

This paper reviews the role of CDK4/6 in tumorigenesis and summarizes the clinical trial experience with inhibitors of these kinases in breast cancer, including key efficacy and toxicity data. The results with oral CDK4/6 inhibitors to date have offered promising glimpses of significant activity in hormone-sensitive breast cancer. This activity, combined with a favorable toxicity profile, makes this family of novel therapies very exciting.
Keywords: Breast neoplasms, Inhibitors of cyclin-dependent kinase 4 proteins, Palbociclib, LEE011, Abemaciclib, Flavopiridol
Abstract
Imbalance of the cyclin D and cyclin-dependent kinase (CDK) pathway in cancer cells may result in diversion away from a pathway to senescence and toward a more proliferative phenotype. Cancer cells may increase cyclin D-dependent activity through a variety of mechanisms. Therapeutic inhibition of CDKs in tumors to negate their evasion of growth suppressors has been identified as a key anticancer strategy. In this review, we outline the development of CDK inhibitory therapy in breast cancer, including the initial experience with the pan-CDK inhibitor flavopiridol and the next generation of oral highly selective CDK4 and CDK6 inhibitors PD0332991 (palbociclib), LEE011 (ribociclib), and LY2835219 (abemaciclib). Data from phase I and II studies in estrogen receptor-positive (ER+) breast cancer demonstrate promising efficacy with manageable toxic effects, chiefly neutropenia. We discuss these studies and the phase III studies that are accruing or nearing completion. We describe the application of such therapy to other breast cancer settings, including HER2-positive breast cancer and the adjuvant treatment of early breast cancer. We also discuss potential concerns surrounding the combination of CDK inhibitors with chemotherapy and their effects on repair of double-strand DNA breaks in cancer cells. Oral highly selective CDK inhibitors show great promise in improving the outcomes of patients with ER+ breast cancer, although caution must apply to their combination with other agents and in the early breast cancer setting.
Implications for Practice:
Cyclin-dependent kinases (CDKs) interact with cyclin D proteins to play an integral role in cell cycle progression and represent attractive therapeutic targets in many tumors, including breast cancer. A number of highly selective inhibitors of CDK4 and CDK6 (CDK4/6) are currently in clinical trial development with one agent recently approved by the U.S. Food and Drug Administration for the treatment of advanced ER+, HER2-negative breast cancer. This article reviews the role of CDK4/6 in tumorigenesis and summarizes the clinical trial experience with inhibitors of these kinases in breast cancer. Key efficacy and toxicity data from the clinical trial experience to date have been reviewed. The planned further expansion of their role in breast cancer therapies and potential concerns regarding combinations with other therapies are addressed.
Introduction
Insensitivity to antigrowth signals is a hallmark of cancer. Human cells have developed a complex interplay of pro- and antiproliferative signals, imbalance of which in cancer cells may result in diversion away from a pathway to senescence and toward a more proliferative phenotype. Phosphorylation of antiproliferative retinoblastoma tumor suppressor protein (Rb) and related proteins by cyclin-dependent kinases 4 and 6 (CDK4/6) in complex with cyclin D subunits allows increased synthesis of genes important for DNA replication and thus progression through the cell cycle. Cancer cells may increase cyclin D-dependent activity through a variety of mechanisms. These cell cycle proteins represent attractive targets for therapeutic intervention in human cancers. In this review, we have outlined the development of CDK4/6 inhibitory therapy in breast cancer, including the initial experience with the pan-CDK inhibitor flavopiridol and the next generation of oral highly selective CDK4/6 inhibitors PD0332991 (palbociclib [Ibrance]; Pfizer Inc., New York, NY, http://www.pfizer.com), LEE011 (ribociclib), and LY2835219 (abemaciclib). We describe the clinical studies exploring the combination of these novel compounds with standard endocrine therapies in advanced breast cancer. Potential future directions and combination approaches are also addressed.
Methods
A comprehensive literature review of all relevant clinical trials, original research articles, review articles, and editorials in the PubMed database concerning cyclin-dependent kinase inhibitors in breast cancer was performed. The search terms “CDK inhibitor,” “breast cancer,” “palbociclib,” “LEE011,” “LY2835219,” “abemaciclib,” and “flavopiridol” were used. The search was limited to human subjects and the English language. Reference lists of key papers were also reviewed. In addition, ongoing clinical trials using these agents were identified from the NIH clinical trial registry (http://www.clinicaltrials.gov) using a search incorporating the names of the selective CDK4/6 inhibitors and the term “breast cancer.” Unpublished abstracts were identified by searching relevant abstract resources including American Society of Clinical Oncology, American Association for Cancer Research (AACR), European Society for Medical Oncology databases.
The Cell Cycle and the Balance Between Senescence and Proliferation
Insensitivity to antigrowth signals was identified as one of the hallmarks of cancer by Hanahan and Weinberg in their seminal 2000 paper and its 2011 update [1, 2]. Cells reach a key cell cycle checkpoint (the so-called restriction point) during the G1 phase of the cell cycle, when they decide whether to continue into the S phase and commit to another cycle of cell division or withdraw into the quiescent G0 phase [3]. Antigrowth signals influence the decision reached by the cell at this key point. The cellular response to antigrowth signals from the extracellular environment is largely controlled by the cell cycle machinery, which regulates progression through the G1 phase (and from there into the S phase) of the cell cycle [1]. These antiproliferative signals are communicated through the retinoblastoma tumor suppressor protein (Rb) and its relatives p107 and p130. Rb itself is regulated by cyclin-CDK complexes, an evolutionarily conserved family of serine-threonine protein kinases [4]. Progression through the G1 to S phases requires phosphorylation of Rb by the cyclin-dependent kinase CDK4 or the highly homologous enzyme CDK6 in complex with their activating subunits cyclin D1, D2, or D3 [5]. Hypophosphorylated Rb sequesters and alters the function of the E2F family, leading to decreased expression of genes involved in cell cycle progression from G1 to S phase and then replication and mitotic progression [6, 7]. In contrast, hyperphosphorylation of Rb reduces its ability to repress activity of E2F transcription factors, resulting in increased synthesis of genes with products that are essential for DNA replication.
