

Abstract
Evolutionary relationships among placental mammalian orders have been controversial. Whole genome sequencing and new computational methods offer opportunities to resolve the relationships among 10 genomes belonging to the mammalian orders Primates, Rodentia, Carnivora, Perissodactyla and Artiodactyla. By application of the double cut and join distance metric, where gene order is the phylogenetic character, we computed genomic distances among the sampled mammalian genomes. With a marsupial outgroup, the gene order tree supported a topology in which Rodentia fell outside the cluster of Primates, Carnivora, Perissodactyla, and Artiodactyla. Results of breakpoint reuse rate and synteny block length analyses were consistent with the prediction of random breakage model, which provided a diagnostic test to support use of gene order as an appropriate phylogenetic character in this study. We the influence of rate differences among lineages and other factors that may contribute to different resolutions of mammalian ordinal relationships by different methods of phylogenetic reconstruction.
Keywords: phylogeny, genome rearrangement, breakpoint, gene order, placental mammal
1. Introduction
A well-resolved mammalian tree is essential for annotation of genetic features in their genomes and sequence evolution within this taxonomic class. However, the phylogenetic relationships of the 18 extant placental mammalian orders are highly contentious (Cannarozzi et al., 2007; Cao et al., 1998; Hallstrom and Janke, 2008; Kullberg et al., 2007; Li et al., 1990; Springer and de Jong, 2001; Wildman et al., 2007). Two major alternative hypotheses have been proposed regarding the evolutionary relationship within placental mammals. One maintains that Rodentia is more closely related to Primates than Perissodactyla, Artiodactyla and Carnivora (three orders of Laurasiatheria), (Fig 1A), while the alternative supports Rodentia as an outgroup to the other four orders (Fig 1B).
Figure 1.
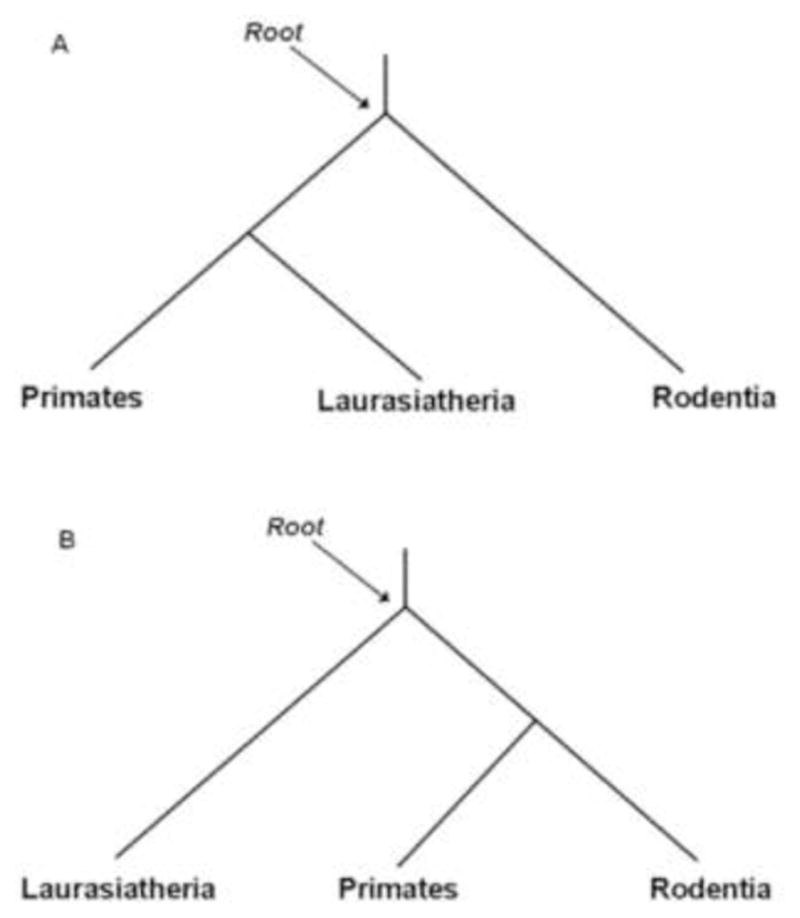
Two alternative hypotheses for the evolutionary relationship among Primates, Laurasiatheria (Artiodactyla, Carnivora, Perissodactyla) and Rodentia. (A) Primates closer to Rodentia than Laurasiatheria; (B) Primates closer to Laurasiatheria than Rodentia.
