

Abstract
The aliphatic side chain plays a pivotal role in determining the cannabinergic potency of tricyclic classical cannabinoids, and we have previously shown that this chain could be substituted successfully by adamantyl or other polycyclic groups. In an effort to explore the pharmacophoric features of these conformationally fixed groups, we have synthesized a series of analogues in which the C3 position is substituted directly with an adamantyl group bearing functionality at one of the tertiary carbon atoms. These substituents included the electrophilic isothiocyanate and photoactivatable azido groups, both of which are capable of covalent attachment with the target protein. Our results show that substitution at the 3′-adamantyl position can lead to ligands with improved affinities and CB1/CB2 selectivities. Our work has also led to the development of two successful covalent probes with high affinities for both cannabinoid receptors, namely, the electrophilic isothiocyanate AM994 and the photoactivatable aliphatic azido AM993 analogues.
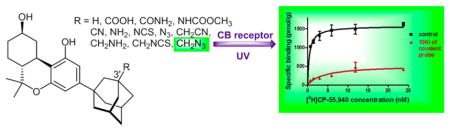
INTRODUCTION
The discovery of the first cannabinoid receptor, CB1, by Howlett and co-workers in 1988 marked the beginning of a renaissance in cannabinoid research.1 CB1, which is implicated in the CNS effects of cannabinoids in humans and other mammals, is found in greatest abundance in the hippocampus, cerebellum, and striatum. Humans, rats, and mice share a 97–99% amino acid sequence homology for CB1. In 1993 Munro and co-workers identified a second cannabinoid receptor.2 This new receptor exhibited 44% amino acid sequence homology overall and 68% homology with the transmembrane region of CB1. This receptor would later come to be known as CB2, and it is associated with the peripheral organs of the immune system. CB2 is found primarily in the spleen, tonsils, and thymus and with low expression in the CNS.3 Activation of CB2 is therefore not expected to lead to any of the CNS effects that are associated with cannabis abuse. Furthermore, its emerging role in immunomodulation makes it an attractive therapeutic target.
The discovery by one of us of the high affinity adamantyl cannabinoid 1a (AM411)4 suggested that rather than excluding the ligand from the receptor active site, the sterically demanding adamantyl group conferred high affinity for CB1 and modest (7-fold) selectivity in favor of CB1 (Figure 1).4 endo-Norbornyl cannabinoid 1b (AM735)5 and exo-norbornyl 1c (AM731)5 underscore some of the subtleties inherent in this structural motif. Both 1c and 1b have greater CB2 affinity than 1a, but 1c also has inverted selectivity compared to 1a, with a 10-fold preference for CB2 over CB1.5 These data suggested that small structural modifications of the adamantyl group may lead to substantial variations in the overall pharmacological profiles within this class of analogues. This motivated us to further explore the pharmacophoric features of these unusual cannabinoids in which the flexible side chain of their classical structure has been substituted by conformationally fixed cyclic substituents.6–8 In this effort, we have now synthesized a series of novel analogues in which the 3-position of the adamantyl ring is substituted with different moieties. For the tricyclic component of the new analogues we used the 9β-hydroxyhexahydrocannabinol structure that we had earlier shown to confer an optimal pharmacophoric profile within the classical cannabinoid structure.6,8,9 In an effort to explore the pharmacophoric features of the cannabinoid binding domain within each of the two receptors’ active site(s), we also included in our series analogues with suitable electrophilic and photoactivatable substituents, as we have done in previous work to obtain structural and functional information on the ligand–receptor complex using an approach involving receptor mutants and LC/MS/MS methods which we named ligand assisted protein structure (LAPS).10–17 To that effect we used the isothiocyanato group as our favored electrophile, having shown that under the experimental conditions of our receptor binding experiments this group reacted exclusively with the cysteine thiol group(s) within the receptor’s ligand binding domain(s).13 Also, as with earlier work, our choice of a photoreactive covalent group was the aliphatic azide moiety that upon photoactivation is transformed into a nitrene group capable of covalent attachment with amino acid residues at or in the vicinity of the active site. This SAR exploration led to a series of analogues with a wide range of affinities and functional CB1/CB2 profiles. Additionally, we obtained four novel covalently reacting analogues, two of which, 17 and 16, exhibit excellent covalent binding profiles. In future work both of these novel analogues will serve as biochemical tools to study the CB1/CB2 ligand–receptor complex using the LAPS approach developed in our laboratory.10–17
Figure 1.
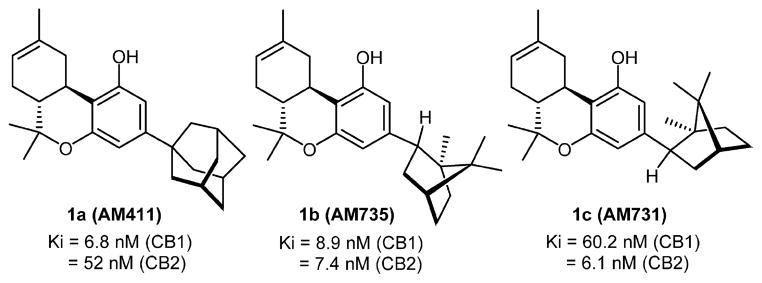
Structures of the adamantyl (1a) and the endo- and exo-norbonyl-(−)-Δ8-THC analogues 1b and 1c.
CHEMISTRY
We considered two distinct strategies to prepare the novel adamantyl analogues. We failed to implement our first approach, which was to functionalize one of the tertiary adamantyl carbon atoms after coupling to 2,6-dimethoxyphenol 1 (Scheme 1); therefore, we explored the coupling reactions of 1 with bifunctional adamantanes 2, 3, and 4. A number of attempts to couple 1 with 1,3-dihydroxyadamantane 2 or with 1-bromo-3-hydroxyadamantane 3 failed to provide the desired products, leading in each case to intractable mixtures. Fortunately, the Friedel–Crafts alkylation of 1 with 3-hydroxyadamantane-1-carboxylic acid 4 took place cleanly and in nearly quantitative yield.4 We found that it was essential to first dissolve 4 at 40–50 °C in the methanesulfonic acid that served as the reaction solvent and to add 2,6-dimethoxyphenol 1 subsequently.18 At higher reaction temperatures we observed a redox process that led to adamantane carboxylic acid as a byproduct.
Scheme 1. Synthesis of C9 Ketone 12 and β-C9 Alcohol 13a.

aReagents and conditions: (a) MeSO3H, 50 °C; 100%; (b) TMSCHN2, PhH, MeOH, rt; Tf2NPh, Et3N, DMAP (cat.), CH2Cl2, reflux; 91%; (c) PdCl2(Ph3P)2, dppp, n-Bu3N, HCOOH, PMHS (cat.), DMF, reflux; 89%; (d) BBr3, CH2Cl2, 0 °C to rt; 89%; (e) p-TSA·H2O, CHCl3/acetone (4:1), 55 °C; 78%; (f) TMSOTf, MeNO2, 0 °C to rt; 96%; (g) NaBH4, MeOH, 0 °C to rt; 95%.
Exposure of 5 to (trimethylsilyl)diazomethane produced the methyl ester that was immediately converted to phenolic triflate 6 in 91% overall yield from 4. Formation of the triflate in the presence of the free carboxylate had proved to be difficult, hence the need to protect the acid as the methyl ester. The catalytic reductive cleavage of the phenolic triflate in the presence of PdCl2(PPh3)2 and dppp19 had been difficult to reproduce, often leading to incomplete reactions and the appearance of precipitated metallic palladium. Careful purification of 6 to remove traces of N-phenyltriflamide byproduct from the preceding reaction was necessary, as was the inclusion of a small amount of polymethylhydrosiloxane (PHMS) in the reaction mixture.20 When this attention was given to the reaction conditions, the reductive cleavage to 7 took place reproducibly in high yield (89%). Global demethylation of 7 with boron tribromide led to resorcinol 8 in 89% yield.21
We chose to apply the excellent procedure that had been developed at the Eli Lilly Company during their synthesis of nabilone for the assembly of the tricyclic cannabinoid nucleus from 8.22 We were aware from our earlier work that the Lilly reaction represents a finely optimized process and that any deviation from the preferred reaction solvent, chloroform, inevitably led to erosion of the yield. The difficulty we faced was the sparing solubility of 8 in chloroform. Indeed, when we performed the condensation of 8 with the mixture of diacetates 9 and 10 in chloroform solvent in the presence of tosic acid, we were able to isolate only ~5% of 11 after several days of reaction. Inspection of the reaction vessel revealed a substantial quantity of insoluble solid material that was determined to be the polar and poorly soluble resorcinol 8. The solution to the problem was to dissolve 8 in the minimum volume of dry acetone and to then add the mixture of diacetates and tosic acid in chloroform. Under these conditions 8 remained in solution and adduct 11 was isolated in 78% yield after 2.5 days. Traces of tricyclic compound 12 were also detected by TLC in the product mixture. The cyclization of 11 to 12 was accomplished in 96% yield by exposure to trimethylsilyl triflate in nitromethane. This reagent has proven to be superior to tin tetrachloride that was used by the Lilly group to assemble the tricyclic cannabinoid nucleus. The isolation of the product from the thick matrix of insoluble tin salts that forms during workup had been problematic. Reduction of the C9 keto group in 12 with sodium borohydride led to alcohol 13 in 95% yield as a 95/5 mixture of β-equatorial and α-axial diastereoisomers. Ketone 12 and acid 13 were the common intermediates for the synthesis of the entire series of functionalized adamantyl cannabinoids that is described in what follows.
Scheme 2 summarizes the conversion of 13 to isothiocyanatomethylene analogue 16 and azidomethylene analogue 17. The carboxylic acid function in 13 was converted to the amide 14 by treatment with aqueous ammonia in THF in the presence of EDCI and HOBT (84% yield).23 Reduction of carboxamide 14 with lithium aluminum hydride followed by workup with either Glauber’s salt or careful addition of water followed by aqueous NaOH did not lead to amine 15 in satisfactory yield. Fortunately, reduction of 14 with borane–dimethyl sulfide complex proceeded cleanly and led to 15 in 90% yield.24 The reaction of amine 15 with carbon disulfide, tosyl chloride, and triethylamine led to isothiocyanate 16 in 89% yield.25 The synthesis of azidoadamantyl cannabinoid 17 from 15 was accomplished through the straightforward application of Wong’s diazo transfer reaction using triflyl azide.26,27 This reaction worked extraordinarily well leading to azide 17 in 93% yield. The absence of any protecting groups at any point in the synthesis is noteworthy.
Scheme 2. Synthesis of Isothiocyanante 16 and Azide 17a.
aReagents and conditions: (a) HOBT, EDCI, NH4OH, THF, rt; 84%; (b) BH3·DMS, 0 °C to rt; 90%; (c) Et3N, CS2, TsCl, THF, 0 °C to rt; 89%; (d) TfN3, Et3N, CuSO4, H2O, CH2Cl2, MeOH, rt; 93%.