Inhibition of CDK activity and G1/S phase progression is mediated through the Cip-Kip family of universal CDK inhibitors and the specific CDK4/6 inhibitors of the INK4 family, including p16INK4a [8]. Many tumors increase cyclin D-dependent activity and thereby escape senescence through multiple mechanisms such as p16 inactivation, CDK4 amplification, CDK4 mutation with loss of INK4 binding, cyclin D1 overexpression or by translocation or amplification of its encoding gene, CCND1 [9]. Cyclin D1 appears to be essential for the development and maintenance of certain breast cancers, with mice lacking cyclin D1 or CDK4 demonstrating resistance to growth of implanted breast cancers driven by the HER2/neu oncogene [10–14]. Furthermore, acute inducible shutdown of cyclin D1 function or inhibition of CDK4 function with the CDK4/6 inhibitor palbociclib in adult mice bearing HER2/neu-driven breast tumors results in tumor cell senescence [15]. CCND1 amplification is a common event in human breast cancers, identified in 38% of the HER2-expressing molecular subtype, 58% of luminal B cancers, and 29% of luminal A cancers [16]; CDK4 gain is seen in 24%, 25%, and 14%, respectively. The interaction of Rb, cyclin D, and CDK4/6 in the cell cycle is illustrated in Figure 1. Cyclin D1 has other non-CDK-dependent functions in tumorigenesis including induction of chromosomal instability (CIN) by transcriptional regulation of CIN-related genes [17]. It is also implicated in enhancement of DNA damage sensing and repair in response to ionizing radiation and DNA damaging drugs, induction of cellular migration and invasion, inhibition of mitochondrial metabolism, and enhancement of angiogenesis [18]. Because many of these functions are independent of its interaction with CDK4/6, they would not be expected to be affected by therapeutic inhibition of these CDKs. Moreover, CDK6 has recently been found to have a kinase-independent function in angiogenesis, representing a potentially separate role for CDK6 inhibition in tumor therapy [19].
Figure 1.

The interaction of Rb, cyclin D, and CDK4/6 in cell cycle progression. Hypophosphorylated Rb sequesters E2F family members, leading to decreased expression of genes involved in cell cycle progression from G1 to S phases. CDK4/6-cyclin D complex phosphorylates Rb leading to loss of repression of E2F factors, resulting in cell cycle progression. Cip-Kip and INK4 family members inhibit cyclin-dependent kinase activity.
Abbreviations: CDK4/6, cyclin-dependent kinases 4 and 6; E2F, E2 transcription factor; P, phosphate group; Rb, retinoblastoma tumor suppressor protein.
CDK6 has recently been found to have a kinase-independent function in angiogenesis, representing a potentially separate role for CDK6 inhibition in tumor therapy.
The importance of the restriction point in determining whether cells continue to divide or become quiescent and the observed frequency of cyclin D-CDK4/6-INK4 pathway alterations in tumor cells have fueled interest in therapies to inhibit cyclin-dependent kinases. The chemical structures of CDK4/6 inhibitors discussed in this review (flavopiridol, palbociclib, ribociclib, and abemaciclib) are illustrated in Figure 2. The multi-CDK inhibitor dinaciclib is also being evaluated in clinical studies in combination with chemotherapy in the triple-negative subset of breast cancer [20, 21]; however, it targets CDKs 1, 2, 5, and 9 rather than CDK4/6 and is reviewed elsewhere [22]. Similarly, roscovitine (seliciclib) has been evaluated in the phase I setting in patients with solid tumors, with minimal disease stabilization seen in monotherapy [23]. It inhibits CDKs 1, 2, 5, and 7 but is a poor inhibitor of CDK4/6 [24]. A search of ClinicalTrials.gov shows no clinical studies currently evaluating this agent in breast cancer therapy [25].
Figure 2.

Chemical structures and formulae of selected CDK4/6 inhibitors.
CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer
Flavopiridol (alvocidib) belongs to a group of relatively nonselective pan-CDK inhibitors that also includes olomoucine and roscovitine and was the first CDK inhibitor to enter human clinical trials [26]. It is an intravenously administered pan-CDK inhibitor with targets including CDKs 1, 2, 4/6, and 7 [27]. It induces G0 and G1 arrest in a variety of tumor cells, whereas chemotherapy-induced S-phase delay may enhance its cytotoxic effect [28]. Flavopiridol has had largely disappointing results as single agent, whereas combination with chemotherapy appears to be more promising, especially in acute leukemia [29]. Some early activity was seen in a phase I study in patients with solid tumors (including breast cancer) treated with docetaxel followed 4 hours later by escalating doses of flavopiridol [30]. Limitations of flavopiridol include its intravenous route of administration, the requirement for complex regimens in which administration must be timed carefully after chemotherapy to achieve effect, and dose-limiting toxicities including diarrhea and neutropenia.
The next generation of CDK inhibitors to reach therapeutic clinical studies in breast cancer are more selective than the pan-CDK inhibitors such as flavopiridol and include highly selective inhibitors of CDK4/6. Preclinical models suggest a particular role for CDK4/6 inhibition in estrogen receptor-positive (ER+) breast cancer cells, including cells retaining estrogen sensitivity and those with acquired estrogen resistance. Cyclin D1 is a transcriptional target of ER, with estrogen-mediated ER signaling resulting in increased expression of cyclin D1 RNA and protein, accompanied by phosphorylation of Rb and G1/S phase transition [31–33]. Conversely, growth arrest induced by antiestrogens in ER+ breast cancer cells is accompanied by decreased cyclin D1 expression [34]. Furthermore, cyclin D1 augments estrogen-dependent gene expression through ERα signaling [35]. Meanwhile, the development of endocrine resistance in ER+ breast cancer cells is associated with persistent cyclin D1 expression and Rb phosphorylation [36]. Direct evidence of a particular role for CDK4/6 inhibition in ER+ cells comes from the evaluation of the CDK4/6 inhibitor palbociclib in vitro in a panel of molecularly characterized breast cancer cell lines that demonstrated most activity in luminal cancers (including those with conditioned estrogen resistance), whereas nonluminal and basal types were most resistant [37].