Earlier studies investigated the placental mammalian relationship with single genes of mitochondrial (Cao et al., 1994; Cao et al., 1998) and nuclear genomes (Easteal, 1988, 1990; Goodman et al., 1985; Li et al., 1990). Single gene analyses frequently created discrepancies in branching order, and it was thought that large molecular data sets of concatenated alignments have the potential to resolve these issues (Madsen et al., 2001; Murphy et al., 2001a; Murphy et al., 2001b). Whole genomic sequences of mammalian mitochondria, each ranging about 16,500 to 17,000 bases (Penny and Hasegawa, 1997), provided more base pairs than single genes. Nevertheless, evidence from mitochondrial genomic analyses is inconsistent. Although a majority of these mitochondrial genomic analyses supported the Primates-Laurasiatheria clade which excluded Rodentia (Arnason et al., 1997; Arnason et al., 1999; Janke et al., 1994; Janke et al., 1997; Mouchaty et al., 2000; Pumo et al., 1998; Reyes et al., 2000; Springer et al., 1997), mitochondrial genomic evidence for Euarchontoglires (Primates-Rodentia clade) also existed (Arnason et al., 2002; Arnason et al., 2008; Reyes et al., 2004). There are other controversies regarding the use of mitochondrial genomic data. A tree derived from first and second codon positions in mitochondrial genes supported the Euarchontoglires hypothesis, whereas another tree of amino acid sequence data suggested a Primates-Laurasiatheria clade (Arnason and Janke, 2002). Further studies showed that complete mitochondrial genomic sequences were valuable for resolving relationships within placental orders, but they appeared inadequate in resolving between-order relationships (Corneli, 2002).
Phylogenomic studies of concatenated alignments of nuclear and mitochondrial genes resolved the 18 extant placental orders into four superordinal groups: Xenarthra, Afrotheria, Laurasiatheria, and Euarchontoglires (Hallstrom et al., 2007; Madsen et al., 2001; Murphy et al., 2001a; Murphy et al., 2001b). Since Artiodactyla, Perissodactyla and Carnivora are three orders of Laurasiatheria and Primates and Rodentia are within Euarchontoglires, these studies favored the Primates and Rodentia as a superordinal clade while excluding Artiodactyla, Perissodactyla and Carnivora (Fig 1B). However, the basal position of Rodentia was again underscored in a recent study of eight housekeeping genes across 22 placental mammals and three marsupials (Kullberg et al., 2006). In addition, another phylogenomic study using distance-, parsimony-, and likelihood-based methods yielded overwhelming support for a Primates-Carnivora clade while excluding Rodentia (Cannarozzi et al., 2007).
The controversial relationships among these mammalian orders have been highlighted by several recent studies. Murphy and others (2001b) claimed that the deep mammalian relationships were resolved by Bayesian-based phylogenetics. Afterwards, Misawa and Nei (2003) showed that Murphy and others’ data (2001b) can lead to two different Bayesian trees, both of which were supported by high posterior probabilities.
Kolaczkowski and Thornton (2004) pointed out that when the substitution rate at a single base or amino acid position varies over evolutionary time (referred to as heterotachy), both Bayesian- and likelihood-based methods are statistically inconsistent, leading to an incorrect partition as the amount of data grows (Nishihara et al., 2007; Wildman et al., 2007). Likewise, Hughes and Friedman (2007) found that genes with different substitution rates yielded a different branching order between Carnivora and Rodentia, but concatenation of these genes yielded a tree supporting Rodentia branching first regardless of the method or model of sequence evolution.
Since several factors can lead to a misleading topology, such as nucleotide or amino acid compositional bias (Nishihara et al., 2007), phylogenetic method (Kullberg et al., 2007), long-branch attraction, and heterotachy, it is desirable to seek whole genome-based phylogenetic characters to resolve the basal relationships in the mammalian tree.
Gene order is a type of rare genomic change, which provides independent ways to evaluate conflicting molecular sequence phylogenies (Rokas and Holland, 2000). It has been demonstrated as a useful phylogenetic character in resolving both shallow and deep prokaryotic relationships (Belda et al., 2005; Luo et al., 2008; Luo et al., 2009). Genome rearrangements include inversion, transposition, block exchange, circularization and linearization, all of which act on a single chromosome, and translocation, fusion, and fission, which act on two chromosomes. All of these operations are subsumed in the double-cut-and-join (DCJ) model, which has formed the basis for much of the algorithmic research on rearrangements over the last few years.
A DCJ operation consists of cutting two adjacencies in the first genome, and rejoining the resulting four unconnected vertices in two new pairs. As a result, it swaps two gene ends in two different vertices of the same genome in the breakpoint graph. Fig. 2 shows an example of a DCJ operation on two adjacencies (a, b) and (c, d). It splits (a, b), (c, d), and can create new adjacencies (a, c), (b, d) or (a, d), (b, c). The DCJ distances between two permutations is defined as the number of minimal DCJ operations needed to transform one permutation into another. If two unichromosomal linear genomes are identical, we can see that there are N cycles in the breakpoint graph (N is the length of the genome). A DCJ operation can increase at most one cycle (Fig. 3), so for unichromosomal linear genomes, DCJ distance is N - C where C is the number of cycles in the breakpoint graph.