The synthesis of isothiocyanate 19 and azide 20 is shown in Scheme 3. Exposure of ketocarboxylate 12 to diphenylphosphoryl azide (DPPA)28 and triethylamine in refluxing toluene led to an intermediate isocyanate through the Curtius rearrangement of the intermediate acyl azide.29 Typical conditions for hydrolysis of the isocyanate to the amine, refluxing in aqueous acid or in aqueous base, led only to decomposition of the isocyanate. After some experimentation we found that exposure of the isocyanate to mercuric acetate in aqueous THF followed by reduction of the mercury salt with sodium borohydride led to amine 18 in 76% overall yield from 12.30 Simultaneous reduction of the C9 keto group during this step led to the to C9-β alcohol group in 18, saving one step. Dimeric urea 21 was formed in ~5% yield as a side product during this reaction. Conversion of the primary amino group in 18 to the isothiocyanate was carried out in the same way as shown in Scheme 2, leading to 19 in 86% yield. Azido transfer from triflyl azide as in the case of 15 converted amine 18 to azide 20 in 98% yield.
Scheme 3. Synthesis of Isothiocyanate 19 and Azide 20a.

aReagents and conditions: (a) DPPA, Et3N, PhMe, reflux; (b) Hg(OAc)2, THF, H2O (1/1), rt; NaBH4, 5% aq KOH, rt; 76% from 12; (c) Et3N, CS2, TsCl, THF, 0 °C to rt; 86%; (d) TfN3, Et3N, CuSO4, H2O, CH2Cl2, MeOH, rt; 98%.
The remaining analogues 22–26 were prepared from ketocarboxylate 12 as summarized in Scheme 4. The challenging task was the preparation of cyanomethyl analogue 23. A model study employing hydroxymethyl adamantane unsurprisingly revealed that SN2 displacement of the derived mesylate by cyanide was uniformly unsuccessful even under forcing conditions. This made it necessary to adapt our strategy, so Arndt–Eistert homologation of carboxylate 12 was carried out. Sequential treatment of 12 with oxalyl chloride and catalytic DMAP in THF to produce the acid chloride was followed by exposure to diazomethane.31 The resulting α-diazoketone was heated to reflux in THF in the presence of catalytic silver benzoate and ammonium hydroxide to give carboxamide 22 in low overall yield (27% from 12).32 The conversion of the crude amide 22 to nitrile 23 was accomplished in two steps. Treatment first with trifluoroacetic anhydride and pyridine in dioxane led to dehydration of the carboxamide to the nitrile and also converted the free phenolic hydroxyl group to the trifluoroacetate ester.33 Hydrolysis of trifluoroacetate with methanolic potassium carbonate followed by addition of sodium borohydride led to 23 in 86% overall yield for the two steps from 22.
Scheme 4. Synthesis of Functionalized Adamantyl Cannabinoids 22 –26a.
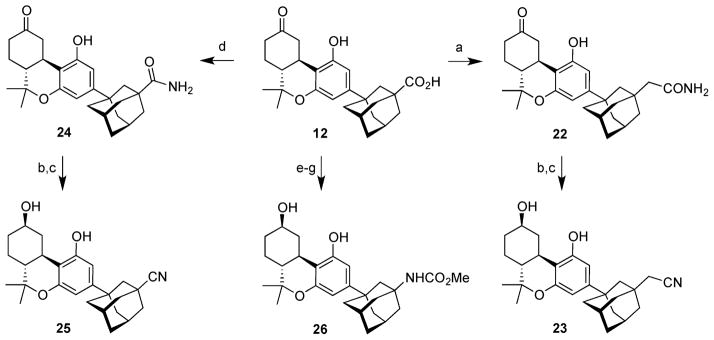
aReagents and conditions: (a) (COCl)2, DMAP (cat.), THF; CH2N2, rt; PhCO2Ag, Et3N, NH4OH, THF, reflux; 27%; (b) TFAA, pyr, dioxane, 0 °C to rt; (c) K2CO3, MeOH, rt; NaBH4, 0 °C to rt; 86% of 23 from 22; 71% of 25 from 24; (d) HOBT, EDCI, NH4OH, THF, rt; 81%; (e) NaBH4, MeOH, 0 °C to rt; 95%; (f) DPPA, Et3N, PhMe, reflux; (g) NaOMe, MeOH, reflux; 65% for two steps.
Ketoacid 12 was converted to carboxamide 24 in 81% yield by exposure to a mixture of EDCI and HOBT in THF in the presence of ammonium hydroxide. The preparation of nitrile 25 was carried out in the same way as for 23. The overall yield of 25 was 71% for the two steps from 24.
Ketoacid 12 was also used to prepare methyl carbamate 26 (Scheme 4). Reduction with sodium borohydride led to C9 alcohol 13. Exposure of 13 to diphenylphosphoryl azide and triethylamine in toluene at reflux led initially to an acyl azide that underwent Curtius rearrangement to an intermediate isocyanate that was immediately treated with sodium methoxide in methanol at reflux. It is noteworthy that no protecting groups were used in any part of the chemistry.
CB1/CB2 BINDING AFFINITIES
These were obtained by measuring the abilities of the new analogues to displace the radiolabeled CB1/CB2 agonist [3H]CP-55,940 from suitably prepared membrane preparations (Table 1). As in previous work for CB1, we used rat brain membranes while for CB2 we used both mouse (mCB2) and human (hCB2) preparations obtained from HEK293 cells expressing these receptors.9,34 The use of two CB2 receptor preparations was aimed at addressing species differences that we observed earlier.35 In addition, because of the covalent nature of the binding of the isothiocyanates 16 and 19, the affinities for CB receptors are presented as “apparent Ki” (Ki*) values, and these are expected to be dependent on the time during which the preparation is pretreated with the ligand. When such time dependent experiments were performed with 19 in CB1 receptor preparations, we observed (data not shown) that increasing the incubation time from 15 to 90 min increases the CB1 affinity of 19 by 3-fold (Ki(15 min) = 96 nM; Ki(30 min) = 58.6 nM; Ki(60 min) = 35.4 nM; Ki(90 min) = 35.7 nM). Thus, under the assay conditions the lower Ki value for 19 (35.4 nM, Table 1) was reached in 60 min.
Table 1.
CB1/CB2 Affinities (Ki) of 3′-Functionalized Adamantyl Cannabinoid Analogues (95% Confidence Limits)
| ||||
|---|---|---|---|---|
|
| ||||
|
Ki (nM)a
|
||||
| compd | R | rCB1 | mCB2 | hCB2 |
| 27 | -H | 23.9b | 39.4b | 40.5b |
| 13 | -COOH | >1000 | >1000 | >1000 |
| 14 | -CONH2 | 297 | 947 | 337 |
| 26 | -NHCOOCH3 | 29.1 | 2.1 | 8.4 |
| 25 | -CN | 45.8 | 2.8 | 4.2 |
| 18 | -NH2 | >1000 | >1000 | >1000 |
| 19 | -NCS | 35.4c | 31.7c | 13.1c |
| 20 | -N3 | 18.6 | 38.4 | 24.8 |
| 23 | -CH2CN | 2.6 | 4.4 | 4.6 |
| 15 | -CH2NH2 | >1000 | >1000 | >1000 |
| 16 | -CH2NCS | 3.0c | 34.6c | 10.3c |
| 17 | -CH2N3 | 4.4 | 26.4 | 9.6 |
Affinities for CB1 and CB2 receptors were determined using rat brain (CB1) preparations or membranes from HEK293 cells expressing mouse or human CB2 receptors and [3H]CP-55,940 as the radioligand following previously described procedures.6,9 Data were analyzed using nonlinear regression analysis. Ki values were obtained from three independent experiments run in triplicate and are expressed as the mean of the three values (SD < ±20%).
Reported previously.8
Apparent Ki (Ki*) value determined in this study.
Our data show that the analogues with the highest affinities for both receptors were those bearing the isothiocyanato, azido, and cyano groups (19, 20, and 25, respectively) as well as their methylene homologues 16, 17, and 23, respectively. Also exhibiting relatively high affinity was the methyl carbamate analogue 26. Conversely, compounds with free carboxylic acid (13) or amine (18, 15) substituents as well as carboxamide 14 had very low affinities for both receptors, suggesting that strong hydrogen bond donating groups could not be accommodated within the binding site(s).
Compounds with the highest affinities were the homologues in which a methylene group is introduced between the adamantyl ring and the NCS, N3, or CN functional groups. Also observed among the analogues was a certain level of CB1 vs CB2 selectivity as exemplified by the methylisothiocyanato 16 which showed 3- or 10-fold selectivity in favor of CB1 and methyl carbamate 26 with a 4- to 15-fold selectivity for CB2. The parent compound (27) bearing an unsubstituted adamantyl group had generally lower affinities for both receptors when compared to the higher affinity substituted analogues, suggesting the existence of favorable interactions of some of the 3′-adamantyl substituents with the CB1/CB2 receptor binding domain.
FUNCTIONAL CHARACTERIZATION
We focused on those analogues possessing the highest CB1/CB2 affinities, and the experiments were carried out by measuring changes in forskolin-stimulated cAMP, as detailed earlier.9,34 Our testing results show that compounds 16, 19, 20, 23, and 25–27 potently decreased the levels of cAMP, indicating that within this signaling mechanism these compounds behaved as potent agonists at the CB1 receptor with the methyl isothiocyanato 16 and methylcyano 23 analogues being more potent than their methylazido counterpart 17 (Table 2). Surprisingly, when tested with hCB2 preparations, all compounds failed to decrease the levels of cAMP with 19, 20, and 17 showing no change in the cAMP measured, thus behaving as neutral antagonists. The remaining analogues showed increases in cAMP levels and behaved as inverse agonists. The dose–response curves of representative analogues for both the CB1 and CB2 receptors are depicted in Figures 2 and 3, respectively, where data for the standard cannabinoid agonist CP-55,940 are also shown for comparison. We observe that 17, 23, and 26 have high binding affinities for CB2 (Table 1), thus resembling the standard CB1/CB2 agonist CP-55,940. However, unlike CP-55,940, which behaves as a full agonist at CB2, the adamantyl analogues described here act as antagonists/inverse agonists at CB2 in the cAMP functional test. We have no immediate explanation for this striking observation.
Table 2.
CB1/CB2 Functional Potencies (EC50) of Selected 3′-Functionalized Adamantyl Cannabinoid Analogues
| rCB1 | hCB2 | |
|---|---|---|
| compd | EC50 (nM),a E(max) (%)b classification | EC50 (nM),a E(max) (%)b classification |
| 27 | 2.7 (2.1–3.3), 69 agonist | NR |
| 26 | 1.6 (1.3–1.9), 71 agonist | 15.8 (12.5–19.1), −74 inverse agonist |
| 25 | 95.2 (75.4–114.9), 82 agonist | 90.7 (71.8–109.6), −93 inverse agonist |
| 19 | 12.0 (9.5–14.5), 68 agonist | NR |
| 20 | 4.4 (3.4–5.5), 73 agonist | NR |
| 23 | 5.4 (4.3–6.5), 78 agonist | 83.1 (65.8–100.4), −78 inverse agonist |
| 16 | 0.8 (0.6–1.0), 94 agonist | >400, −91 inverse agonist |
| 17 | 2.4 (1.9–2.9), 45 agonist | NR |
Functional potencies at rCB1 and hCB2 receptor were determined by measuring the decrease in forskolin-stimulated cAMP levels.9,34 EC50 values were calculated using nonlinear regression analysis. Data are the average of two independent experiments run in triplicate, and 95% confidence intervals for the EC50 values are given in parentheses.
