

Abstract
The elongation condensing enzymes in the bacterial fatty acid biosynthesis pathway represent desirable targets for the design of novel, broad-spectrum antimicrobial agents. A series of substituted benzoxazolinones was identified in this study as a novel class of elongation condensing enzyme (FabB and FabF) inhibitors using a two-step virtual screening approach. Structure activity relationships were developed around the benzoxazolinone scaffold showing that N-substituted benzoxazolinones were most active. The benzoxazolinone scaffold has high chemical tractability making this chemotype suitable for further development of bacterial fatty acid synthesis inhibitors.
Keywords: virtual screening, fatty acid synthesis, condensing enzymes, antibiotics
Bacterial fatty acid biosynthesis has emerged as an appealing target for the development of novel antibacterial chemotherapeutics.1 Mammals utilize a single multifunctional enzyme complex that is structurally distinct from the dissociated bacterial system (FASII).2 The catalytic steps are the same in both systems, but significant structural differences between the mammalian and bacterial system can be exploited to design specific inhibitors.1,2 Multiple inhibitors of the enoyl-acyl carrier protein (ACP) reductase step (FabI) have been described.3 For example, AFN-1252 is a nanomolar FabI inhibitor that eradicates Staphylococcus aureus infections.4,5,6 However, FabI inhibitors are not broad spectrum agents because many important pathogens express structurally distinct enoyl-ACP reductases (FabK, FabL or FabV) that are refractory to FabI inhibitors.3 Our work is focused on the elongation condensing enzymes (3-ketoacyl-ACP synthase) because they are ubiquitously expressed in bacteria and the two subgroups (FabB and FabF) have superimposable active sites.7 These targets are essential in Gram-negative bacteria. Although this group of bacteria can incorporate extracellular fatty acids into phospholipid, FASII is required to produce the acyl chains in the lipopolysaccharide of the outer membrane.1 Some Gram-positive bacteria (Streptococci) can circumvent FASII inhibitors by incorporating extracellular fatty acids, but others (Staphylococci) require FASII even when environmental fatty acids are present8 The natural products cerulenin, thiolactomycin (TLM) and platensimycin target both FabB and FabF.9,10 These inhibitors are broad-spectrum agents with in vivo efficacy against Gram-positive and Gram-negative bacteria. However, they have severe limitations, including substandard pharmacokinetic properties and limited synthetic access.11 New chemical scaffolds are clearly needed, and this paper describes a virtual screening approach to discover a novel class of elongation condensing enzyme inhibitors using E. coli FabB as the model.
The high resolution crystal structure of the FabB-TLM binary complex12 was used as the template to identify the key pharmacophore features to be incorporated into the design of new condensing enzyme inhibitors. TLM binds non-covalently adjacent to the active site residue Cys163 (Fig. 1).12 The carbonyl group forms hydrogen bonds with both His298 and His333 in the active site, and the isoprenoid moiety slides into a tight hydrophobic pocket sandwiched between Gly391/Phe392 and Ala271/Pro272. A standard molecular dynamics simulation using AMBER13 with a production run of 5 ns was carried out to gain a dynamic picture of TLM binding as well to optimize hydrogen positions. The chain A in the co-crystal structure of TLM bound to E. coli FabB (PDB: 2VB8)14 was used as the starting conformation. The complex system was solvated in explicit water molecules with counter ions to neutralize the system. Energy minimization was performed first with solute constrained then released. The system temperature was slowly heated from 0 to 300K followed by equilibration and production simulation. The observed binding mode of TLM in the crystal structure was highly stable and all key interactions were maintained through simulation. A free energy analysis was executed to provide residue-based energy contribution to the TLM binding (Fig. 2A).15, 16 This analysis showed that His298, Phe392, Thr302, Phe390, Val270, Pro272, Thr300, Gly391 and His333 contribute to TLM binding.
Figure 1.

Binding modes of TLM and compound 14 to FabB. (A) Cocrystal structure of TLM in complex with FabB from E. coli (PDB: 2vb8). TLM is shown in spheres. (B) A close-up view of the interactions between TLM and the binding site. (C) Compound 14 (green) docked into the binding site superimposed with TLM (purple).
Figure 2.

The TLM pharmacophore model. (A) Decomposed free energy contribution per residue to TLM binding calculated from MD simulation. (B) Pharmacophore model developed in UNITY.