Palbociclib
Palbociclib, formerly PD0332991, is the most clinically advanced CDK4/6 inhibitor. This agent was selected from a group of pyridopyrimidine compounds based on its selectivity for CDK4, its ability to induce clean G1 arrest in MDA-MB-435 tumor cells, and its superior physical and pharmaceutical properties [38]. It is a highly selective inhibitor of CDK4 and the highly homologous enzyme CDK6 (which also complexes with cyclin D subunits and phosphorylates Rb at the same sites). In preclinical studies, palbociclib demonstrated in vitro and in vivo activity against Rb-positive tumors, including significant tumor regression in certain xenograft models [37–40]. This was somewhat surprising because it had been postulated that cell cycle-targeting agents would result in growth arrest without tumor regression. Palbociclib demonstrated most activity in ER-positive breast cancer cell lines with luminal features in a panel of molecularly characterized breast cancer cell lines [37]. Activity was also observed in a high proportion of HER2-positive breast cancer cell lines, particularly those with luminal features.
Palbociclib demonstrated most activity in ER-positive breast cancer cell lines with luminal features in a panel of molecularly characterized breast cancer cell lines. Activity was also observed in a high proportion of HER2-positive breast cancer cell lines, particularly those with luminal features.
The key clinical studies of palbociclib in breast cancer are summarized in Table 1. Palbociclib was first investigated in humans in an open-label phase I dose-finding study in patients with Rb-positive solid tumors or lymphoma [41]. Palbociclib was administered orally on a 2-week-on, 1-week-off (2/1) schedule at doses ranging from 100 to 225 mg daily. It demonstrated low to moderate interpatient pharmacokinetic variability, with a general dose-dependent increase in exposure over the range of 100–225 mg daily and was excreted slowly with a mean t1/2 of 26.7 hours. The 200-mg daily dose was established as the maximum tolerated dose, with myelosuppression (particularly neutropenia) being the only dose-limiting toxicitiy (DLT). This myelosuppressive effect is consistent with the expected effect of CDK inhibition in rapidly dividing cell types and with preclinical models. A nadir in absolute neutrophil count (ANC) and platelets was observed at the end of the 2-week dosing period in cycle 1. This was followed by a rebound in platelet and neutrophil counts, although neutrophils did not tend to recover fully to pretreatment levels.
Table 1.
Selected phase II and III studies of palbociclib in breast cancer
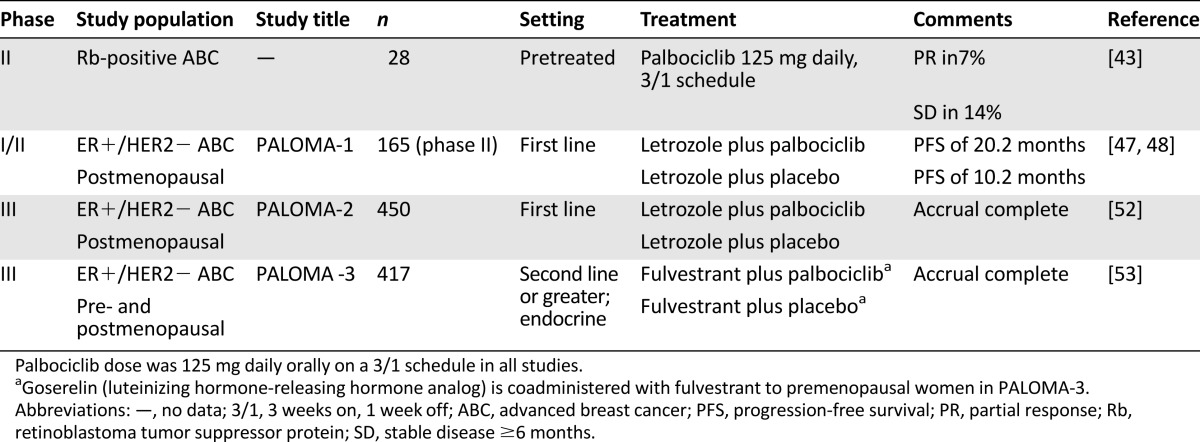
A separate 3-week-on, 1-week-off (3/1) schedule also underwent phase I evaluation [42]. In that trial, 41 patients with Rb-positive solid tumors or lymphoma were treated at daily doses ranging from 25 to 150 mg daily. A half-life for palbociclib of approximately 26 hours was confirmed. On this 3/1 schedule, a nadir of ANC and platelets was observed at the end of the dosing schedule, followed, as before, by a rebound in counts. The DLT of neutropenia was seen starting at the 75-mg dose, but dose escalations continued cautiously because this uncomplicated neutropenia was considered tolerable. The recommended phase II dose was established as 125 mg daily for the 3/1 schedule. The most common nonhematologic adverse events were fatigue, diarrhea, nausea, and constipation and were mainly low grade. No patients in these two phase I studies had clinically significant electocardiographic QT-interval changes.
Single-agent palbociclib at 125 mg daily orally on the 3/1 schedule was evaluated in a phase II study in patients with Rb-positive advanced breast cancer [43]. Partial responses were seen in 2 of 28 (7%) evaluable patients, with stable disease >6 months in 14%. No responses or prolonged disease stabilizations were seen in patients with ER-negative tumors. The disappointing single-agent activity observed in this chemotherapy-pretreated population suggests potential cross-resistance with chemotherapy and increases the attractiveness of combining palbociclib with other agents, notably endocrine therapy. As in the phase I studies, myelotoxicity predominated, with grade 3/4 neutropenia in 50% and thrombocytopenia in 21%, whereas no nonhematologic grade 3/4 toxicities were seen. This study was the first in which a patient experienced a clinically significant neutropenia: febrile neutropenia occurred during cycle 1 in a patient who had been heavily pretreated with six prior chemotherapy regimens.