Forskolin stimulated cAMP levels were normalized to 100%. E(max) is the maximum inhibition of forskolin stimulated cAMP levels and is presented as the percentage of CP-55,940 response at 500 nM. NR: no response up to 3 μM.
Figure 2.
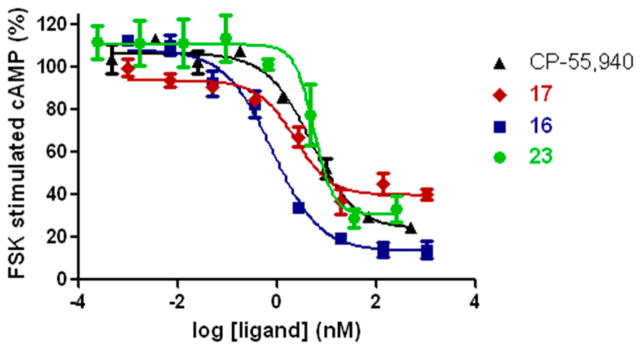
Concentration-dependent inhibition of forskolin-stimulated cAMP accumulation in HEK293 cells expressing rCB1 receptors by representative agonists. All compounds behave as full agonists.
Figure 3.
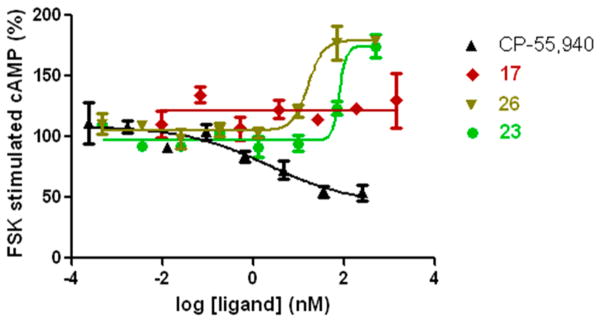
Concentration-dependent response of forskolin-stimulated cAMP accumulation in HEK293 cells expressing hCB2 receptors by representative ligands. The cannabinoid standard CP-55,940 behaves as a potent agonist decreasing the levels of cAMP, while compounds 23 and 26 behave as inverse agonists. Compound 17 showed no response up to a 3 μM concentration.
COVALENT LABELING OF THE CB1/CB2 RECEPTORS
The ability of the compounds carrying covalently reacting groups to label each of the two receptors was obtained by measuring reductions in the binding for the radioligand [3H]CP-55,940 when the preparation was pretreated with the covalent ligand, compared to the untreated sample (details in Experimental Section). Following earlier work from our laboratory, the experiment was conducted by pretreating the sample with a concentration equal to 10-fold the compound’s Ki value for the receptor in question and subsequently measuring the decrease of Bmax obtained from a saturation curve using [3H]CP-55,940. Our results clearly show that all four of our covalent ligands (16, 17, 19, 20) are capable of covalently labeling both receptors with the best results being obtained by the 3′-isothiocyanatomethyl (16) and the 3′-azidomethyl (17) analogues (data shown in Figure 4 and Table 3, while saturation binding curves for analogues 19 and 20 are provided in Supporting Information). Thus, 16 and 17 appear to be optimal probes for studying the structures of the ligand–receptor binding complexes to be carried out in future work.
Figure 4.

Saturation binding curves using [3H]CP-55,940 for CB1 (left panel) and CB2 (right panel) receptors preincubated with the 3′-azidomethyl (red, 17) and 3′-isothiocyanatomethyl (blue, 16) analogues. Control membranes processed in parallel but without prior exposure to covalent ligands are shown in black. Data represent the mean values ± SEM of at least two independent experiments performed in triplicate. Both compounds inhibit the specific binding of the radioligand to CB1 and CB2 receptors.
Table 3.
Reductions in the Specific Binding of [3H]CP-55,940 after Covalent Ligand Pretreatment
| covalent labeling (%)a | ||
|---|---|---|
|
|
||
| compd | CB1 | CB2 |
| 20 | 61 (58–67) | 26 (21–32) |
| 19 | 54 (49–60) | 25 (22–29) |
| 17 | 67 (60–75) | 60 (53–75) |
| 16 | 63 (57–72) | 74 (69–82) |
The receptor membranes were pretreated with a concentration equal to 10-fold the compound’s Ki value. Percent covalent labeling was calculated as {[Bmax(control) − Bmax(ligand)]/Bmax(control)} × 100. Bmax(ligand) was calculated as described in Experimental Section. The Bmax(control) for [3H]CP-55,940 was determined by conducting the assay as described but in the absence of the test ligand. Confidence intervals (95%) are shown in parentheses.
As negative controls we used the cyano analogue 23 and its azido counterpart 17 prior to irradiation, both of which were not expected to interact covalently with the CB1/CB2 receptor preparations. We observed no reductions in the levels of specific binding (Bmax) of [3H]CP-55,940 under conditions identical to those used for the covalent labeling (data not shown).
To better understand the receptor labeling characteristics of the electrophilic isothiocyanato analogues, we carried out a time-dependent study using CB1 receptor membranes, a 10Ki nM concentration of 19 and varied the incubation time (Figure 5). According to our experimental conditions, the termination of the ligand–receptor covalent reaction is accomplished by centrifugation. Unreacted ligand was removed by repeatedly pelleting and washing the membrane preparation after the incubation period was completed. Our studies showed that with a 15 min incubation 26% of the receptor sites were occupied irreversibly. By increasing the incubation time to 30 min, covalent binding increased to 54% with no further increase seen if the incubation time was extended to 60 min. Thus, at the ligand concentration used, covalent binding had reached its plateau at 30 min of incubation time. The control experiment was a 60 min incubation with no added 19. However, we note that the true kinetic behavior of the covalent reaction between 19 and the cannabinoid receptor is not necessarily reflected by this time-dependent receptor labeling, since the incubation time does not represent the precise reaction time of the ligand with the receptor.12
Figure 5.
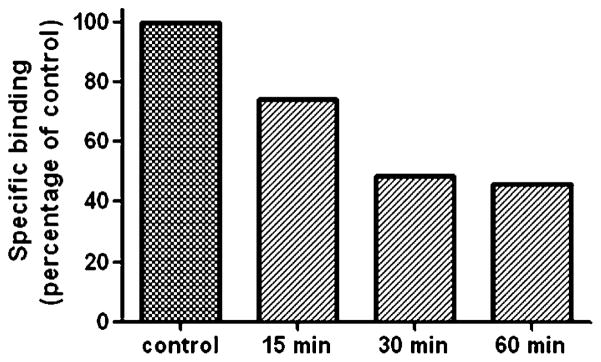
Time course for CB1 receptor labeling with isothiocyanato probe 19. The membranes were preincubated with 10-fold the Ki value of compound 19 for 15, 30, and 60 min. Membranes in the control experiment were preincubated for 60 min without compound 19. The [3H]CP-55,940 specific bindings were determined by the saturation binding method reported in Experimental Section.
CONCLUSION
In our current effort to explore the pharmacophoric requirements of the “adamantyl subsite” within the CB1/CB2 binding domain we have synthesized a family of (1-adamanyl)-9β-hydroxyhexahydrocannabinols carrying C3′-substituents on the adamantyl ring. Our studies show that within this subsite, properly chosen C3′-substituents are capable of engaging in favorable interactions with both CB1 and CB2 receptors with some substantial level of CB1/CB2 selectivity. Arguably, such substitutions could be further extended to develop more selective CB1 or CB2 analogues. All successful compounds when tested in vitro in the cAMP assay were potent agonists for the CB1 receptor. Conversely, all of these ligands behaved as CB2 inverse agonists or neutral antagonists in the same functional test. This unexpected discovery opens the door for developing highly functionally selective CB1 agonists. Importantly, our study has led to the development of a new pair of potentially very successful covalent ligands 17 and 16. In future work these will be tested by using our LAPS methodology to obtain structural information on the active CB1 and inactive CB2 receptor conformations. The two covalent probes are also currently being explored for X-ray crystallographic work involving the CB1/CB2 ligand–receptor complexes and for labeling work with other cannabinoid-related GPCRs.
EXPERIMENTAL SECTION
Chemistry
1H NMR and 13C spectra were recorded either at 300 MHz (1H) and 75 MHz (13C) or at 500 MHz (1H) and 126 MHz (13C). Chemical shifts are reported in parts per million (δ) and are referenced to the solvent, i.e., 7.26/77.0 for CDCl3. Multiplicities are indicated as br (broadened), s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sept (septet), or m (multiplet). Coupling constants (J) are reported in hertz (Hz). Thin layer chromatography (TLC) was performed on glass plates 250 μm, particle size 5–17 μm, pore size 60 Å. Flash column chromatography was performed on silica gel, 200–400 mesh, or premium silica gel, 60 Å, 40–75 μm. All moisture sensitive reactions were performed under a static atmosphere of nitrogen or argon in oven-dried or flame-dried glassware. Purity and homogeneity of all materials were determined to be at least 95% from TLC, 1H NMR, and 13C NMR spectra as well as by LC/MS analysis using a Waters MicroMass ZQ system [electrospray ionization (ESI) with Waters-2525 binary gradient module coupled to a photodiode array detector (Waters-2996) and ELS detector (Waters-2424) using a XTerra MS C18, 5 μm, 4.6 mm × 50 mm column and acetonitrile/water]. All optical rotations were measured on a JASCO digital polarimeter in a 0.1 dL cell.
Methyl (1r,3r)-3-(4-Hydroxy-3,5-dimethoxyphenyl)-adamantane-1-carboxylate (5)
To a solution of 3-hydroxyada-mantane-1-carboxylic acid 4 (5.0 g, 25.5 mmol) in methanesulfonic acid (23.0 mL) heated to 40 °C and stirred under argon atmosphere in a reaction flask equipped with stir bar and reflux condenser was added of 2,6-dimethoxyphenol 1 (3.6 g, 23.2 mmol), and the mixture was stirred at 50 °C for 3.0 h. The reaction was allowed to cool to room temperature, diluted with CH2Cl2 (100 mL), poured onto ice–water (300 mL), and was stirred overnight until both organic and aqueous layers were clear. The organic layer was removed, and the aqueous layer was back-extracted with CH2Cl2 (2 × 50 mL). The organic layers were combined, washed with water (2 × 50 mL) followed by brine (20 mL), dried over MgSO4, filtered, and concentrated in vacuo to give 5 as a lavender solid in quantitative yield (7.706 g, 23.20 mmol). The crude product was taken on to the next step without purification. An analytical sample was obtained via flash chromatography on silica gel (10–30% acetone in hexanes). 1H NMR (300 MHz, CD3OD) δ 6.49 (s, 2H), 3.71 (s, 6H), 2.06 (br, 2H), 1.85 (br, 2H), 1.79 (br, 4H), 1.75 (br, 2H), 1.62 (br, 2H); 13C NMR (75 MHz, CD3OD) δ 181.4, 148.9, 142.7, 134.7, 103.5, 56.8, 45.8, 43.6, 42.8, 39.4, 37.5, 36.7, 30.3; mp 174–176 °C; IR (neat; cm−1) 3371, 2907, 2854, 1693, 1605, 1521, 1451, 1214, 1115; HRMS (ESI+) calculated for C19H24O5 [M + H+] 333.1703, found 333.1717.
