either immediately after 2 min of light exposure to warm white fluorescent light (38.5 klux) or at indicated times after dark adaptation following such exposure, and eyes were excised with curved scissors. Eyes were deep-frozen in liquid nitrogen immediately after removal and stored at −80°C until used. For analyses, tubes with eyes were transferred to dry ice, and initially frozen eyes were homogenized with a glass/glass homogenizer in 1 ml of retinoid analysis buffer (50 mm MOPS, 10 mm NH4OH and 50% ethanol in H2O, pH 7.0). Retinoids were extracted twice by addition of 4 ml hexane, vortexing and centrifugation. Hexane fractions with retinoids were collected and dried in a SpeedVac concentrator, resuspended in 400 μl hexane and then separated by normal-phase HPLC (Ultrasphere-Si, 4.6 × 250 mm; Beckman Coulter, Brea, CA, USA) in 10% ethyl acetate and 90% hexane at a flow rate of 1.4 ml/min (25,35,36).
A2e quantification
Mouse eyes were obtained as described in the previous paragraph. A2E was extracted twice from both frozen eyes of each mouse in 1 ml of acetonitrile after homogenization with a Brinkmann Politron homogenizer (Kinematica, Lucerne, Switzerland). After evaporation of solvent, extracts were dissolved in 150 μl acetonitrile with 0.1% TFA and then filtered through a Teflon syringe filter. Samples (100 μl) were loaded on C18 columns (Phenomenex, Torrance, CA, USA) and analyzed by reverse-phase HPLC with a mobile phase gradient of acetonitrile:H2O from 100:0 to 80:20 with 0.1% TFA for 20 min. Quantification of A2E was performed by comparison with known concentrations of a pure synthetic A2E standards prepared according to Parish et al. (53).
Two-photon microscopy
TPM images of the RPE in intact eyes of 3-month-old mice were obtained with a Leica TCS SP5 upright confocal microscope immediately after eye enucleation as previously described (19). The microscope was equipped with a 1.0 NA water immersion objective and a femtosecond laser Vision S (Coherent, Santa Clara, CA, USA) tunable from 690–1020 nm. RPE images were obtained with 730 and 850 nm excitation light using 5 mW of laser power and the same detector gain for both wavelengths. To avoid artifacts introduced by different transparencies of the sclera and choroid, we used fluorescence ratios to quantify differences among mice with different genetic backgrounds. Ratios of fluorescence excited with 850-nm light to that excited with 730 nm were calculated off-line by using pixel gray values of raw retinal images obtained with Leica LAS AF 3.0.0 software. Sigma Plot 11.0 software (Systat Software, Inc., San Jose, CA, USA) was employed for statistical analyses.
Expression and purification of ABCA4
WT human ABCA4 as well as P, V and PV mutants encoded in pCEP4 plasmids were transiently expressed in adherent HEK293 or COS-7 cells and isolated as we reported previously (13). Briefly, cell membranes were solubilized with n-dodecyl-β-d-maltopyranoside (DDM) (Anatrace, Maumee, OH, USA) in the presence of porcine brain polar lipids (Avanti Polar Lipids, Alabaster, AL, USA), and the proteins were isolated with a combination of lectin affinity chromatography on agarose-conjugated lectin from Galanthus nivalis (GNL, Vector Laboratories) and immunoaffinity chromatography on agarose-conjugated Rim3F4 monoclonal antibody. Protein purity was monitored with sodium dodecyl sulfate-polyacrylamide gel electrophoresis. For structural analyses, DDM was replaced with Amphipol A8–35 (Anatrace) (13).
ABCA4 activity measurements
Basal and all-trans-retinal-stimulated ATPase activities were evaluated after reconstitution of ABCA4 into lipid vesicles following our published protocol (13). Experiments were repeated three times with different protein preparations, and each of these involved at least three independent measurements.
Immunofluorescent labeling of cells
HEK293 or COS-7 cells were grown on glass coverslips and transfected with X-tremeGENE 9 DNA transfection reagent (Roche) according to the manufacturer's protocol. After 48 h, cells were washed with PBS, fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS. After blocking with 5% goat serum in PBS, cells were incubated with monoclonal anti-ABCA4 antibody TMR1 and a rabbit polyclonal anti-calreticulin antibody (Millipore, Billerica, MA, USA). Goat anti-mouse IgG labeled with Alexa-647 and goat anti-rabbit IgG labeled with Cy3 were used as secondary antibodies. Stained cells were visualized with a laser scanning confocal microscope (Leica TCS SP2).