Letrozole is a nonsteroidal aromatase inhibitor approved for the treatment of advanced hormone receptor-positive (HR+) breast cancer in postmenopausal women [44]. A multicenter randomized phase I/II study (NCT00721409, known as PALOMA-1) evaluated the combination of palbociclib (at 125 mg orally each day on the 3/1 schedule) with letrozole 2.5 mg daily continuously versus letrozole alone in the first-line treatment of ER+/HER2− advanced breast cancer in postmenopausal women. An initial phase I study evaluated safety and tolerability of the combination in an unselected population. This phase I study demonstrated no significant pharmacokinetic interaction between palbociclib and letrozole. It also yielded a promising early efficacy signal, with a partial response rate of 33% in evaluable patients and a clinical benefit rate of 67% [45]. The phase II component of the study was divided into two parts: the first evaluated the recommended phase II dosing (palbociclib 125 mg daily orally on the 3/1 schedule plus letrozole 2.5 mg daily continuously versus letrozole alone) in patients selected only by ER/HER2 status, and the second used the same treatment allocations in patients further selected for CCND1 amplification and/or p16 loss. A total of 66 patients were enrolled in part 1, and 99 were enrolled in part 2. Exploratory analysis revealed no additional predictive value of CCND1 status or p16 loss for palbociclib efficacy over ER status alone; consequently, parts 1 and 2 were combined for an overall efficacy analysis. The addition of palbociclib resulted in a statistically significant prolongation of the primary endpoint of progression-free survival (PFS), which was more than tripled in the combination arm at the second interim analysis (26.1 vs. 7.5 months; hazard ratio: 0.37; 95% confidence interval: 0.21–0.63; p < .001) [46, 47]. Combination treatment was extremely well tolerated. The most frequent treatment-related adverse events were neutropenia, leukopenia, anemia, and fatigue, and no cases of febrile neutropenia were observed in this study. There were also increased incidences of grade 3 or 4 infections and pulmonary embolism in the combination arm (5% vs. 0% for both toxic effects).These promising efficacy results led the U.S. Food and Drug Administration (FDA) to assign a “breakthrough therapy” designation to palbociclib for breast cancer treatment in April 2013.
Results of the final PFS efficacy analysis of PALOMA-1 were presented at the AACR annual meeting in April 2014 and have been published subsequently [48]. An impressive improvement in PFS was maintained in the palbociclib arm (20.2 vs. 10.2 months; p = .0004). A significant difference in overall survival (OS) had not yet been seen at this analysis. Following the AACR presentation, Pfizer submitted a new drug application (NDA) with the FDA for palbociclib in combination with letrozole for the first-line treatment of women with ER+/HER2− advanced or metastatic breast cancer [49]. An NDA is the vehicle through which drug manufacturers formally propose that the FDA approve a novel therapy for sales and marketing. On February 3, 2015, the FDA granted accelerated approval for palbociclib for this indication [50, 51]. Under the accelerated approval regulations, results from additional well-controlled studies are required to verify and describe the clinical benefit in more detail. In the case of palbociclib, these required results will include the PFS and OS results from a large randomized phase III study, PALOMA-2 (NCT01740427) [52]. The design of this study mirrors the phase II component of the PALOMA-1 study, with randomization between letrozole plus palbociclib versus letrozole alone for women with advanced, previously untreated ER+/HER2− breast cancer. A total of 450 patients will be enrolled worldwide, with final data collection for the primary outcome measure of PFS expected in March 2015.
Meanwhile, the phase III PALOMA-3 study (NCT01942135) is investigating hormonal therapy plus palbociclib in pre- and postmenopausal patients whose disease has progressed after prior endocrine therapy [53]. The endocrine therapy used in PALOMA-3 is the estrogen receptor antagonist fulvestrant (Faslodex; AstraZeneca, London, U.K., http://www.astrazeneca.com), with coadministration of the luteinizing hormone-releasing hormone analog goserelin in premenopausal patients to induce chemical menopause. A search of ClinicalTrials.gov shows that further studies are evaluating palbociclib in other breast cancer clinical settings (adjuvant therapy, residual disease after neoadjuvant chemotherapy) and in combination with other agents including paclitaxel and trastuzumab emtansine (T-DM1) [54].
Combinations with HER2-targeted therapy are of key interest, given the sensitivity observed in HER2-positive breast cancer (especially with luminal features) in preclinical models [37]. There is a plausible biologic rationale for CDK inhibition in HER2-positive breast cancer, given the frequent activation of the cyclin D-CDK4 pathway in this breast cancer subtype [10]. Preclinical models demonstrate activity of palbociclib against HER2-positive breast cancer, both as monotherapy and in combination with HER2-targeting agents [40, 55, 56]. In particular, the mode of action of CDK4/6 inhibitors appears complementary to that of T-DM1, a recently approved antibody drug conjugate targeting the HER2 pathway [57]. In tumor explants, palbociclib suppressed the proliferation of residual clones of disease after T-DM1 therapy, indicating a potential use in sequential therapy [56]. A currently accruing phase Ib study will assess the combination of palbociclib with T-DM1 (NCT01976169) [58].
LEE011 (Ribociclib)
LEE011 is an orally bioavailable small molecule inhibitor of CDK4/6 that exhibits highly specific inhibitory activity against CDK4/cyclin D1 and CDK6/cyclin D3 complexes in isolated enzyme assays [59]. In preclinical tumor models, LEE011 has demonstrated a dose-dependent antitumor activity that tracks well with CDK4/6 inhibition [60]. As with palbociclib, tumor regression was seen in some in vivo models. In mouse models, LEE011 showed single-agent activity in melanomas with activating mutations of BRAF or NRAS and in breast cancers with intact estrogen receptor and/or activating aberrations of PIK3CA and/or HER2 [60].
Animal pharmacokinetic studies indicate that the elimination of LEE011 may potentially be affected by coadministered drugs that inhibit or induce CYP3A4, whereas LEE011 may itself inhibit CYP3A4, CYP1A2, and BSEP in a dose-dependent manner [60]. As of January 17, 2014, 132 patients had been treated with single-agent LEE011 in the first-in-human phase I study (NCT01237236) [61]. Doses ranging from 50 to 1,200 mg daily were evaluated on a 3/1 schedule. In addition, continuous dosing of LEE011 at 600 mg was evaluated (once daily for 28 days of a 28-day cycle). The most frequently reported adverse events (AEs), regardless of study treatment relationship, included neutropenia (40%), leukopenia (36%), nausea (35%), and fatigue (27%). The majority of adverse events were grade 1 or 2 and reversible. Asymptomatic grade 2 Fridericia corrected QT prolongation was observed with increasing frequency starting at 600 mg. No new cardiac abnormalities were observed in any patient. The recommended phase II dosing was 600 mg/day on 21 days of the 28-day schedule.