Methyl (1r,3r)-3-[3,5-Dimethoxy-4-(trifluoromethylsulfonyloxy)phenyl]adamantane-1-carboxylate (6)
To a solution of 5 (7.7 g, 23.2 mmol) in benzene (92.8 mL) and MeOH (23.2 mL) was added TMSCHN2 (12.8 mL of 2.0 M in hexanes, 25.5 mmol), and the mixture was stirred at room temperature under air atmosphere for 30 min. The progress of the reaction was monitored by TLC (Rf = 0.45 in 30% acetone in hexanes). The crude product was concentrated in vacuo to yield a yellow oil, which was directly taken on to the subsequent reaction. To a solution of the methyl ester in CH2Cl2 (128 mL) in a reaction flask equipped with stir bar and reflux condenser were added PhNTf2 (9.1 g, 25.5 mmol), Et3N (3.6 mL, 25.5 mmol), and catalytic DMAP. The reaction was heated to reflux overnight, cooled to room temperature, then washed with a 3 N NaOH solution (2 × 30 mL). The aqueous layer was back-extracted with CH2Cl2 (2 × 20 mL). The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and adsorbed onto Celite. The product was purified via flash chromatography on silica gel (0% to 5% to 10% acetone in hexanes, TLC Rf = 0.30 in 15% acetone in hexanes) to give triflate 6 as a white solid in 96% yield (10.6 g, 22.2 mmol) over two steps. 1H NMR (500 MHz, CDCl3) δ 6.59 (s, 2H), 3.89 (s, 6H), 3.68 (s, 3H), 2.25 (br, 2H), 2.00 (br, 2H), 1.93 (m, 4H), 1.87 (br, 4H), 1.74 (m, 2H); 13C (125 MHz, CDCl3) δ 177.6, 151.9, 151.1, 126.0, 118.7 (q, JC–F = 320.1 Hz, 1C), 101.8, 56.2, 51.8, 44.1, 42.0, 41.8, 38.0, 37.1, 35.4, 28.5; mp 148.9–150.1 °C; IR (neat; cm−1) 2936, 1729, 1607, 1454, 1417, 1248, 1205, 1136, 1080, 886, 872; HRMS (ESI+) calculated for C21H26F3O7S [M + H+] 479.1352, found 479.1369.
Methyl (1r,3r)-3-(3,5-Dimethoxyphenyl)adamantane-1-carboxylate (7)
To solution of triflate 6 (5.2 g, 10.9 mmol) in DMF (44.0 mL) were added PdCl2(PPh3)2 (418 mg, 0.6 mmol), dppp (450 mg, 1.1 mmol), n-Bu3N (13.0 mL, 54.5 mmol), HCOOH (88% w/v in H2O, 1.03 mL, 27.3 mmol), and catalytic polymethylhydrosiloxane (PMHS) in a reaction flask equipped with stir bar, reflux condenser, and a three-way tap, and the mixture was stirred for 5 min under argon atmosphere at room temperature for 5 min. The reaction vessel was subjected to three cycles of vacuum/argon, then heated to 95 °C under argon pressure for 16 h. The reaction mixture was cooled to room temperature, diluted with Et2O (25.0 mL) and 1 N HCl (10 mL), stirred for 30 min, and then filtered through a plug of Celite to remove insoluble palladium. The organic layer was extracted, and the aqueous layer was back-extracted with Et2O (3 × 20 mL). The organic layers were combined, washed with 1 N HCl (2 × 10 mL) followed by brine (20 mL), dried over MgSO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (0% to 5% to 10% EtOAc in hexanes) to give 7 as an off-white solid in 89% yield (3.2 g, 9.7 mmol). 1H NMR (300 MHz, CDCl3) δ 6.52 (d, J = 2.2 Hz, 2H), 3.32 (t, J = 2.2 Hz, 1H), 3.80 (s, 6H), 3.66 (s, 3H), 2.22 (br, 2H), 2.01 (br, 2H), 1.91 (m, 8H), 1.72 (br, 2H); 13C NMR (75 MHz, CDCl3) δ 177.8, 160.6, 152.7, 103.5, 97.2, 55.2, 51.7, 44.1, 42.0, 41.8, 38.1, 36.7, 35.5, 20.6; mp 60.7–63.2 °C; IR (neat; cm−1) 2907, 2854, 1728, 1596, 1455, 1423, 1309, 1242, 1204, 1153, 1087, 1068, 1013, 830; HRMS (ESI+) calculated for C20H27O4 [M + H+] 331.1909, found 331.1922.
(1r,3r)-3-(3,5-Dihydroxyphenyl)adamantane-1-carboxylic Acid (8)
A solution of dimethoxy aromatic 7 (3.5 g, 10.6 mmol) in CH2Cl2 (53.0 mL) under N2 atmosphere was cooled to 0 °C and stirred for 5 min. To the cooled solution was added BBr3 (5.12 mL, 53.2 mmol) dropwise, and the mixture was stirred and gradually warmed to room temperature over 3 h. The reaction was cooled to 0 °C before quenching with ice cold H2O (20 mL) and was taken up in Et2O (50 mL). The organic layer was washed with 1 N HCl (2 × 10 mL), and the aqueous layer was back-extracted with Et2O (2 × 20 mL). The organic layers were combined, washed with brine (10 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (20% to 30% to 40% acetone in hexanes) to give resorcinol 8 as a white foam in 89% yield (2.7 g, 9.5 mmol). 1H NMR (300 MHz, CD3OD) δ 6.32 (d, J = 2.1 Hz, 2H), 6.10 (t, J = 2.1 Hz, 1H), 2.17 (br, 2H), 1.95 (br, 2H), 1.90 (br, 4H), 1.83 (br, 4H), 1.74 (br, 2H); 13C NMR (75 MHz, CD3OD) δ 181.5, 159.3, 154.1, 104.5, 101.0, 45.6, 43.4, 42.8, 39.5, 37.5, 36.8, 30.3; mp 194.8–195.9 °C; IR (neat; cm−1) 3234 (br), 2907, 2853, 1690, 1602, 1513, 1450, 1278, 1156, 1005; HRMS (ESI+) calculated for C17H21O4, [M + H+] 289.1440, found 289.1449.
(1R,3r)-3-{4-[(1R,2R,5R)-6,6-Dimethyl-4-oxobicyclo[3.1.1]-heptan-2-yl]-3,5-dihydroxyphenyl}adamantane-1-carboxylic Acid (11)
A solution of resorcinol 8 (609 mg, 2.11 mmol) dissolved in a minimal volume of acetone (~2 mL) by heating to 55 °C in a reaction flask equipped with a stir bar and reflux condenser was degassed with argon, and to it was added a mixture of p-TSA·H2O (442 mg, 2.32 mmol) and diacetates 9 and 10 (609 mg, 2.54 mmol) in CHCl3 (6.0 mL). The reaction was degassed with argon for an additional 10 min and then stirred at 55 °C for 2.5 days. The progress of the reaction was monitored by multiple elution TLC. Upon completion, the reaction was allowed to cool to room temperature and taken up in with Et2O (20 mL) and washed with 1 N HCl (10 mL). The aqueous layer was back-extracted with Et2O (2 × 10 mL), and organic phases were combined, washed with brine, dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (10% to 20% to 30% to 50% acetone in hexanes) to give condensation product 11 as an off-white foam in 78% yield (698 mg, 1.64 mmol). 1H NMR (300 MHz, CD3OD) δ 6.32 (s, 2H), 4.00 (t, J = 8.0 Hz, 1H), 3.73 (dd, J = 18.8, 7.6 Hz, 1H), 2.65–2.58 (m, 1H), 2.49–2.46 (m, 2H), 2.42 (dd, J = 18.8, 8.7 Hz, 1H), 2.18–2.16 (m, 3H), 1.94–1.73 (m, 12H), 1.36 (s, 3H), 0.96 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 219.9, 181.6, 157.6, 151.0, 114.5, 105.0, 59.3, 48.6, 45.4, 43.4, 43.2, 42.7, 39.5, 38.5, 37.2, 36.8, 30.4, 30.3, 26.6, 25.0, 22.5; [α]23D +56.2 (c 1.99, CH3OH); IR (neat; cm−1) 3387 (br), 2930, 2855, 1693, 1621, 1421, 1024; HRMS (ESI+) calculated for C26H33O5 [M + H+] 425.2329, found 425.2338.
(1R,3r)-3-[(6aR,10aR)-1-Hydroxy-6,6-dimethyl-9-oxo-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-adamantane-1-carboxylic Acid (12)
To a solution of condensation product 11 (50 mg, 0.12 mmol) in MeNO2 (12.0 mL) cooled to 0 °C under N2 atmosphere was added TMSOTf (0.108 mL, 0.59 mmol) dropwise. The reaction was allowed to gradually warm to room temperature and stirred for 2 h. Upon completion of the reaction, monitored by TLC, 1 N HCl (3 mL) was added and stirred for 5 min and the reaction mixture was concentrated in vacuo. The product was taken up in Et2O (3 mL), and the organic phase was washed with 1 N HCl (2 × 2 mL). The aqueous layer was back-extracted with Et2O (2 × 3 mL), and the organic layers were combined, washed with brine (1 × 2 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (10% to 20% to 30% acetone in hexanes) to give the ketone 12 as an off-white foam in 96% yield (48 mg, 0.11 mmol). 1H NMR (300 MHz, CD3OD) δ 6.34 (d, J = 1.8 Hz, 1H), 6.28 (d, J = 1.8 Hz, 1H), 3.85 (dd, J = 14.9, 3.4 Hz, 1H), 2.80 (td, J = 12.8, 3.5 Hz, 1H), 2.49 (dd, J = 9.5, 5.1 Hz, 2H), 2.22–1.69 (m, 16H), 1.60–1.24 (m, 2H), 1.44 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CD3OD) δ 214.1, 181.2, 157.1, 155.5 151.6 109.7, 106.1, 104.8, 77.4, 46.0, 45.2, 43.0, 42.4, 41.3, 39.2, 37.0, 36.9, 36.5, 35.8, 30.0, 27.9, 27.6, 18.8; [α]23D −85.2 (c 0.95, CH3OH); IR (neat; cm−1) 3360 (br), 2907, 2855, 1694, 1621, 1452, 1360; HRMS (ESI+) calculated for C26H33O5 [M + H+] 425.2329, found 425.2322.