EM and single-particle analyses
Negatively stained grids were prepared as we described earlier (13). Micrographs were recorded with a FEI Tecnai F20 microscope (FEI, Eindhoven, Netherlands) operated at 200 kV and equipped with a Gatan US4000 UHS CCD camera (4K × 4 K) at a magnification of ×71 600. The pixel size at the specimen level was 2.26 Å/pixel. Particles were picked manually with e2boxer from the EMAN2 software package (54). The data sets for WT ABCA4 and the P, V and PV mutants contained 10 215, 11 489, 6979 and 15 075 particles, respectively. After contrast transfer function correction, images were aligned and classified based on correspondence analysis in SPIDER (55). The initial 3D model of WT ABCA4 was obtained with the e2initial model module of EMAN2. This model was used as the reference for the referenced-based projection alignment and 3D reconstruction in SPIDER. The reconstructed structure was then subjected to multiple rounds of angular refinement. The final structure of the WT protein was used as the reference to obtain the reconstruction of the V mutant. For the P and PV mutants, the reference for the reference-based projection alignment and 3D reconstruction was obtained by segmentation of the WT ABCA4 map using UCSF Chimera (56).
Database
RNA-Seq data are available in the GEO database under accession number GSE63772.
Supplementary Material
Funding
This work was supported by funding from the National Institutes of Health [K08EY019031 (A.M.), EY009339 (K.P.), EY021126 (K.P.), EY13203 (A.V.C.), EY022658 (A.M.), AG043645 (G.P.), EY019312 (V.J.K.), EY021126 (V.J.K.) and P30 EY11373], the Research to Prevent Blindness Foundation, Foundation Fighting Blindness, and Fight for Sight, the Ohio Lions Eye Research Foundation. N.Z. was supported by the Knights Templar Eye Foundation. A.V.C. is a RPB Senior Scientific Scholar. K.P. is John H. Hord Professor of Pharmacology.
Supplementary Material
Acknowledgements
We thank Dr Leslie T. Webster Jr. and members of Palczewski laboratory for helpful comments on this manuscript and Drs Mee Jee Lee, Nathan Alexander, Ernest R. Chan, Hisashi Fujioka, Jasmine Feng, Debarshi Mustafi, Tivadar Orban, Brian Kevany and Thomas Sundermeier (all from CWRU) for support and advice during this study.
Conflict of Interest statement. None declared.
References
- 1.Gregersen N., Bross P., Vang S., Christensen J.H. (2006) Protein misfolding and human disease. Annu. Rev. Genomics Hum. Genet., 7, 103–124. [DOI] [PubMed] [Google Scholar]
- 2.Quazi F., Lenevich S., Molday R.S. (2012) ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun., 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiser P.D., Golczak M., Palczewski K. (2014) Chemistry of the retinoid (visual) cycle. Chem. Rev., 114, 194–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zernant J., Xie Y.A., Ayuso C., Riveiro-Alvarez R., Lopez-Martinez M.A., Simonelli F., Testa F., Gorin M.B., Strom S.P., Bertelsen M., et al. (2014) Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet., 23, 6797–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cideciyan A.V., Swider M., Aleman T.S., Tsybovsky Y., Schwartz S.B., Windsor E.A., Roman A.J., Sumaroka A., Steinberg J.D., Jacobson S.G., et al. (2009) ABCA4 disease progression and a proposed strategy for gene therapy. Hum. Mol. Genet., 18, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivera A., White K., Stohr H., Steiner K., Hemmrich N., Grimm T., Jurklies B., Lorenz B., Scholl H.P., Apfelstedt-Sylla E., et al. (2000) A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet., 67, 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiszniewski W., Zaremba C.M., Yatsenko A.N., Jamrich M., Wensel T.G., Lewis R.A., Lupski J.R. (2005) ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum. Mol. Genet., 14, 2769–2778. [DOI] [PubMed] [Google Scholar]
- 8.Tsybovsky Y., Wang B., Quazi F., Molday R.S., Palczewski K. (2011) Posttranslational modifications of the photoreceptor-specific ABC transporter ABCA4. Biochemistry, 50, 6855–6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun H., Smallwood P.M., Nathans J. (2000) Biochemical defects in ABCR protein variants associated with human retinopathies. Nat. Genet., 26, 242–246. [DOI] [PubMed] [Google Scholar]
- 10.Zhong M., Molday L.L., Molday R.S. (2009) Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J. Biol. Chem., 284, 3640–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quazi F., Molday R.S. (2013) Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J. Biol. Chem., 288, 34414–34426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahn J., Beharry S., Molday L.L., Molday R.S. (2003) Functional interaction between the two halves of the photoreceptor-specific ATP binding cassette protein ABCR (ABCA4). Evidence for a non-exchangeable ADP in the first nucleotide binding domain. J. Biol. Chem., 278, 39600–39608. [DOI] [PubMed] [Google Scholar]