LEE011 is currently being evaluated in ER+ breast cancer and other tumor types including BRAF-mutant melanoma and other solid tumors, as shown in a search of ClinicalTrials.gov [62]. The breast cancer studies include a phase Ib/II open-label study in combination with letrozole and the phosphatidylinositol 3-kinase (PI3K) inhibitor BYL719 (NCT01872260) [63] and another phase Ib/II study with the steroidal aromatase inhibitor exemestane and the mammalian target of rapamycin inhibitor everolimus (NCT01857193) [64]. These combinations are attractive, given preclinical evidence that CDK4/6 inhibition may overcome intrinsic and adaptive resistance to PI3K inhibition [65]. Preliminary data from 16 patients enrolled in the exemestane/everolimus/LEE011 combination study indicate that the triplet combination appears feasible [66]. The phase II component will compare safety and efficacy of the triplet regimen versus two doublets (exemestane plus everolimus; exemestane plus LEE011). Similarly, phase I data for the combination of LEE011 plus letrozole (n = 10) and BYL719 (n = 7) have been presented [67]. Both arms demonstrated acceptable safety profiles, with neutropenia observed, as expected with LEE011. Accrual to the third arm (LEE011, BYL719 plus letrozole) will proceed, followed by planned phase II randomization between the triplet regimen and the two doublets. Two studies are evaluating the combination of LEE011 with letrozole in ER+/HER2− breast cancer: a large phase III randomized double-blind placebo-controlled study in women with previously untreated breast cancer (MONALEESA-2, NCT01958021) and a smaller open-label randomized presurgical pharmacodynamic study targeting 120 patients (MONALEESA-1, NCT01919229) [68, 69].
LY2835219 (Abemaciclib)
Abemaciclib is another selective oral CDK4/6 inhibitor that is in early clinical trial development. In preclinical assessment it was found to selectively inhibit CDK4 and CDK6 with half maximal inhibitory concentration values of 2 and 10 nM, respectively [70, 71]. It is a potent inhibitor of Rb phosphorylation in vitro and in vivo and induces G1-specific arrest and inhibition of tumor growth [72]. It was evaluated as monotherapy in a phase I study in patients with multiple tumor types at doses of 50–225 mg daily and 75–275 mg twice daily, followed by expansion in 5 tumor types [73–75]. In the dose-escalation phase (n = 55), the maximum tolerated dose for every 12-hour dosing was 200 mg with dose-limiting toxicity of grade 3 fatigue at 200 mg (1 of 6 evaluable patients) and 275 mg (2 of 3 evaluable patients) [73]. This study included a monotherapy cohort of 47 women with heavily pretreated metastatic breast cancer. Abemaciclib monotherapy achieved partial response in 9 of 47 women (19%), whereas 51% had stable disease [74]. All of these breast cancer responses were in women with ER+ disease, with a response rate of 25% (9 of 36) in this subset. The disease control rate (defined for this study as partial responses plus stable disease of any duration) for the entire study population was 70%, and 81% among the 36 women with ER+ breast cancer. Common treatment-related adverse events included diarrhea, nausea, fatigue, vomiting, and neutropenia. Neutropenia was the only grade 3/4 adverse event seen in >5% of patients, occurring in 11%. A separate metastatic breast cancer cohort (n = 13) evaluated the combination of abemaciclib 200 mg twice daily plus fulvestrant [75]. The most common possibly treatment-related AEs were diarrhea (8% grade 3), fatigue (8% grade 3), neutropenia (33% grade 3), nausea (no grade 3), vomiting (no grade 3), and leukopenia (23% grade 3). There were no grade 4 events and no episodes of febrile neutropenia. Eight confirmed and 3 unconfirmed partial responses were observed.
Ongoing studies are assessing abemaciclib in the ER+ breast cancer population, both as monotherapy (MONARCH 1, NCT02102490) and in combination with endocrine agents, according to ClinicalTrials.gov [76]. These studies include a planned phase III randomized double-blind placebo-controlled trial of nonsteroidal aromatase inhibitors with or without abemaciclib in previously untreated advanced hormone-sensitive breast cancer (MONARCH 3, NCT02246621) [77]. In addition, an open-label safety study is assessing this CDK inhibitor in combination with various standard hormone therapies (letrozole, anastrozole, tamoxifen, exemestane, or exemestane plus everolimus) with a primary outcome measure of the number of patients with one or more drug-related adverse events (NCT02057133) [78]. Finally, a randomized double-blind placebo-controlled phase III study will compare the combination of abemaciclib 200 mg twice daily orally on days 1–28 of each 28-day cycle with fulvestrant 500 mg intramuscularly on days 1 and 15 of cycle 1 and then on day 1 of cycle 2 and beyond versus fulvestrant alone (Monarch 2, NCT02107703) [79]. The primary endpoint will be PFS.
Palbociclib in Combination With Chemotherapy
Evaluation of palbociclib in triple-negative breast cancer cell lines indicates potential antagonism between the cytostatic effect of CDK4/6 inhibition and the cytotoxic effects of chemotherapy agents [80]. Palbociclib maintained viability of Rb-proficient (but not Rb-deficient) cells treated with doxorubicin, resulting in recurrent cell growth after doxorubicin exposure. Continuous coadministration of palbociclib with paclitaxel also resulted in antagonization of the cytotoxic effects of this chemotherapy agent. Similarly, coadministration of palbociclib with doxorubicin or carboplatin in mouse models of Rb-proficient breast cancer resulted in lower cytotoxicity than either chemotherapy agent alone [40]. As with flavopiridol, the timing of palbociclib administration may be important with chemotherapy partners, with acute administration for 24 hours to synchronize cell populations prior to paclitaxel administration leading to increased paclitaxel cytotoxicity [80]. A potential deleterious effect of combining such agents with chemotherapy is suggested by an observed shift in the repair of DNA double-strand breaks induced by ionizing radiation from the homologous recombination (HR) pathway to the nonhomologous end joining (NHEJ) pathway [80]. HR is a relatively error-free mechanism of double-strand break repair because it relies on the sister chromatid as a repair template, whereas NHEJ is a more error-prone mechanism that results in greater genomic instability and is implicated in tumorigenesis [81]. The combination of CDK inhibitors with radiation and/or DNA-damaging chemotherapy could theoretically result in tumor progression in the advanced cancer setting. Similarly, caution must be used in the application of such therapies in the adjuvant or neoadjuvant settings, in which induction of genomic instability could result in the potentially devastating development of secondary malignancies. It is essential that any study applying CDK inhibitors to the adjuvant or neoadjuvant settings take these potential negative interactions with chemotherapy and radiation into account in study design. Such concerns do not extend to combinations of CDK4/6 inhibitors with endocrine therapy in the adjuvant setting, and such combinations will be the focus of upcoming clinical studies in early breast cancer.