(1R,3r)-3-[(6aR,9R,10aR)-1,9-Dihydroxy-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-adamantane-1-carboxylic Acid (13)
To a solution of keto acid 12 (183 mg, 0.043 mmol) in MeOH (2.1 mL) cooled to 0 °C was added NaBH4 (16 mg, 0.43 mmol). The reaction flask was warmed to room temperature, stirred for 1.5 h, cooled to 0 °C, quenched with 1 N HCl (0.5 mL), and concentrated in vacuo. The crude product was taken up in Et2O (5 mL) and washed with 1 N HCl (2 × 1 mL). The aqueous layer was back-extracted with Et2O (2 × 3 mL), and the organic layers were combined, washed with brine (1 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash column chromatography on silica gel (10–40% acetone in hexanes) to give C9-β hydroxy acid 13 as an off-white amorphous solid in 95% yield (17 mg, 0.041 mmol). 1H NMR (300 MHz, CD3OD) δ 6.33 (d, J = 1.9 Hz, 1H), 6.23 (d, J = 1.9 Hz, 1H), 3.72 (dt, J = 15.3, 5.4 Hz, 1H), 3.52 (m, 1H), 2.44 (td, J = 11.2, 2.4 Hz, 1H), 2.21–2.07 (m, 3H), 1.96–1.71 (m, 14H), 1.48–1.11 (m, 4H), 1.35 (s, 3H), 1.03 (s, 3H); 13C (125 MHz, CD3OD) δ 181.5, 157.5, 155.9, 151.2, 110.8, 106.3, 105.0, 77.7, 71.4, 50.3, 45.5, 43.3, 42.7, 39.9, 39.5, 37.2, 36.8, 36.7, 34.9, 30.3, 28.3, 27.2, 19.2; [α]22D −87.9 (c 2.83, MeOH); IR (neat; cm−1) 3215 (br), 2932, 2856, 2661, 1695, 1621, 1514, 1055, 830; HRMS (ESI+) calculated for C26H35O5 [M + H+] 427.2484, found 427.2489. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 98% and retention time of of 4.5 min for the title compound.
(1R,3r)-3-[(6aR,9R,10aR)-1,9-Dihydroxy-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-adamantane-1-carboxamide (14)
To a solution of hydroxy acid 13 (86 mg, 0.20 mmol) and HOBT (32 mg, 0.24 mmol) in THF (2.0 mL), stirred at room temperature for 5 min under ambient atmosphere, was added a suspension of EDCI (41 mg, 0.24 mmol), and the reaction mixture was stirred overnight. After 16 h, NH4OH (0.5 mL) was added dropwise over 5 min, and the reaction mixture was stirred for an additional 1 h. Additional EDCI (41 mg, 0.24 mmol) was added to the reaction and was stirred overnight. The reaction was quenched with 1 N HCl (1 mL) and extracted with EtOAc (3 × 1 mL). The organic layers were combined, washed with phosphate buffer (1 mL) followed by brine (1 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (5–10% EtOH in CH2Cl2 with NH4OHsat) to give amide 14 as an off-white semisolid in 81% yield (69 mg, 0.072 mmol), mp 174–176 °C. 1H NMR (300 MHz, CD3OD) δ 6.32 (d, J = 1.9 Hz, 1H), 6.25 (d, J = 1.9 Hz, 1H), 3.74 (m, 1H), 3.57–3.48 (m, 1H), 2.48–2.42 (m, 1H), 2.21 (m, 2H), 2.12 (m, 1H), 1.90–1.74 (m, 12H), 1.48–1.28 (m, 3H), 1.34 (s, 3H), 1.24–1.15 (m, 1H), 1.18 (m, 1H), 1.04 (s, 3H), 0.95 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 183.8, 157.5, 155.9, 151.2, 110.8, 106.3, 105.1, 77.7, 71.4, 50.3, 45.7, 43.3, 42.9, 40.0, 39.6, 37.4, 36.8, 36.7, 34.9, 30.4, 28.3, 27.2, 19.2; [α]25D −90.6 (c 0.64, CH3OH); IR (neat; cm−1) 3354 (br), 2937, 2861, 1625, 1421, 1035; HRMS (ESI+) calculated for C26H36NO4 [M + H+] 426.2645, found 426.2650. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 95% and retention time of 4.2 min for the title compound.
(6aR,9R,10aR)-3-[(1r,3R)-3-(Aminomethyl)adamantan-1-yl]-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]-chromene-1,9-diol (15)
To a solution of amide 14 (27 mg, 0.064 mmol) in THF (1.3 mL) cooled to 0 °C under N2 was added BH3·S(CH3)2 in THF (0.064 mL, 0.64 mmol) dropwise, and the reaction mixture was stirred. The reaction mixture was allowed to warm to room temperature over 3 h. Then the reaction was quenched by addition of EtOH (1 mL) dropwise and refluxed overnight. The crude reaction mixture was concentrated in vacuo, taken up in EtOAc (2 mL), and washed with pH 7 phosphate buffer (1 mL). The aqueous layer was back-extracted with EtOAc (2 × 1 mL). The organic layers were combined and washed with brine (2 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (5–10% EtOH in CH2Cl2 with sat. aq NH4OH) to give homologated amine 15 as a clear glass in 90% yield (24 mg, 0.058 mmol). 1H NMR (300 MHz, CD3OD) δ 6.32 (d, J = 1.9 Hz, 1H), 6.23 (d, J = 1.9 Hz, 1H), 3.73 (m, 1H), 3.52 (m, 1H), 3.05 (s, 2H), 2.43 (td, J = 11.2, 2.4 Hz, 1H), 2.19–1.08 (m, 19H), 1.34 (s, 3H), 1.02 (s, 3H), 0.93 (m, 1H); 13C NMR (75 MHz, CD3OD) δ 157.4, 155.8, 151.8, 110.6, 106.4, 105.2, 77.6, 71.4, 64.6, 50.3, 47.5, 43.8, 41.3, 40.0, 37.6, 37.4, 36.6, 36.5, 34.9, 30.7, 28.3, 27.2, 19.2; [α]25D −41.9 (c 0.51, CH3OH); IR (neat; cm−1) 3395 (br), 2944, 1581, 1574, 1477, 1402, 1279, 1131, 1001; HRMS (ESI+) calculated for C26H38NO3 [M + H+] 412.2852, found 412.2846. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 95% and retention time of 4.1 min for the title compound.
(6aR,9R,10aR)-3-[(1r,3R)-3-(Isothiocyanatomethyl)-adamantan-1-yl]-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromene-1,9-diol (16)
To a solution of homologated amine 15 (5 mg, 0.012 mmol) and Et3N (~8 μL, 0.061 mmol) in THF (1 mL) cooled to 0 °C under N2 was added CS2 (3 μL, 0.049 mmol), and the reaction mixture was stirred for 2 h at 0 °C. To the reaction flask was added TsCl (8 mg, 0.043 mmol), and the solution was stirred for an additional 2 h while gradually warming to room temperature. The reaction was quenched with pH 7 phosphate buffer (1 mL) and the mixture diluted with Et2O (5 mL). The organic phase was washed with phosphate buffer (2 × 2 mL), and the aqueous phase was back-extracted with Et2O (2 × 3 mL). The organic layers were combined, washed with brine (4 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (10–20% acetone in hexanes) to give isothiocyanate 16 as a white amorphous solid in 89% yield (5 mg, 0.011 mmol). 1H NMR (500 MHz, CD3OD) δ 6.33 (d, J = 1.9 Hz, 1H), 6.23 (d, J = 1.9 Hz, 1H), 3.73 (m, 1H), 3.52 (m, 1H), 3.28 (s, 2H), 2.44 (td, J = 11.3, 2.5 Hz, 1H), 2.20 (m, 2H), 2.11 (m, 1H), 1.91–1.54 (m, 13H), 1.45–1.29 (m, 2H), 1.35 (s, 3H), 1.18 (m, 1H), 1.04 (s, 3H), 0.94 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 157.6, 155.9, 151.1, 131.3, 110.9, 106.3, 105.1, 77.7, 71.4, 57.6, 50.3, 46.4, 43.5, 40.1, 39.9, 37.5, 37.0, 36.9, 36.7, 34.9, 30.4, 28.3, 27.2, 19.2; [α]23D −54.0 (c 0.98, CH3CN); IR (neat; cm−1) 3333 (br), 2925, 2850, 2181, 2104, 1701, 1620, 1573, 1516, 1448, 1416, 1384, 1273, 1141, 1040; HRMS (EI+) calculated for C27H35NO3S [M+] 453.2338, found 453.2323. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 97% and retention time of 5.3 min for the title compound.
(6aR,9R,10aR)-3-[(1r,3R)-3-(Azidomethyl)adamantan-1-yl]-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]-chromene-1,9-diol (17)
To a mixture of amine 15 (~6 mg, 0.014 mmol), Et3N (~6 μL, 0.041 mmol), and CuSO4 (cat.) in H2O (0.50 mL) at room temperature under air was added dropwise a 0.1 M solution of TfN3 (0.27 mL, 0.027 mmol) in CH2Cl2 and to it was added additional CH2Cl2 (0.25 mL) followed by MeOH (1.5 mL) until a homogeneous solution was obtained. The reaction mixture was stirred for 12 h, then concentrated in vacuo and taken up in Et2O (5 mL). The organic layer was washed with pH 7 phosphate buffer (2 × 3 mL) and the aqueous layer back-extracted with Et2O (2 × 5 mL). The organic layers were combined, washed with brine (3 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (10–20% acetone in hexanes) to give azide 17 as an orange amorphous solid in 93% yield (~6 mg, 0.013 mmol). 1H NMR (500 MHz, CD3OD) δ 6.31 (d, J = 1.8 Hz, 1H), 6.21 (d, J = 1.8 Hz, 1H), 3.73 (m, 1H), 3.52 (m, 1H), 3.06 (s, 2H), 2.44 (td, J = 11.2, 2.2 Hz, 1H), 2.15 (m, 2H), 2.10 (m, 1H), 1.91–0.90 (m, 17H), 1.35 (s, 3H), 1.03 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 157.5, 155.8, 151.4, 110.7, 106.3, 105.1, 77.7, 71.4, 65.1, 50.3, 46.6, 43.6, 40.4, 39.9, 37.4, 37.1, 36.7, 36.7, 34.9, 30.4, 28.3, 27.2, 19.2; [α]23D −73.2 (c 0.56, CH3CN); IR (neat; cm−1) 3356 (br), 2925, 2850, 2098, 1703, 1621, 1573, 1449, 1416, 1363, 1275, 1143, 1054; HRMS (EI+) calculated for C26H35N3O3 [M+] 437.2678, found 423.2688. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 98% and retention time of 5.1 min for the title compound.