Figure 2.
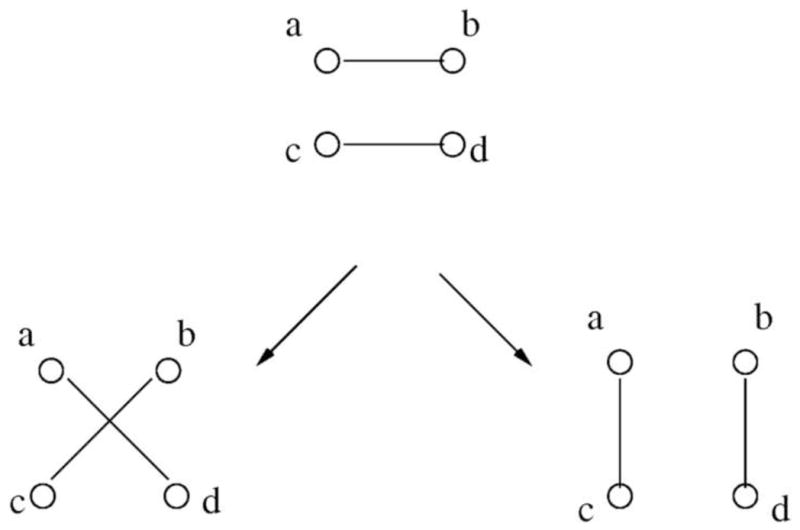
A DCJ operation on adjacencies (a, b) and (c, d) can create two possible results: (a, c), (b, d) or (a, d), (b, c).
Figure 3.
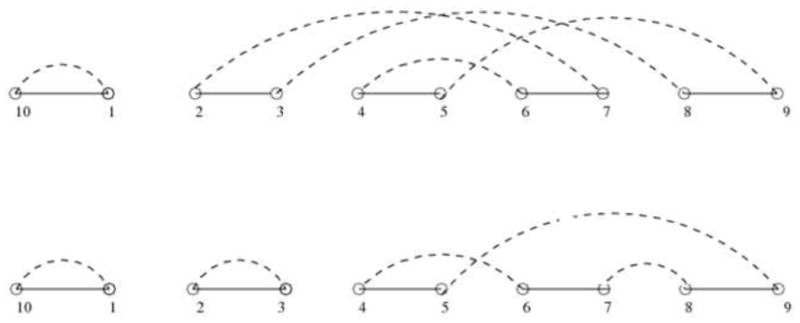
On the top is the breakpoint graph of genome G = 1, 2,-3, 4,-5, with respect to the identity genome G = 1, 2, 3, 4, 5. We can see there are two cycles in the graph. An optimal DCJ operation removes adjacencies (2,7), (3,8) and creates adjacencies (2,3), (7,8) so that the number of cycles increases by one.
Genome rearrangements occur at a much lower rate than that of nucleotide substitution, hence it has the potential to resolve ancient interordinal divergences among mammals (Boore, 2006; Murphy et al., 2004). Applying genome rearrangement data to the Primates-Rodentia-Carnivora controversy, a recent study used the Multiple Genome Rearrangements and Ancestors (MGRA) algorithm, based on a multiple breakpoint graph, which favored the sister relationship between Primates and Carnivora, but did not exclude the possibility of a Primates-Rodentia clade (Alekseyev and Pevzner, 2009). In this present study, we constructed a distance-based gene order phylogeny to resolve the phylogenetic relationship among Primates, Rodentia, Artiodactyla, Perissodactyla and Carnivora.
2. Materials and Methods
2.1 Genome data
10 genomes were used in the analysis, including Homo sapiens (Human), Macaca mulatta (Macaque), Pan troglodytes (Chimpanzee), Pongo pygmaeus (Orangutan), Equus caballus (Horse), Bos Taurus (Bovine), Mus musculus (Mouse), Rattus norvegicus (Rat), Canis familiaris (Canine), and Monodelphis domestica (Opossum). The supersets of all gene translations, both known and predicted, were obtained from Ensembl release 52. Non-nuclear genes and genes that were not mapped to a chromosome were discarded. Some genes encode alternative spliced proteins, and only the longest translation was kept. The final number of protein in each of the 10 mammalian genomes that was used for ortholog prediction was summarized in Table 1. Although some assembly errors occur in the Macaque genome (Karere et al. 2008) and likely to occur in other genomes, simulation studies have shown that gene order phylogenetic approaches are robust against those errors (Lin et al. 2011a and Lin et al. 2011b). By the time of this study, no genomes from Afrotheria (e.g. elephant) were properly assembled. We also analyzed two relevant but unpublished genomes, including Oryctolagus cuniculus (Rabbit) and Sus scrofa (Swine). These lineages are associated with long branches with unknown reasons (Supplementary Figure 1 and Supplementary Table 2).