Conclusion
The restoration of antigrowth signaling through inhibition of cyclin-dependent kinases has been established as a promising therapeutic strategy for human breast cancers and other tumor types. Studies with first-generation inhibitors were hampered by relative nonselectivity for CDK types and complex administration schedules that would be difficult to translate into clinical practice. Preclinical studies with next-generation oral selective CDK4/6 inhibitors demonstrated expected effects on target proteins and the ability to induce G1 arrest. Early therapeutic clinical trials in HR+ breast cancer have provided heartening efficacy signals. In particular, the randomized phase II study of letrozole with or without the CDK4/6 inhibitor palbociclib indicated a doubling of progression-free survival for the combination therapy, which—if borne out in the confirmatory phase III study—would represent a major therapeutic advance for metastatic HR+ breast cancer. Similar studies with other selective CDK4/6 inhibitors are under way. The oral route of administration and the relative absence of burdensome toxic effects would make this therapeutic family very attractive to patients if the early efficacy signals are replicated in these phase III studies.
The decision by the FDA to grant accelerated approval to palbociclib in combination with letrozole was based on the results from only 165 patients enrolled in the PALOMA-1 study. It remains to be seen whether the impressive PFS benefits observed in this small randomized study will be replicated in the phase III PALOMA-2 study. The FDA approval adds another option to the selection of first-line therapies available for the management of advanced HR+ breast cancer. Reasonable alternatives still include endocrine monotherapy with aromatase inhibitors, fulvestrant, or tamoxifen. Selection of treatment for each patient should take individual patient factors into account including comorbidities, anticipated compliance with treatment, blood count monitoring, the burden of metastatic disease, and treatment-related toxicity. In addition, pharmacoeconomic implications must be considered. The cost of combination therapy with palbociclib will exceed that of endocrine monotherapy many times over, given a planned retail cost of Ibrance in the U.S. of $9,850 per month [82].
Ongoing studies are expanding the potential application of these agents outside the ER-positive advanced breast cancer setting, including adjuvant and neoadjuvant treatment of patients with resectable breast cancer, treatment of patients with residual cancer post neoadjuvant therapy, and combinations with chemotherapy and other targeted therapies.
The results of clinical trials with oral CDK4/6 inhibitors to date have offered promising glimpses of significant activity in hormone-sensitive breast cancer. This activity, combined with a favorable toxicity profile, makes this family of novel therapies very exciting. It is to be hoped that the currently accruing phase III studies will build on this promise and confirm the place of these therapies in the breast cancer armamentarium.
Author Contributions
Conception/Design: Conleth G. Murphy, Maura N. Dickler
Collection and/or assembly of data: Conleth G. Murphy, Maura N. Dickler
Data analysis and interpretation: Conleth G. Murphy, Maura N. Dickler
Manuscript writing: Conleth G. Murphy, Maura N. Dickler
Final approval of manuscript: Conleth G. Murphy, Maura N. Dickler
Disclosures
Maura N. Dickler: Roche/Genentech, Novartis, Pfizer, Astra Zeneca (C/A), Roche/Genentech, Eli Lilly, Seragon, Novartis (RF). The other author indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Pardee AB. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- 4.Morgan DO. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20:187–190. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- 6.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 7.Dean JL, Thangavel C, McClendon AK, et al. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene. 2010;29:4018–4032. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 8.Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 9.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 10.Bowe DB, Kenney NJ, Adereth Y, et al. Suppression of neu-induced mammary tumor growth in cyclin D1 deficient mice is compensated for by cyclin E. Oncogene. 2002;21:291–298. doi: 10.1038/sj.onc.1205025. [DOI] [PubMed] [Google Scholar]
- 11.Landis MW, Pawlyk BS, Li T, et al. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 12.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- 13.Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Reddy HK, Mettus RV, Rane SG, et al. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005;65:10174–10178. doi: 10.1158/0008-5472.CAN-05-2639. [DOI] [PubMed] [Google Scholar]
- 15.Choi YJ, Li X, Hydbring P, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22:438–451. doi: 10.1016/j.ccr.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casimiro MC, Crosariol M, Loro E, et al. ChIP sequencing of cyclin D1 reveals a transcriptional role in chromosomal instability in mice. J Clin Invest. 2012;122:833–843. doi: 10.1172/JCI60256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pestell RG. New roles of cyclin D1. Am J Pathol. 2013;183:3–9. doi: 10.1016/j.ajpath.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kollmann K, Heller G, Schneckenleithner C, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2013;24:167–181. doi: 10.1016/j.ccr.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinaciclib and epirubicin hydrochloride in treating patients with metastatic triple-negative breast cancer. Available at http://clinicaltrials.gov/ct2/show/NCT01624441. Accessed November 4, 2014.
- 21.Phase I/Ib dose-escalation of dinaciclib with weekly paclitaxel for advanced solid tumor malignancies & assessment of MYC oncogene overexpression. Available at http://www.clinicaltrials.gov/ct2/show/NCT01676753. Accessed November 4, 2014.
- 22.Criscitiello C, Viale G, Esposito A, et al. Dinaciclib for the treatment of breast cancer. Expert Opin Investig Drugs. 2014;23:1305–1312. doi: 10.1517/13543784.2014.948152. [DOI] [PubMed] [Google Scholar]
- 23.Benson C, White J, De Bono J, et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br J Cancer. 2007;96:29–37. doi: 10.1038/sj.bjc.6603509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cicenas J, Valius M. The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol. 2011;137:1409–1418. doi: 10.1007/s00432-011-1039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Search results: Roscovitine AND breast cancer. Available at http://clinicaltrials.gov/ct2/results?term=roscovitine+AND+breast+cancer. Accessed November 4, 2014.