A two-step virtual screen was performed against E.coli FabB using a total of ≈1.1 million compounds from the Enamine (Advanced Collection) and Chembridge (EXPRESS-Pick Collection Stock and CORE Library Stock) libraries. A single-conformation UNITY17 database was created and 3D conformations were generated for each compound by Concord. Compound sets were filtered for a molecular weight cut off of 350 to search for lead-like inhibitors18 that allows for facile further modification. Using the key binding elements identified from MD simulation, a UNITY pharmacophore query was established including a hydrogen bond acceptor atom connected to a five-member ring that could form a bidentate interaction with His298 and His333 (Fig. 2B).19 Spatial constrains were applied to constrain the three-dimensional conformation of pharmacophore features. This pharmacophore query search was applied to remove inactives and to simplify the subsequent computationally expensive docking step. Compounds that successfully passed the filter (≈250, 000 compounds) were advanced into docking experiments using the Virtual Screening Workflow from Schrödinger.20 Compounds were filtered to remove those with reactive functional groups. Compounds were docked with Glide HTVS followed by Glide SP.21 Preference was given in docking to compounds that could form the desired hydrogen bonds with His298 and His333. The top ranking 500 compounds were preserved for further examination. After consideration of binding pose by eye, chemical diversity and tractability, a total of 31 compounds were prioritized and acquired for in vitro testing (see Supplementary Fig. 1). TLM was incorporated in the virtual screening library from the beginning as a positive control. This compound passed the pharmacophore search filter and was ranked very high in the subsequent docking study.
The purchased compounds were all analyzed by LCMS to confirm purity and identity. All 31 compounds passed this step and were then screened using NMR wLOGSY assay22 to evaluate FabB binding (see Supplementary Fig. 1). The eight compounds that exhibited binding in the NMR wLOGSY assay were further tested as FabB inhibitors using the condensing enzyme radiochemical assay previously described12 except myristoyl-ACP, which was substituted by lauryl-ACP synthesized using the Vibrio harveyii acyl-ACP synthetase enzyme.23 Initial analysis of inhibition was conducted at a single final inhibitor concentration of 200 µM. Both screens revealed that compound 1 exhibited binding in NMR and showed 24% inhibition at 200 µM in the enzymatic assay. Dose response experiments established an IC50 of 800 µM (Table 1). The IC50 for compound 1 was confirmed by resynthesis and retesting in the enzyme assay.
Table 1.
The structures and measured FabB inhibition of compounds from procurement and chemical synthesis arranged for SAR purposes. All compounds with stereochemistry were tested as racemates.
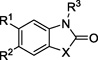 |
||||||
|---|---|---|---|---|---|---|
| # | X | R1 | R2 | R3 | %Inhibition @ 200 µM |
IC50(µM) |
| 1 | O | H | Cl | 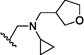 |
24.1 | 800 |
| 2 | O | H | H | 24 | ND | |
| 3 | O | Cl | H | 17.7 | ND | |
| 4 | O | H | Br | 17.7 | ND | |
| 5 | O | CH3 | H | 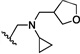 |
40.1 | ND |
| 6 | O | Cl | H | 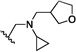 |
36.7 | 350 |
| 7 | O | H | Cl | 31.7 | ND | |
| 8 | O | H | Cl | 24.3 | ND | |
| 9 | O | H | Br | 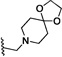 |
32.7 | 600 |
| 10 | O | H | Cl | 38.8 | ND | |
| 11 | O | Cl | H | 37.7 | 335 | |
| 12 | O | H | Cl | 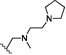 |
26.4 | ND |
| 13 | O | H | Cl | 23.8 | ND | |
| 14 | O | H | H | 33.5 | 235 | |
| 15 | O | H | Cl | 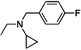 |
32.1 | ND |
| 16 | O | H | H | 5 | ND | |
| 17 | O | H | Cl | 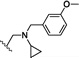 |
28.3 | ND |
| 18 | O | Cl | H | 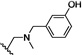 |
41.4 | ND |
| 19 | O | H | Cl | 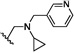 |
23.6 | ND |
| 20 | O | H | Cl | 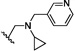 |
30 | ND |
| 21 | O | H | H | 30 | ND | |
| 22 | O | H | H | 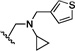 |
37.8 | ND |
| 23 | O | H | Cl | 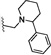 |
38 | ND |
| 24 | O | Cl | H | 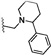 |
30.4 | 415 |
| 25 | O | H | Cl | 36.4 | ND | |
| 26 | O | Cl | H | 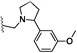 |
37.1 | 305 |
| 27 | O | Cl | H | 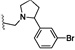 |
55.4 | 260 |
| 28 | O | Cl | H | 31.9 | ND | |
| 29 | O | CN | H | 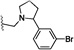 |
42.1 | 253 |
| 30 | S | Cl | H | 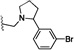 |
36.1 | 303 |
A similarity search performed in SciFinder uncovered 11 analogues of compound 1 that were ordered and tested. The scifinder analysis was complemented by a medicinal chemistry effort to fill in the SAR through synthesis of 18 additional compounds. Compounds were synthesized in one step from appropriately substituted benzoxazolinones and structurally unique secondary amines using Mannich chemistry (Supplementary Scheme 1).24 The inhibitory activities of compounds from both sets are listed and grouped by structure in Table 1.