(6aR,9R,10aR)-3-[(1r,3R)-3-Aminoadamantan-1-yl]-6,6-di-methyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromene-1,9-diol (18)
To a solution of keto acid 12 (0.11 g, 0.27 mmol) and Et3N (45 μL, 0.032 mmol) in PhMe (2.7 mL) at room temperature under air in a reaction flask equipped with a stir bar and reflux condenser was added DPPA (70 μL, 0.032 mmol) dropwise, and the solution was refluxed for 3 h. Upon completion of the reaction, the reaction mixture was concentrated in vacuo and taken up in THF/H2O (1:1, v/v) (1.3 mL) and stirred. A solution of Hg(OAc)2 (0.10 g, 0.32 mmol) in THF/H2O (1:1, v/v) (1.3 mL) was added to the reaction mixture, and stirring was continued overnight. To the reaction mixture was added a solution of NaBH4 (51 mg, 1.4 mmol) in 5% NaOH (w/v), and stirring was continued for an additional 3 h. The reaction was quenched by the addition of 1 N HCl (~1 mL) until gas evolution ceased, and the solution was concentrated in vacuo. The crude product was taken up in EtOAc (2 mL) and washed with pH 7 phosphate buffer (2 mL). The aqueous phase was back-extracted with EtOAc (2 × 1 mL), and the organic layers were combined, washed with brine (2 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (5–10% EtOH in CH2Cl2 with sat. aq NH4OH) to give amine 18 as a clear glass in 76% yield (82 mg, 0.21 mmol). 1H NMR (500 MHz, CD3OD) δ 6.31 (d, J = 1.8 Hz, 1H), 6.22 (d, J = 1.8 Hz, 1H), 3.72 (m, 1H), 3.52 (m, 1H), 2.42 (td, J = 11.2, 2.3 Hz, 1H), 2.20–2.05 (m, 3H), 1.90–1.83 (m, 1H), 1.78–1.47 (m, 11H), 1.45–1.11 (m, 4H), 1.33 (s, 3H), 1.01 (s, 3H), 0.94 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 157.5, 155.8, 150.9, 110.7, 106.3, 105.2, 77.6, 71.3, 51.5, 50.3, 49.5, 45.2, 43.1, 39.9, 39.2, 36.7, 36.5, 34.9, 31.5, 28.3, 27.2, 19.2; [α]23D −57.4 (c 0.67, CH3OH); IR (neat; cm−1) 3415 (br), 2973, 2848, 1651, 1593, 1574, 1437, 1384, 1234, 1203, 1042; HRMS (ESI+) calculated for C25H36NO3 [M + H+] 398.2696, found 398.2690. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 95% and retention time of 4.0 min for the title compound.
(6aR,9R,10aR)-3-[(1r,3R)-3-Isothiocyanatoadamantan-1-yl]-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]-chromene-1,9-diol (19)
To a solution of amine 18 (~5 mg, 0.012 mmol) and Et3N (8 μL, 0.058 mmol) in THF (1 mL) cooled to 0 °C under N2 was added CS2 (~3 μL, 0.046 mmol), and the solution was stirred for 2 h at 0 °C. To the reaction mixture was added TsCl (8 mg, 0.041 mmol), and stirring was continued for an additional 2 h while gradually warming to room temperature. The reaction was quenched with pH 7 phosphate buffer (1 mL) and the mixture diluted with Et2O (5 mL). The organic phase was washed with phosphate buffer (2 × 2 mL) and the aqueous phase back-extracted with Et2O (2 × 3 mL). The organic layers were combined, washed with brine (4 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (10–20% acetone in hexanes) to give isothiocyanate 19 as a white amorphous solid in 86% yield (5 mg, 0.010 mmol). 1H NMR (500 MHz, CD3OD) δ 6.30 (d, J = 1.8 Hz, 1H), 6.22 (d, J = 1.8 Hz, 1H), 3.74 (m, 1H), 3.52 (m, 1H), 2.44 (td, J = 11.3, 2.3 Hz, 1H), 2.26 (m, 2H), 2.15–1.55 (m, 13H), 1.49–1.26 (m, 3 H), 1.35 (s, 3H), 1.18 (m, 1H), 1.03 (s, 3H), 0.94 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 157.6, 156.0, 149.7, 129.3, 111.1, 106.2, 104.9, 77.7, 71.3, 60.7, 50.3, 50.0, 44.0, 42.4, 39.9, 39.2, 36.6, 35.8, 34.9, 31.3, 28.2, 27.2, 19.2; [α]23D −67.0 (c 0.88, CH3CN); IR (neat; cm−1) 3338 (br), 2930, 2857, 2093(br), 1701, 1620, 1573, 1452, 1416, 1384, 1273, 1054; HRMS (EI+) calculated for C26H33NO3S [M+] 439.2181, found 439.2183. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 99% and retention time of 5.2 min for the title compound.
(6aR,9R,10aR)-3-[(1r,3R)-3-Azidoadamantan-1-yl]-6,6-di-methyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromene-1,9-diol (20)
To a mixture of amine 18 (5 mg, 0.012 mmol), Et3N (~5 μL, 0.037 mmol), and CuSO4 (cat.) in H2O (0.5 mL) at room temperature under air was added dropwise a 0.1 M solution of TfN3 (0.25 mL, 0.025 mmol) in CH2Cl2, and to it was added additional CH2Cl2 (0.25 mL) followed by MeOH (1.5 mL) until a homogeneous solution was obtained. The reaction was stirred for 12 h, then concentrated in vacuo and taken up in Et2O (5 mL). The organic layer was washed with pH 7 phosphate buffer (2 × 3 mL) and the aqueous layer back-extracted with Et2O (2 × 5 mL). The organic layers were combined, washed with brine (2 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (10–20% acetone in hexanes) to give azide 20 as an orange amorphous solid in 98% yield (5 mg, 0.012 mmol). 1H NMR (500 MHz, CD3OD) δ 6.31 (d, J = 1.8 Hz, 1H), 6.21 (d, J = 1.8 Hz, 1H), 3.74 (m, 1H), 3.52 (m, 1H), 2.44 (td, J = 11.2, 2.2 Hz, 1H), 2.29 (m, 2H), 2.11 (m, 1H), 1.95–1.67 (m, 12H), 1.48–1.13 (m, 4H), 1.35 (s, 3H), 1.03 (s, 3H), 0.94 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 158.0, 156.0, 150.1, 111.1, 106.0, 105.1, 77.7, 71.3, 60.8, 50.3, 48.0, 42.8, 41.8, 39.9, 39.6, 36.7, 36.2, 34.9, 31.7, 28.2, 27.2, 19.2; [α]23D −33.3 (c 1.02, CH3CN); IR (neat; cm−1) 3312 (br), 2924, 2754, 2088, 1704, 1620, 1574, 1455, 1416, 1364, 1249, 1138, 1055; HRMS (EI+) calculated for C25H33N3O3 [M+] 423.2522, found 423.2510. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 98% and retention time of 5.0 min for the title compound.
2-{(1R,3r)-3-[(6aR,9R,10aR)-1,9-Dihydroxy-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-adamantan-1-yl}acetonitrile (23)
To a solution of ketoacid 12 (42 mg, 0.099 mmol) in THF (2.0 mL) cooled to 0 °C were added a catalytic amount of DMF and (COCl)2 (42 μL, 0.49 mmol), and the reaction mixture was allowed to warm to room temperature over 45 min. The reaction mixture was again cooled to 0 °C, and to it was added a solution of CH2N2 in ether (1.0 mL, 0.5 M). The reaction mixture was stirred for 2 h while gradually warming to room temperature. Excess CH2N2 was quenched with acetic acid. The contents were dissolved in Et2O (5 mL) and extracted with 1 N HCl (2 × 1 mL). The aqueous layer was back-extracted with Et2O (2 × 1 mL). The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and concentrated. The crude α-diazoketone was carried onto the next reaction.
To a solution of the intermediate α-diazoketone (~0.099 mmol) in THF (5.0 mL) were added silver benzoate (0.090 g, 0.39 mmol), Et3N (0.055 mL, 0.39 mmol), and NH4OH (0.13 mL, 0.97 mmol), and the reaction mixture was stirred at room temperature for 2 h. The contents were dissolved in Et2O (5 mL) and extracted with 1 N HCl (2 × 1 mL). The aqueous layer was back-extracted with Et2O (2 × 1 mL), and the organic layers were combined, washed with brine, dried over Na2SO4, filtered, and absorbed onto Celite. The crude product was purified via flash chromatography on silica gel (20–40% acetone in hexanes) to give the homologated amide 22 as an amorphous solid in 27% yield (12 mg, 0.026 mmol) over the two steps.
To a solution of amide 22 (11 mg, 0.025 mmol) in dioxane (1 mL) at room temperature under N2 atmosphere was added pyridine (40 μL, 0.50 mmol), and the mixture was stirred for 10 min and followed by the addition of TFAA (34 μL, 0.25 mmol). The reaction mixture was stirred for 16 h and quenched with the addition of pH 7 phosphate buffer (0.5 mL). The reaction mixture was taken up in Et2O (1 mL) and the organic phase washed with phosphate buffer (2 × 1 mL) and the aqueous phase back-extracted with Et2O. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was redissolved in MeOH (1 mL), and to the mixture was added K2CO3 (35 mg, 0.25 mmol). The mixture was stirred at room temperature under ambient atmosphere for 3 h. The reaction mixture was cooled to 0 °C, and to it was added NaBH4 (92 mg, 0.25 mmol). The mixture was stirred for 3 h. The reaction mixture was quenched with 1 N HCl (1 mL) and was concentrated in vacuo and taken up in Et2O (3 mL). The organic phase was extracted with 1 N HCl (2 mL), and the aqueous phase was back-extracted with Et2O (2 × 2 mL). The organic layers were combined, washed with brine (2 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (10% to 20% to 30% acetone in hexanes) to yield nitrile 23 as a clear colorless film in 86% yield (9.1 mg, 0.021 mmol) from amide 22. 1H (300 MHz, CD3OD) δ 6.33 (d, J = 1.8 Hz, 1H), 6.23 (d, J = 1.8 Hz, 1H), 3.71 (m, 1H), 3.51 (m, 1H), 2.43 (td, J = 11.2, 2.2 Hz, 1H), 2.30 (s, 2H), 2.23–2.02 (m, 3H), 1.95–1.53 (m, 12H), 1.48–1.10 (m, 4H), 1.35 (s, 3H), 1.03 (s, 3H), 1.05–0.85 (m, 1H); 13C NMR (75 MHz, CD3OD) δ 155.9, 151.0, 119.1, 111.2, 110.9, 106.3, 105.0, 77.7, 71.4, 50.3, 48.3, 43.2, 43.2, 41.9, 39.9, 37.7, 36.7, 34.9, 34.0, 32.0, 30.6, 28.2, 27.2, 19.2; [α]24D −73.1 (c 0.41, CH3OH); IR (neat; cm−1) 3377 (br), 2975, 2909, 2852, 2247, 1620, 1574, 1448, 1416, 1055, 840; HRMS (ESI+) calculated for C27H36NO [M + H+] 422.2695, found 422.2700. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 97% and retention time of 4.8 min for the title compound.