- 13.Tsybovsky Y., Palczewski K. (2014) Expression, purification and structural properties of ABC transporter ABCA4 and its individual domains. Protein Expr. Purif., 97, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsybovsky Y., Orban T., Molday R.S., Taylor D., Palczewski K. (2013) Molecular organization and ATP-induced conformational changes of ABCA4, the photoreceptor-specific ABC transporter. Structure, 21, 854–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bringmann A., Pannicke T., Grosche J., Francke M., Wiedemann P., Skatchkov S.N., Osborne N.N., Reichenbach A. (2006) Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res., 25, 397–424. [DOI] [PubMed] [Google Scholar]
- 16.Zhang N., Kolesnikov A.V., Jastrzebska B., Mustafi D., Sawada O., Maeda T., Genoud C., Engel A., Kefalov V.J., Palczewski K. (2013) Autosomal recessive retinitis pigmentosa E150 K opsin mice exhibit photoreceptor disorganization. J. Clin. Invest., 123, 121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calvert P.D., Krasnoperova N.V., Lyubarsky A.L., Isayama T., Nicolo M., Kosaras B., Wong G., Gannon K.S., Margolskee R.F., Sidman R.L., et al. (2001) Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha-subunit (vol 97, pg 13913, 2000). Proc. Natl Acad. Sci. USA, 98, 10515–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolesnikov A.V., Tang P.H., Parker R.O., Crouch R.K., Kefalov V.J. (2011) The mammalian cone visual cycle promotes rapid M/L-cone pigment regeneration independently of the interphotoreceptor retinoid-binding protein. J. Neurosci., 31, 7900–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda A., Palczewska G., Golczak M., Kohno H., Dong Z., Maeda T., Palczewski K. (2014) Two-photon microscopy reveals early rod photoreceptor cell damage in light-exposed mutant mice. Proc. Natl Acad. Sci. USA, 111, E1428–E1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palczewska G., Dong Z., Golczak M., Hunter J.J., Williams D.R., Alexander N.S., Palczewski K. (2014) Noninvasive two-photon microscopy imaging of mouse retina and retinal pigment epithelium through the pupil of the eye. Nat. Med., 20, 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palczewska G., Maeda T., Imanishi Y., Sun W., Chen Y., Williams D.R., Piston D.W., Maeda A., Palczewski K. (2010) Noninvasive multiphoton fluorescence microscopy resolves retinol and retinal condensation products in mouse eyes. Nat. Med., 16, 1444–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsybovsky Y., Molday R.S., Palczewski K. (2010) The ATP-binding cassette transporter ABCA4: structural and functional properties and role in retinal disease. Adv. Exp. Med. Biol., 703, 105–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maeda A., Maeda T., Golczak M., Palczewski K. (2008) Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem., 283, 26684–26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maeda A., Golczak M., Chen Y., Okano K., Kohno H., Shiose S., Ishikawa K., Harte W., Palczewska G., Maeda T., et al. (2012) Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat. Chem. Biol., 8, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakami S., Maeda T., Bereta G., Okano K., Golczak M., Sumaroka A., Roman A.J., Cideciyan A.V., Jacobson S.G., Palczewski K. (2011) Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem., 286, 10551–10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiang W.C., Kroeger H., Sakami S., Messah C., Yasumura D., Matthes M.T., Coppinger J.A., Palczewski K., LaVail M.M., Lin J.H. (2014) Robust endoplasmic reticulum-associated degradation of rhodopsin precedes retinal degeneration. Mol. Neurobiol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charbel Issa P., Barnard A.R., Singh M.S., Carter E., Jiang Z., Radu R.A., Schraermeyer U., MacLaren R.E. (2013) Fundus autofluorescence in the Abca4(-/-) mouse model of Stargardt disease—correlation with accumulation of A2E, retinal function, and histology. Invest. Ophthalmol. Vis. Sci., 54, 5602–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cideciyan A.V., Aleman T.S., Swider M., Schwartz S.B., Steinberg J.D., Brucker A.J., Maguire A.M., Bennett J., Stone E.M., Jacobson S.G. (2004) Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum. Mol. Genet., 13, 525–534. [DOI] [PubMed] [Google Scholar]
- 29.Eldred G.E., Lasky M.R. (1993) Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature, 361, 724–726. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z., Gerstein M., Snyder M. (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet., 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weng J., Mata N.L., Azarian S.M., Tzekov R.T., Birch D.G., Travis G.H. (1999) Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell, 98, 13–23. [DOI] [PubMed] [Google Scholar]