Compared with the initial hit (1), many compounds in table 1 showed improved enzymatic inhibition at a single inhibitor concentration of 200 µM. Selected compounds were measured in dose response experiments producing IC50 values ranging from 235 – 625 µM (Table 1). Various benzoxazolinone substitutions were tested with R1 and R2 (5 and 6 position) halogen substitution producing the most potent analogs. It is notable that moving the chlorine atom from the 6 position in 1 to the 5 position in 6 substantially increased inhibitory activity. Most structural divergence in this series was introduced via R3 N-substitution to the benzoxazolinone core, and divergent secondary and cyclic amines were used to probe for improved sidechain moieties in three subseries. First, a series of similar aliphatic amine side chains (2–13) were examined. Substituting tetrahydrofuran with a cyclopentyl ring in compounds 3 and 4 resulted in loss of activity. Tetrahydropyran system (7 and 8) was well tolerated as well as corresponding spiro-tetrahydropyran moieties ( 10 and 11) which maintained inhibitory activity. Compound 11 had an IC50 of 335 µM, on par with the best tetrahydrofuran analogue (6). The pyrrolidine ring in 13 was well tolerated in comparison with tetrahydrofuran compound 1. In the second sub set (14–22), an aryl group was introduced into the side chain in place of the tetrahydrofuran group found in 1. The most potent analog of this set was the simple phenyl compound 14 and 18 with a hydroxyl group on the meta-position. Replacing phenyl group with 2-pyridyl (20) and 3-pyridyl (19) groups did not improve potency. However, bioisosteric phenyl replacement of 14 with a thiophene ring (21 and 22) was well tolerated. The third subseries was designed to conformationally restrict the ter-amine side chain (set 23–30) in which the benzyl group of compound 14 was attached by a 4- to 6-membered ring. Compounds 27 and 29 both with a 5-membered pyrrolidine ring and a bromo substitution on meta-position of the phenyl ring showed the best activity with IC50’s of 260 µM and 253 µM, respectively. The cyclopropane group (22) was also a better substitutent than methyl (21). Substituting 5-chloro of 27 with 5-cyano in 29, or changing benzoxazole with benzothiozole 30 yielded comparable IC50 values. The expectation that the benzoxazolinones would also be FabF inhibitors was tested by examining the IC50 of compound 14 against FabF from S. aureus. The IC50 of compound 14 against E. coli FabB was 235 µM compared to an IC50 of 387 µM against S. aureus FabF, indicatingthe potential for broad-spectrum activity of this scaffold. (Fig. 3)
Figure 3.

Compound 14 inhibition of E. coli FabB and S. aureus FabF in vitro. Maximum activities were: FabB, 4.7 nmoles/min/mg; FabF, 4.0 nmoles/min/mg.
The docking of compound 14 into the TLM binding site (Fig. 1C) provides a rationale for understanding the structural basis of benzoxazolinones binding. The superimposition of compound 14 with TLM bound to the E. coli FabB enzyme showed that the ketone group on the oxazole ring mimics the bidentate binding mode with the catalytic His298 and His333, and the methylaniline group occupied the same binding pocket as the isoprene side chain of TLM forming favorable van der Waals interactions. Other active compounds were also modeled to study their potential binding conformations. Compound 6 and 27 adopted very similar poses to each other and 14 (Supplementary Fig. 2 A,B). They both formed two hydrogen bonds with His298 and His333. The respective cyclopropane (6) and pyrrolidine (27) side chains occupy the same cavity above H298 that was originally partially filled by the chiral 5-methyl group on thiophene ring in TLM, while the tetrahydrofuran (6) and bromobenzene (27) rings are located in the same binding pocket as the methylaniline group in 14. Although an active inhibitor, compound 11 showed less favorable docking, as binding requires reorientation of the TLM binding site to fit the bigger azaspiro side chain, losing one hydrogen bond with His298 (Supplementary Fig. 2 C).