(1R,3r)-3-[(6aR,10aR)-1-Hydroxy-6,6-dimethyl-9-oxo-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-adamantane-1-carboxamide (24)
To a solution of keto acid 12 (14 mg, 0.20 mmol) and HOBT (6 mg, 0.048 mmol) in THF (1.6 mL), stirred at room temperature for 5 min under ambient atmosphere, was added a suspension of EDCI (9 mg, 0.048 mmol), and the mixture was stirred overnight. After 16 h, NH4OH (0.5 mL) was added dropwise over 5 min and the reaction mixture was stirred for an additional 1 h. EDCI (9 mg, 0.048 mmol) was added, and the reaction mixture was stirred overnight. The reaction was quenched with 1 N HCl (1 mL) and extracted with EtOAc (3 × 1 mL). The organic layers were combined, washed once with phosphate buffer (1 mL) followed by brine (1 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (5–10% EtOH in CH2Cl2 with sat. aq NH4OH) to yield amide 24 as an off-white semisolid in 76% yield (10 mg, 0.024 mmol). 1H NMR (300 MHz, CD3OD) δ 6.34 (d, J = 1.8 Hz, 1H), 6.28 (d, J = 1.8 Hz, 1H), 3.85 (dd, J = 14.9, 3.4 Hz, 1H), 2.80 (m, 1H), 2.51 (m, 2H), 2.24–1.75 (m, 16H), 1.60–1.24 (m, 2H), 1.45 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CD3OD) δ 214.4, 183.7, 157.4, 155.8, 151.9, 110.0, 106.4, 105.1, 77.7, 46.3, 45.7, 43.2, 43.1, 42.9, 41.6, 39.6, 37.5, 36.8, 36.1, 30.4, 28.2, 27.9, 19.0; [α]25D −93.3 (c 0.31, CH3OH); IR (neat; cm−1) 3478, 3345, 2925, 2854, 1679, 1634, 1572, 1415, 1355; HRMS (ESI+) calculated for C25H34NO4 [M + H+] 424.2489, found 424.2488.
(1R,3r)-3-[(6aR,9R,10aR)-1,9-Dihydroxy-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-adamantane-1-carbonitrile (25)
To a solution of ketoamide 24 (6 mg, 0.014 mmol) in dioxane (2.5 mL) at room temperature under N2 atmosphere was added pyridine (~24 μL, 0.28 mmol), and the mixture was stirred for 10 min followed by the addition of TFAA (~19 μL, 0.14 mmol). The reaction mixture was stirred for 16 h and quenched with the addition of pH 7 phosphate buffer (0.5 mL) and diluted with Et2O (1 mL). The organic phase was washed with phosphate buffer (2 × 1 mL) and the aqueous phase back-extracted with Et2O (1 mL). The organic layers were combined, washed with brine (2 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was redissolved in MeOH (1 mL), and to it was added K2CO3 (20 mg, 0.14 mmol). The mixture was stirred at room temperature under ambient atmosphere for 16 h. The reaction mixture was cooled to 0 °C, and NaBH4 (5 mg, 0.14 mmol) was added. The mixture was stirred for 3 h. The reaction was quenched with 1 N HCl (1 mL), and the reaction mixture was concentrated in vacuo. The reaction mixture was taken up in Et2O (3 mL). The organic phase was extracted with 1 N HCl (2 mL), and the aqueous phase was back-extracted with Et2O (2 × 2 mL). The organic layers were combined, washed with brine (2 mL), dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (10% to 20% to 30% acetone in hexanes) to yield nitrile 25 as a clear colorless film in 71% yield (5 mg, 0.010 mmol) from ketoamide 24. 1H NMR (300 MHz, CDCl3) δ 6.30 (d, J = 1.8 Hz, 1H), 6.23 (d, J = 1.8 Hz, 1H), 3.73 (m, 1H), 3.51 (m, 1H), 2.44 (td, J = 11.2, 2.2 Hz, 1H), 2.24–1.69 (m, 15H), 1.50–1.10 (m, 4H), 1.35 (s, 3H), 1.03 (s, 3H), 0.94 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 155.1, 155.0, 148.6, 124.9, 109.8, 106.3, 103.9, 71.0, 70.9, 48.2, 44.8, 41.3, 39.2, 38.5, 35.7, 35.5, 34.9, 33.4, 31.2, 27.9, 27.8, 26.0, 19.0; [α]24D −91.8 (c 0.74, CH3OH); IR (neat; cm−1) 3380 (br), 2931, 2859, 2235, 1700, 1620, 1575, 1452, 1416, 1273, 1192, 1141, 1056, 827; HRMS (ESI+) calculated for C26H34NO3 [M + H+] 408.2539, found 408.2532. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 98% and retention time of 4.7 min for the title compound.
Methyl {(1R,3r)-3-[(6aR,9R,10aR)-1,9-Dihydroxy-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-adamantan-1-yl}carbamate (26)
To a solution of hydroxy acid 13 (19 mg, 0.045 mmol) in PhMe (1.6 mL) were added Et3N (~9 μL, 0.068 mmol) and DPPA (14 μL, 0.0068 mmol), and the reaction was heated to reflux for 2 h. The reaction was concentrated in vacuo, and the crude isocyanate was taken up in MeOH (1.6 mL). To it was added NaOMe (24 mg, 0.45 mmol). The reaction mixture was heated to reflux overnight. Upon completion, as determined by TLC, the reaction mixture was concentrated in vacuo. The contents were taken up in Et2O (5 mL) and extracted with 1 N HCl (2 × 1 mL). The aqueous layer was back-extracted with Et2O (2 × 1 mL). The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and adsorbed onto Celite. The crude product was purified via flash chromatography on silica gel (20–40% acetone in hexanes) to give the methyl carbamate 26 as a yellow amorphous solid in 65% yield (13 mg, 0.029 mmol). 1H NMR (300 MHz, CD3OD) δ 6.32 (d, J = 1.8 Hz, 1H), 6.23 (d, J = 1.8 Hz, 1H), 3.73 (m, 1H), 3.56 (s, 3H), 3.52 (m, 1H), 2.44 (td, J = 11.2, 2.2 Hz, 1H), 2.21 (m, 2 H), 2.12 (m, 1H), 2.03–1.67 (m, 12H), 1.48–1.13 (m, 4H), 1.35 (s, 3H), 1.03 (s, 3H), 0.94 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 157.6, 157.5, 155.9, 150.9, 110.8, 106.3, 105.1, 77.6, 71.3, 52.5, 51.7, 50.3, 47.9, 43.2, 41.9, 39.9, 39.0, 36.66, 36.70, 34.9, 31.4, 28.3, 27.2, 19.2; [α]23D −64.7 (c 0.58, CH3OH); IR (neat; cm−1) 3362, 2910, 2856, 1703, 1621, 1574, 1519, 1453, 1416, 1361, 1249, 1135, 1071; HRMS (ESI+) calculated for C27H38NO5 [M + H+] 456.2750, found 456.2749. LC/MS analysis (Waters MicroMass ZQ system) showed purity of 98% and retention time of 4.6 min for the title compound.
Radioligand Binding Assays
The affinities (Ki) of the new compounds for rat CB1 receptor as well as for mouse and human CB2 receptors were obtained by using membrane preparations from rat brain or HEK293 cells expressing either mCB2 or hCB2 receptors, respectively, and [3H]CP-55,940 as the radioligand, as previously described.9,34 Results from the competition assays were analyzed using nonlinear regression to determine the IC50 36 values for the ligand; Ki values were calculated from the IC50 (Prism by GraphPad Software, Inc.). Each experiment was performed in triplicate, and Ki values were determined from three independent experiments and are expressed as the mean of the three values. Results for the covalently reacting isothiocyanato electrophiles are expressed as pseudoaffinities (Ki*).
cAMP Assay.9,34
HEK293 cells stably expressing rCB1 receptor were used for the studies. The cAMP assay was carried out using PerkinElmer’s Lance Ultra cAMP kit following the protocol of the manufacturer. Briefly, the assays were carried out in 384-well plates using 1000–1500 cells/well. The cells were harvested with non-enzymatic cell dissociation reagent Versene, washed once with HBSS, and resuspended in the stimulation buffer. The various concentrations of the test compound (5 μL) in forskolin (2 μM final concentration) containing stimulation buffer were added to the plate followed by the cell suspension (5 μL). Cells were stimulated for 30 min at room temperature. Eu-cAMP tracer working solution (5 μL) and Ulight-anti-cAMP working solution (5 μL) were then added to the plate and incubated at room temperature for 60 min. The data were collected on a PerkinElmer Envision instrument. The EC50 values were determined by nonlinear regression analysis using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA).
Photoaffinity Covalent Labeling
Rat brain membranes (rCB1, Pel-Freez Biologicals, Rogers, AR) were prepared following the previously described and appropriately modified procedures.37 Membranes from human CB2 receptors (hCB2) expressed in HEK29338 were incubated with the azido ligands (20 and 17) in concentrations of 10-fold their Ki values for 30 min at 37 °C in a water bath with gentle agitation and then exposed to UV (254 nm) for 1 min to activate the ligand.39 Unbound excess ligand was washed out twice with 1% BSA in TME. This was followed by an additional washing to remove residual BSA, and the membranes were isolated by centrifugation (Beckman Coulter, JA 20, 17 000 rpm, 10 min, 25 °C). A blank membrane sample was treated in parallel using the same procedure and used as a control.
Electrophilic Covalent Labeling
CB1 and CB2 membranes were incubated with the isothiocyanato ligands (19 and 16) in a concentration of 10-fold their Ki values at 37 °C in a water bath with gentle agitation for 1 h and treated as above without the photoirradiation step. Unbound excess ligand was washed out twice with 1% BSA in TME and once with TME alone to remove BSA, and the membranes were isolated by centrifugation.
Saturation Binding Assay
Protein concentrations were determined by using a Bio-Red Bradford protein assay kit,40 and saturation binding assays were performed in a 96-well format.16 Membrane pellets were resuspended in TME containing 0.1% BSA. A total of 25 μg of protein was added to each well, and [3H]CP-55,940 was diluted in 0.1% BSA/TME buffer to yield ligand concentrations ranging from 0.5 to 23.8 nM. Nonspecific binding was determined in the presence of 4 μM unlabeled CP-55,940. The assay plates were incubated at 30 °C with gentle agitation for 1 h. The resultant mixture was then transferred to Unifilter GF/B filter plates, and the bound ligand was separated from unbound using a Packard Filtermate-96 cell harvester (PerkinElmer Packard, Shelton, CT). Filter plates were washed five times with ice-cold wash buffer (50 mM Tris-base, 5 mM magnesium chloride with 0.5% BSA, pH 7.4). Bound radioactivity was quantitated in a Packard TopCount scintillation counter. Nonspecific binding was subtracted from the total bound radioactivity to calculate the specific binding of [3H]CP-55,940 (measured as pmol/g in saturation curves). Saturation assays were performed in triplicate, and data points were presented as the mean ± SEM. Bmax and Kd values were calculated by nonlinear regression using GraphPad Prism 4.0 (one-site binding analysis equation Y = BmaxX/(Kd + X), GraphPad Software, San Diego, CA).
Supplementary Material
Acknowledgments
Acknowledgment is made to The National Institute on Drug Abuse (Grants 5R01 DA07215, 2P01 DA09158, 5 R01 DA03801) for generous support of this research.