- 26.Cicenas J, Kalyan K, Sorokinas A, et al. Highlights of the latest advances in research on CDK inhibitors. Cancers (Basel) 2014;6:2224–2242. doi: 10.3390/cancers6042224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sedlacek H, Czech J, Naik R, et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int J Oncol. 1996;9:1143–1168. doi: 10.3892/ijo.9.6.1143. [DOI] [PubMed] [Google Scholar]
- 28.Matranga CB, Shapiro GI. Selective sensitization of transformed cells to flavopiridol-induced apoptosis following recruitment to S-phase. Cancer Res. 2002;62:1707–1717. [PubMed] [Google Scholar]
- 29.Gallorini M, Cataldi A, di Giacomo V. Cyclin-dependent kinase modulators and cancer therapy. BioDrugs. 2012;26:377–391. doi: 10.1007/BF03261895. [DOI] [PubMed] [Google Scholar]
- 30.Fornier MN, Rathkopf D, Shah M, et al. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clin Cancer Res. 2007;13:5841–5846. doi: 10.1158/1078-0432.CCR-07-1218. [DOI] [PubMed] [Google Scholar]
- 31.Altucci L, Addeo R, Cicatiello L, et al. 17beta-Estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G(1)-arrested human breast cancer cells. Oncogene. 1996;12:2315–2324. [PubMed] [Google Scholar]
- 32.Altucci L, Addeo R, Cicatiello L, et al. Estrogen induces early and timed activation of cyclin-dependent kinases 4, 5, and 6 and increases cyclin messenger ribonucleic acid expression in rat uterus. Endocrinology. 1997;138:978–984. doi: 10.1210/endo.138.3.5002. [DOI] [PubMed] [Google Scholar]
- 33.Prall OW, Sarcevic B, Musgrove EA, et al. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem. 1997;272:10882–10894. doi: 10.1074/jbc.272.16.10882. [DOI] [PubMed] [Google Scholar]
- 34.Watts CK, Brady A, Sarcevic B, et al. Antiestrogen inhibition of cell cycle progression in breast cancer cells in associated with inhibition of cyclin-dependent kinase activity and decreased retinoblastoma protein phosphorylation. Mol Endocrinol. 1995;9:1804–1813. doi: 10.1210/mend.9.12.8614416. [DOI] [PubMed] [Google Scholar]
- 35.Casimiro MC, Wang C, Li Z, et al. Cyclin D1 determines estrogen signaling in the mammary gland in vivo. Mol Endocrinol. 2013;27:1415–1428. doi: 10.1210/me.2013-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thangavel C, Dean JL, Ertel A, et al. Therapeutically activating RB: Reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. 2011;18:333–345. doi: 10.1530/ERC-10-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- 39.Michaud K, Solomon DA, Oermann E, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228–3238. doi: 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts PJ, Bisi JE, Strum JC, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104:476–487. doi: 10.1093/jnci/djs002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwartz GK, LoRusso PM, Dickson MA, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1) Br J Cancer. 2011;104:1862–1868. doi: 10.1038/bjc.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flaherty KT, Lorusso PM, Demichele A, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18:568–576. doi: 10.1158/1078-0432.CCR-11-0509. [DOI] [PubMed] [Google Scholar]
- 43.DeMichele A, Sanders Clark A, Heitian D, et al. A phase II trial of an oral CDK 4/6 inhibitor, PD0332991, in advanced breast cancer. J Clin Oncol. 2013;31(suppl):519a. [Google Scholar]
- 44.Cohen MH, Johnson JR, Li N, et al. Approval summary: Letrozole in the treatment of postmenopausal women with advanced breast cancer. Clin Cancer Res. 2002;8:665–669. [PubMed] [Google Scholar]
- 45.Slamon DJ, Hurvitz SA, Applebaum S, et al. Phase I study of PD-0332991, cyclin-D kinase (CDK) 4/6 inhibitor in combination with letrozole for first-line treatment of patients with ER-positive, HER2-negative breast cancer. J Clin Oncol. 2010;28(suppl):3060a. [Google Scholar]
- 46.Finn RS, Crown JP, Boer K et al. Preliminary results of a randomized phase II study of PD 0332991, a cyclin-dependent kinase 4/6 inhibitor, in combination with letrozole for first-line treatment of patients with post-menopausal, ER-positive, HER2-negative advanced breast cancer. Poster presented at: San Antonio Breast Cancer Symposium; December 6–10, 2011; San Antonio, TX. [Google Scholar]
- 47.Finn RS, Crown JP, Boer K, et al. Results of a randomized phase II study of PD-0332991, a cyclin dependent kinase (CDK) 4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2- advanced breast cancer (BC) [abstract 1000] Ann Oncol. 2012;23(suppl 2):ii43–ii45. [Google Scholar]
- 48.Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 49.Pfizer to submit palbociclib New Drug Application with FDA based on final results of PALOMA-1. Available at http://www.pfizer.com/news/press-release/press-release-detail/pfizer_to_submit_palbociclib_new_drug_application_with_fda_based_on_final_results_of_paloma_1. Accessed November 4, 2014.
- 50.NDA 207103: Accelerated approval. Available at http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2015/207103Orig1s000ltr.pdf. Accessed February 24, 2015.
- 51.Ibrance (palbociclib) [prescribing information]. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207103s000lbl.pdf. Accessed February 24, 2015.
- 52.A study of palbociclib (PD-0332991) + letrozole vs. letrozole for 1st line treatment of postmenopausal women with ER+/HER2- advanced breast cancer (PALOMA-2). Available at http://clinicaltrials.gov/ct2/show/NCT01740427. Accessed November 4, 2014.
- 53.Palbociclib (PD0332991) combined with fulvestrant in hormone receptor+ HER2-negative metastatic breast cancer after endocrine failure (PALOMA-3). Available at http://clinicaltrials.gov/ct2/show/NCT01942135. Accessed November 4, 2014.
- 54.Search results: Palbociclib AND breast cancer. Available at http://clinicaltrials.gov/ct2/results?term=palbociclib+AND+breast+cancer. Accessed November 4, 2014.