- 32.Quazi F., Molday R.S. (2014) ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc. Natl Acad. Sci. USA, 111, 5024–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J.S., Kefalov V.J. (2011) The cone-specific visual cycle. Prog. Retin. Eye Res., 30, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J.S., Kefalov V.J. (2009) An alternative pathway mediates the mouse and human cone visual cycle. Curr. Biol., 19, 1665–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y., Okano K., Maeda T., Chauhan V., Golczak M., Maeda A., Palczewski K. (2012) Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J. Biol. Chem., 287, 5059–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maeda A., Maeda T., Golczak M., Chou S., Desai A., Hoppel C.L., Matsuyama S., Palczewski K. (2009) Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem., 284, 15173–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamb T.D., Pugh E.N., Jr. (2004) Dark adaptation and the retinoid cycle of vision. Prog. Retin. Eye Res., 23, 307–380. [DOI] [PubMed] [Google Scholar]
- 38.Sakami S., Kolesnikov A.V., Kefalov V.J., Palczewski K. (2014) P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum. Mol. Genet., 23, 1723–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cideciyan A.V., Swider M., Aleman T.S., Feuer W.J., Schwartz S.B., Russell R.C., Steinberg J.D., Stone E.M., Jacobson S.G. (2012) Macular function in macular degenerations: repeatability of microperimetry as a potential outcome measure for ABCA4-associated retinopathy trials. Invest. Ophthalmol. Vis. Sci., 53, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cideciyan A.V., Swider M., Aleman T.S., Roman M.I., Sumaroka A., Schwartz S.B., Stone E.M., Jacobson S.G. (2007) Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J. Opt. Soc. Am. A Opt. Image Sci. Vis., 24, 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calvert P.D., Krasnoperova N.V., Lyubarsky A.L., Isayama T., Nicolo M., Kosaras B., Wong G., Gannon K.S., Margolskee R.F., Sidman R.L., et al. (2000) Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha -subunit. Proc. Natl Acad. Sci. USA, 97, 13913–13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pittler S.J., Baehr W. (1991) Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc. Natl Acad. Sci. USA, 88, 8322–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehalow A.K., Kameya S., Smith R.S., Hawes N.L., Denegre J.M., Young J.A., Bechtold L., Haider N.B., Tepass U., Heckenlively J.R., et al. (2003) CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet., 12, 2179–2189. [DOI] [PubMed] [Google Scholar]
- 44.Trapnell C., Pachter L., Salzberg S.L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B., Salzberg S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol., 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Q., Sun W., Okano K., Chen Y., Zhang N., Maeda T., Palczewski K. (2011) Sponge transgenic mouse model reveals important roles for the microRNA-183 (miR-183)/96/182 cluster in postmitotic photoreceptors of the retina. J. Biol. Chem., 286, 31749–31760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oprian D.D., Molday R.S., Kaufman R.J., Khorana H.G. (1987) Expression of a synthetic bovine rhodopsin gene in monkey kidney cells. Proc. Natl Acad. Sci. USA, 84, 8874–8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Illing M., Molday L.L., Molday R.S. (1997) The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J. Biol. Chem., 272, 10303–10310. [DOI] [PubMed] [Google Scholar]
- 50.Nikonov S.S., Kholodenko R., Lem J., Pugh E.N., Jr. (2006) Physiological features of the S- and M-cone photoreceptors of wild-type mice from single-cell recordings. J. Gen. Physiol., 127, 359–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sillman A.J., Ito H., Tomita T. (1969) Studies on the mass receptor potential of the isolated frog retina. I. General properties of the response. Vision Res., 9, 1435–1442. [DOI] [PubMed] [Google Scholar]
- 52.Nymark S., Heikkinen H., Haldin C., Donner K., Koskelainen A. (2005) Light responses and light adaptation in rat retinal rods at different temperatures. J. Physiol., 567, 923–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parish C.A., Hashimoto M., Nakanishi K., Dillon J., Sparrow J. (1998) Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc. Natl Acad. Sci. USA, 95, 14609–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang G., Peng L., Baldwin P.R., Mann D.S., Jiang W., Rees I., Ludtke S.J. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol., 157, 38–46. [DOI] [PubMed] [Google Scholar]
- 55.Shaikh T.R., Gao H., Baxter W.T., Asturias F.J., Boisset N., Leith A., Frank J. (2008) SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc., 3, 1941–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem., 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.