ABBREVIATIONS USED
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- (−)-Δ9-THC
(−)-Δ9-tetrahydrocannabinol
- CNS
central nervous system
- HEK293
human embryonic kidney cell line
- SAR
structure–activity relationship
- NMR
nuclear magnetic resonance
- HOBT
hydroxybenzotriazole
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HPLC
high-performance liquid chromatography
- LAPS
ligand assisted protein structure
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- DPPA
diphenylphosphorylazide
- TFAA
trifluoromethylacetic anhydride
- TLC
thin layer chromatography
- TMSOTf
trimethylsilyl trifluoromethanesulfonate
- p-TSA
p-toluenesulfonic acid
- TsCl
p-toluenesulfonyl chloride
Footnotes
Notes
The authors declare no competing financial interest.
Saturation binding curves using [3H]CP-55,940 for CB1 and CB2 receptors preincubated with the 3′-azido (20) and 3′-isothiocyanato (19) analogues. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 2.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 3.Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 4.Lu D, Meng Z, Thakur GA, Fan P, Steed J, Tartal CL, Hurst DP, Reggio PH, Deschamps JR, Parrish DA, George C, Jarbe TU, Lamb RJ, Makriyannis A. Adamantyl cannabinoids: a novel class of cannabinergic ligands. J Med Chem. 2005;48:4576–4585. doi: 10.1021/jm058175c. [DOI] [PubMed] [Google Scholar]
- 5.Lu D, Guo J, Duclos RI, Jr, Bowman AL, Makriyannis A. Bornyl-and isobornyl-Delta8-tetrahydrocannabinols: a novel class of cannabinergic ligands. J Med Chem. 2008;51:6393–6399. doi: 10.1021/jm8005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dixon DD, Sethumadhavan D, Benneche T, Banaag AR, Tius MA, Thakur GA, Bowman A, Wood JT, Makriyannis A. Heteroadamantyl cannabinoids. J Med Chem. 2010;53:5656–5666. doi: 10.1021/jm100390h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Goanvic D, Tius MA. Oxaza adamantyl cannabinoids. A new class of cannabinoid receptor probes. J Org Chem. 2006;71:7800–7804. doi: 10.1021/jo061352c. [DOI] [PubMed] [Google Scholar]
- 8.Thakur GA, Bajaj S, Paronis C, Peng Y, Bowman AL, Barak LS, Caron MG, Parrish D, Deschamps JR, Makriyannis A. Novel adamantyl cannabinoids as CB1 receptor probes. J Med Chem. 2013;56:3904–3921. doi: 10.1021/jm4000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikas SP, Alapafuja SO, Papanastasiou I, Paronis CA, Shukla VG, Papahatjis DP, Bowman AL, Halikhedkar A, Han X, Makriyannis A. Novel 1′,1′-chain substituted hexahydrocannabinols: 9beta-hydroxy-3-(1-hexylcyclobut-1-yl)-hexahydrocannabinol (AM2389) a highly potent cannabinoid receptor 1 (CB1) agonist. J Med Chem. 2010;53:6996–7010. doi: 10.1021/jm100641g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu C, Ramamurthy A, Makriyannis A, Tius MA. Synthesis of covalent probes for the radiolabeling of the cannabinoid receptor. J Org Chem. 2003;68:55–61. doi: 10.1021/jo0264978. [DOI] [PubMed] [Google Scholar]
- 11.Guo Y, Abadji V, Morse KL, Fournier DJ, Li X. Makriyannis A (−)-11-Hydroxy-7′-isothiocyanato-1′, 1′-dimethylheptyl-delta 8-THC: a novel high-affinity irreversible probe for the cannabinoid receptor in the brain. J Med Chem. 1994;37:3867–3870. doi: 10.1021/jm00049a002. [DOI] [PubMed] [Google Scholar]
- 12.Li C, Xu W, Vadivel SK, Fan P, Makriyannis A. High affinity electrophilic and photoactivatable covalent endocannabinoid probes for the CB1 receptor. J Med Chem. 2005;48:6423–6429. doi: 10.1021/jm050272i. [DOI] [PubMed] [Google Scholar]
- 13.Makriyannis A. 2012 Division of Medicinal Chemistry Award Address. Trekking the cannabinoid road: a personal perspective. J Med Chem. 2014;57:3891–3911. doi: 10.1021/jm500220s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mercier RW, Pei Y, Pandarinathan L, Janero DR, Zhang J, Makriyannis A. hCB2 ligand-interaction landscape: cysteine residues critical to biarylpyrazole antagonist binding motif and receptor modulation. Chem Biol. 2010;17:1132–1142. doi: 10.1016/j.chembiol.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pei Y, Mercier RW, Anday JK, Thakur GA, Zvonok AM, Hurst D, Reggio PH, Janero DR, Makriyannis A. Ligand-binding architecture of human CB2 cannabinoid receptor: evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem Biol. 2008;15:1207–1219. doi: 10.1016/j.chembiol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picone RP, Khanolkar AD, Xu W, Ayotte LA, Thakur GA, Hurst DP, Abood ME, Reggio PH, Fournier DJ, Makriyannis A. (−)-7′-Isothiocyanato-11-hydroxy-1′, 1′-dimethylheptylhexahydrocannabinol (AM841) a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol Pharmacol. 2005;68:1623–1635. doi: 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- 17.Szymanski DW, Papanastasiou M, Melchior K, Zvonok N, Mercier RW, Janero DR, Thakur GA, Cha S, Wu B, Karger B, Makriyannis A. Mass spectrometry-based proteomics of human cannabinoid receptor 2: covalent cysteine 6.47(257)-ligand interaction affording megagonist receptor activation. J Proteome Res. 2011;10:4789–4798. doi: 10.1021/pr2005583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dominianni SJ, Ryan CW, DeArmitt CW. Synthesis of 5-(tert-alkyl)resorcinols. J Org Chem. 1977;42:344–346. doi: 10.1021/jo00422a037. [DOI] [PubMed] [Google Scholar]
- 19.Saa JM, Dopico M, Martorell G, Garciaraso A. Deoxygenation of highly hindered phenols. J Org Chem. 1990;55:991–995. [Google Scholar]
- 20.Martin MT, Liu B, Cooley BE, Eaddy JF. Open air palladium catalyzed cyanation—the use of PMHS to protect from oxygen. Tetrahedron Lett. 2007;48:2555–2557. [Google Scholar]
- 21.McOmie JFW, West DE. 3,3′-Dihydroxybiphenyl. Org Synth, Collect. 1973;V:412–414. [Google Scholar]
- 22.Archer RA, Blanchard WB, Day WA, Johnson DW, Lavagnino ER, Ryan CW, Baldwin JE. Cannabinoids. 3. Synthetic approaches to 9-ketocannabinoids. Total synthesis of nabilone. J Org Chem. 1977;42:2277–2284. doi: 10.1021/jo00433a020. [DOI] [PubMed] [Google Scholar]
- 23.Nikitenko A, Alimardanov A, Afragola J, Schmid J, Kristofova L, Evrard D, Hatzenbuhler NT, Marathias V, Stack G, Lenicek S, Potoski J. First scale-up synthesis of WAY-262398, a novel, dual-acting SSRI/5HT1a antagonist. Org Process Res Dev. 2009;13:91–97. [Google Scholar]
- 24.Lewis FW, Eichler MC, Grayson DH. Synthesis of beta-amino alcohols via the reduction of lactamides derived from ethyl (2S)-lactate with borane-methyl sulfide. Synlett. 2009:1923–1928. [Google Scholar]
- 25.Wong R, Dolman SJ. Isothiocyanates from tosyl chloride mediated decomposition of in situ generated dithiocarbamic acid salts. J Org Chem. 2007;72:3969–3971. doi: 10.1021/jo070246n. [DOI] [PubMed] [Google Scholar]
- 26.Nyffeler PT, Liang CH, Koeller KM, Wong CH. The chemistry of amine-azide interconversion: catalytic diazotransfer and regioselective azide reduction. J Am Chem Soc. 2002;124:10773–10778. doi: 10.1021/ja0264605. [DOI] [PubMed] [Google Scholar]
- 27.Alper PB, Hung SC, Wong CH. Metal catalyzed diazo transfer for the synthesis of azides from amines. Tetrahedron Lett. 1996;37:6029–6032. [Google Scholar]
- 28.Shioiri T, Ninomiya K, Yamada S. Diphenylphosphoryl azide. A new convenient reagent for a modified Curtus reaction and for the peptide synthesis. J Am Chem Soc. 1972;94:6203–6205. doi: 10.1021/ja00772a052. [DOI] [PubMed] [Google Scholar]
- 29.Carda M, Gonzalez F, Sanchez R, Marco JA. Stereo-selective synthesis of (−)-cytoxazone. Tetrahedron: Asymmetry. 2002;13:1005–1010. [Google Scholar]
- 30.Malanga C, Urso A, Lardicci L. Mercuration demercuration of aliphatic isocyanates—a new mild route to primary amines. Tetrahedron Lett. 1995;36:8859–8860. [Google Scholar]
- 31.Gademann K, Ernst M, Hoyer D, Seebach D. Synthesis and biological evaluation of a cyclo-beta-tetrapeptide as a somatostatin analogue. Angew Chem, Int Ed. 1999;38:1223–1226. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1223::AID-ANIE1223>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 32.Newman MS, Beal PF., III An improved Wolff rearrangement in homogeneous medium. J Am Chem Soc. 1950;72:5163–5165. [Google Scholar]
- 33.Campagna F, Carotti A, Casini G. Convenient synthesis of nitriles from primary amides under mild conditions. Tetrahedron Lett. 1977;18:1813–1816. [Google Scholar]
- 34.Sharma R, Nikas SP, Paronis CA, Wood JT, Halikhedkar A, Guo JJ, Thakur GA, Kulkarni S, Benchama O, Raghav JG, Gifford RS, Jarbe TU, Bergman J, Makriyannis A. Controlled-deactivation cannabinergic ligands. J Med Chem. 2013;56:10142–10157. doi: 10.1021/jm4016075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khanolkar AD, Lu D, Ibrahim M, Duclos RI, Jr, Thakur GA, Malan TP, Jr, Porreca F, Veerappan V, Tian X, George C, Parrish DA, Papahatjis DP, Makriyannis A. Cannabilactones: a novel class of CB2 selective agonists with peripheral analgesic activity. J Med Chem. 2007;50:6493–6500. doi: 10.1021/jm070441u. [DOI] [PubMed] [Google Scholar]
- 36.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 37.Charalambous A, Yan G, Houston DB, Howlett AC, Compton DR, Martin BR, Makriyannis A. 5′-Azido-delta 8-THC: a novel photoaffinity label for the cannabinoid receptor. J Med Chem. 1992;35:3076–3079. doi: 10.1021/jm00094a023. [DOI] [PubMed] [Google Scholar]
- 38.Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A. Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem. 1999;42:769–776. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- 39.Cavalla D, Neff NH. Chemical mechanisms for photoaffinity labeling of receptors. Biochem Pharmacol. 1985;34:2821–2826. doi: 10.1016/0006-2952(85)90001-2. [DOI] [PubMed] [Google Scholar]
- 40.Bradford MM. Rapid and sensitive method for quantitation of microgram guantities of protein utilizing principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.