Compared to the FabB natural product inhibitors, the benzoxazolinones show selective inhibition of the condensing enzymes along with improved physicochemical properties and easy synthetic access. This new inhibitor class is being further explored to increase target affinity.
Supplementary Material
Acknowledgements
We would like to thank Laura Wells and Protein Production Shared Resource for their expert technical assistance. This study was supported by the National Institutes of Health grant 5R01GM034496, Cancer Center Support grant CA21765 and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found in the online version
References
- 1.Parsons JB, Rock CO. Current opinion in microbiology. 2011;14:544. doi: 10.1016/j.mib.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White SW, Zheng J, Zhang YM, Rock Annual review of biochemistry. 2005;74:791. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 3.Lu H, Tonge PJ. Acc Chem Res. 2008;41:11. doi: 10.1021/ar700156e. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, Dorsey M, Hafkin B, Ramnauth J, Romanov V, Schmid MB, Thalakada R, Yethon J, Pauls HW. Antimicrobial agents and chemotherapy. 2012;56:5865. doi: 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banevicius MA, Kaplan N, Hafkin B, Nicolau DP. J Chemother. 2013;25:26. doi: 10.1179/1973947812Y.0000000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao J, Maxwell JB, Rock CO. J Biol Chem. 2013 doi: 10.1074/jbc.M113.512905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price AC, Rock CO, White SW. J Bacteriol. 2003;185:4136. doi: 10.1128/JB.185.14.4136-4143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. Proc Natl Acad Sci U S A. 2011;108:15378. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heath RJ, White SW, Rock CO. Appl Microbiol Biotechnol. 2002;58:695. doi: 10.1007/s00253-001-0918-z. [DOI] [PubMed] [Google Scholar]
- 10.Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. Proc Natl Acad Sci U S A. 2011;108:15378. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim P, Zhang YM, Shenoy G, Nguyen QA, Boshoff HI, Manjunatha UH, Goodwin MB, Lonsdale J, Price AC, Miller DJ, Duncan K, White SW, Rock CO, Barry CE, 3rd, Dowd CS. J Med Chem. 2006;49:159. doi: 10.1021/jm050825p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price AC, Choi KH, Heath RJ, Li Z, White SW, Rock CO. J Biol Chem. 2001;276:6551. doi: 10.1074/jbc.M007101200. [DOI] [PubMed] [Google Scholar]
- 13.Case DA, T. A. D., Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts BP, Wang B, Hayik S, Roitberg A, Seabra G, Kolossvai I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. University of California; San Francisco: 2010. [Google Scholar]
- 14.Pappenberger G, Schulz-Gasch T, Kusznir E, Muller F, Hennig M. Acta Crystallogr D Biol Crystallogr. 2007;63:1208. doi: 10.1107/S0907444907049852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massova I, Kollman PA. Perspect Drug Discov. 2000;18:113. [Google Scholar]
- 16.Zhu T, Lee H, Lei H, Jones C, Patel K, Johnson ME, Hevener KE. J Chem Inf Model. 2013;53:560. doi: 10.1021/ci300502h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homer RW, Swanson J, Jilek RJ, Hurst T, Clark RD. J Chem Inf Model. 2008;48:2294. doi: 10.1021/ci7004687. [DOI] [PubMed] [Google Scholar]
- 18.Wunberg T, Hendrix M, Hillisch A, Lobell M, Meier H, Schmeck C, Wild H, Hinzen B. Drug discovery today. 2006;11:175. doi: 10.1016/S1359-6446(05)03700-1. [DOI] [PubMed] [Google Scholar]
- 19.Martin YC. Journal of Medicinal Chemistry. 1992;35:2145. doi: 10.1021/jm00090a001. [DOI] [PubMed] [Google Scholar]
- 20.Schrodinger LLC. New York, NY; 2013. [Google Scholar]
- 21.Repasky MP, Shelley M, Friesner RA. Curr Protoc Bioinformatics. 2007 doi: 10.1002/0471250953.bi0812s18. Chapter 8, Unit 8 12. [DOI] [PubMed] [Google Scholar]
- 22.Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B. J Biomol NMR. 2001;21:349. doi: 10.1023/a:1013302231549. [DOI] [PubMed] [Google Scholar]
- 23.Jiang Y, Chan CH, Cronan JE. Biochemistry. 2006;45:10008. doi: 10.1021/bi060842w. [DOI] [PubMed] [Google Scholar]
- 24.Gokhan N, Koksal M, Kupeli E, Yesilada E, Erdogan H. Turk J Chem. 2005;29:445. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.