- 55.Knudsen E, Cox D, Franco J, et al. Targeting CDK4/6 in HER2 positive breast cancer: Therapeutic effect, markers and combination strategies. Ann Oncol. 2014;25(suppl 1):i21. [Google Scholar]
- 56.Witkiewicz AK, Cox D, Knudsen ES. CDK4/6 inhibition provides a potent adjunct to Her2-targeted therapies in preclinical breast cancer models. Genes Cancer. 2014;5:261–272. doi: 10.18632/genesandcancer.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phase Ib study of PD-0332991 in combination with T-DM1 (trastuzumab-DM1). Available at http://clinicaltrials.gov/ct2/show/NCT01976169. Accessed November 4, 2014.
- 59.Pediatric oncology subcommittee of the oncologic drugs advisory committee briefing document: LEE011. Available at http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM373175.pdf. Accessed November 4, 2014.
- 60.Kim S, Loo A, Chopra R, et al. LEE011: An orally bioavailable, selective small molecule inhibitor of CDK4/6–reactivating Rb in cancer. Mol Cancer Ther. 2013;12(suppl):PR02a. [Google Scholar]
- 61.Infante JR, Shapiro G, Witteveen P, et al. A phase I study of the single-agent CDK4/6 inhibitor LEE011 in pts with advanced solid tumors and lymphomas. J Clin Oncol. 2014;32(suppl):2528a. [Google Scholar]
- 62.Search results: LEE011 | open studies. Available at http://clinicaltrials.gov/ct2/results?term=LEE011&recr=Open. Accessed November 4, 2014.
- 63.Study of LEE011, BYL719 and letrozole in advanced ER+ breast cancer. Available at http://clinicaltrials.gov/ct2/show/NCT01872260. Accessed November 4, 2014.
- 64.Phase Ib/II trial of LEE011 with everolimus (RAD001) and exemestane in the treatment of ER+ HER2- advanced breast cancer. Available at http://clinicaltrials.gov/ct2/show/NCT01857193. Accessed November 4, 2014.
- 65.Vora SR, Juric D, Kim N, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–149. doi: 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bardia A, Modi S, Chavez-Mac Gregor M, et al. Phase Ib/II study of LEE011, everolimus, and exemestane in postmenopausal women with ER+/HER2-metastatic breast cancer. J Clin Oncol. 2014;32(suppl):535a. [Google Scholar]
- 67.Munster PN, Paige Hamilton E, Franklin C, et al. Phase lb study of LEE011 and BYL719 in combination with letrozole in estrogen receptor-positive, HER2-negative breast cancer (ER+, HER2– BC) J Clin Oncol. 2014;32(suppl):533a. [Google Scholar]
- 68.Study of efficacy and safety of LEE011 in postmenopausal women with advanced breast cancer.(MONALEESA-2). Available at http://clinicaltrials.gov/ct2/show/NCT01958021. Accessed November 4, 2014.
- 69.A pharmacodynamics pre-surgical study of LEE011 in early breast cancer patients (MONALEESA-1). Available at http://clinicaltrials.gov/ct2/show/NCT01919229. Accessed November 4, 2014.
- 70.Gelbert LM, Cai S, Lin X, et al. Identification and characterization of LY2835219: A potent oral inhibitor of the cyclin-dependent kinases 4 and 6 (CDK4/6) with broad in vivo antitumor activity. Mol Cancer Ther. 2011;10(suppl):B233a. [Google Scholar]
- 71.Sanchez-Martinez C, Gelbert LM, Shannon H, et al. LY2835219, a potent oral inhibitor of the cyclin-dependent kinases 4 and 6 (CDK4/6) that crosses the blood-brain barrier and demonstrates in vivo activity against intracranial human brain tumor xenografts. Mol Cancer Ther. 2011;10(suppl):B234a. [Google Scholar]
- 72.Dempsey JA, Chan EM, Burke TF, et al. LY2835219, a selective inhibitor of CDK4 and CDK6, inhibits growth in preclinical models of human cancer. Cancer Res. 2013;73(suppl):LB-122a. [Google Scholar]
- 73.Shapiro G, Rosen LS, Tolcher AW. A first-in-human phase I study of the CDK4/6 inhibitor, LY2835219, for patients with advanced cancer. J Clin Oncol. 2013;31(suppl):2500a. [Google Scholar]
- 74.Patnaik A, Rosen LS, Tolaney SM et al. Clinical activity of LY2835219, a novel cell cycle inhibitor selective for CDK4 and CDK6, in patients with metastatic breast cancer [abstract CT232]. Presented at: American Association for Cancer Research annual meeting; April 5–9, 2014; San Diego, CA. [Google Scholar]
- 75.Patnaik A, Rosen LS, Tolaney SM, et al. LY2835219, a novel cell cycle inhibitor selective for CDK4/6, in combination with fulvestrant for patients with hormone receptor positive (HR+) metastatic breast cancer. J Clin Oncol. 2014;32(suppl):534a. [Google Scholar]
- 76.Search results: LY2835219 | open studies. Available at http://clinicaltrials.gov/ct2/results?term=LY2835219&recr=Open. Accessed November 4, 2014.
- 77.A study of nonsteroidal aromatase inhibitors plus abemaciclib (LY2835219) in postmenopausal women with breast cancer (MONARCH 3). Available at http://clinicaltrials.gov/ct2/show/NCT02246621. Accessed November 4, 2014.
- 78.A study of LY2835219 (abemaciclib) in combination with therapies for breast cancer that has spread. Available at http://clinicaltrials.gov/ct2/show/NCT02057133. Accessed November 4, 2014.
- 79.A study of abemaciclib (LY2835219) combined with fulvestrant in women with hormone receptor positive HER2 negative breast cancer (MONARCH 2). Available at http://clinicaltrials.gov/ct2/show/NCT02107703. Accessed November 4, 2014.
- 80.Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. 2012;287:29075–29087. doi: 10.1074/jbc.M112.365494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pierce AJ, Stark JM, Araujo FD, et al. Double-strand breaks and tumorigenesis. Trends Cell Biol. 2001;11:S52–S59. doi: 10.1016/s0962-8924(01)02149-3. [DOI] [PubMed] [Google Scholar]
- 82.Pfizer breast cancer drug gets early FDA approval. Available at http://finance.yahoo.com/news/pfizer-breast-cancer-drug-gets-225132522.html. Accessed February 24, 2015.