Table 1.
Size of mammalian proteome
| Mammal | Proteome size 1 |
|---|---|
| bovine | 19030 |
| canine | 19014 |
| horse | 20170 |
| human | 21165 |
| macaque | 21023 |
| mouse | 23228 |
| orangutan | 18868 |
| chimpanzee | 19199 |
| rat | 22490 |
| opossum | 18641 |
The proteome does not include translations of genes that are not mapped to nuclear chromosomes. For alternative spliced genes with multiple translations, only the longest protein was counted.
2.1. Ortholog identification and data preparation
All pairs of proteomes were assembled by a reciprocal all-versus-all BLASTP search with an E value smaller than 0.1 and by filtering sequenced regions with low complexity. The output file was prepared to include mapped position and strandedness. MSOAR software was then used to identify common orthologs in every pair of genomes. MSOAR is a high-throughput genome-scale ortholog assignment system; it is a two-step procedure where homologous genes were first identified by sequence similarity search and then orthologous genes were identified by corresponding to each other in the most parsimonious evolutionary scenario involving both genome rearrangements and gene duplications (Chen et al., 2005; Jiang, 2007). Then the pairwise ortholog sets were used to identify the common ortholog sets in the subsets of the 10 genomes. The same procedure was applied in a previous study (Luo et al., 2009).
MSOAR identifies orthologs by minimizing the number of rearrangement events, which will have an impact on the distance-based gene order phylogeny only if the distance obtained severely underestimates the true number of events. Based on various simulation studies (Moret et al. 2002; Shi and Tang 2010), distance correction must be performed if the number of events between two genomes exceeds 70% of the number of genes. In this study, the largest DCJ distance is 1450 (between rate and opossum), far smaller than the number of orthologues (9212), thus using DCJ distance based on orthologs identified by MSOAR is still valid.
The genomic positions of all protein-coding regions were extracted by Perl scripts. The order of orthologs in each genome was determined based upon their chromosomal position and strandedness. In this way, each genome was represented by a set of signed permutations for each chromosome where sign indicates strandedness (Moret et al., 2002).
2.2. Gene order phylogeny construction
Evolutionary events that change gene orders include inversion and transposition which act on a single chromosome, and translocation, fusion, and fission which act across chromosomes. Although combining events such as inversion, translocation, fusion and fission have been well studied, handling transpositions is still beyond reach. The “double-cut-and-join” (DCJ) operation was proposed to provide a “universal” operation to account for all rearrangement events. A DCJ operation occurs when two breaks are created in the chromosomes of a genome and the fresh telomeres are reconnected in a new arrangement. Two supplementary DCJ operations are the separation of an adjacency into two telomeres and the attachment of two telomeres to form a single adjacency. As a result, the DCJ operation can simulate each of the rearrangement events in one or two steps.
Bergeron et al. (Bergeron et al., 2006) provided a linear algorithm to compute the edit DCJ distance between two genomes, which can be used to reconstruct phylogenies using distance-based methods such as neighbor-joining (Saitou and Nei, 1987) and FastME (Desper and Gascuel, 2002). Experiments on simulated datasets showed that inversion and DCJ distances return very similar results even on data generated using only transpositions (Kothari and Moret, 2007). Lin and Moret later showed that it is possible to estimate the true number of evolutionary events from the DCJ model (Lin and Moret, 2008), making the DCJ model attractive in phylogenetic reconstruction. The Double Cut and Join (DCJ) distance metric (Yancopoulos et al., 2005) is implemented in GRAPPA (Moret et al., 2002; Zhang et al., 2009), which computed the pairwise DCJ and breakpoint distances from the gene order data and generated a pairwise distance matrix. Next, the FastME (Desper and Gascuel, 2002) software constructed the DCJ and breakpoint phylogenetic trees. The tree topologies were visualized by MEGA4 (Tamura et al., 2007).
To calculate the statistical reliability of the branches of the phylogeny, we applied a jackknife resampling technique that, in each iteration, randomly removed 50% of the initial orthologous gene sets. Note that bootstrapping is not applicable here, because gene order is one character with multiple states (Shi et al., 2010).
1000 jackknife random samples were generated to compute 1000 matrices for both DCJ and breakpoint distances. Each of these 1000 matrices were imported into the FastME program to obtain 1000 DCJ and breakpoint distance-based trees. Finally, the CONSENSE program in the PHYLIP software package (Felsenstein, 1989) calculated a majority-rule consensus tree with percent values at each node. Each value represents the percentage of trees supporting a clade defined by a node. This procedure was applied in a prior study (Luo et al., 2009), and the usefulness of the jackknife technique in gene order phylogeny was illustrated in a recent study (Shi et al., 2010). The elephant genome has not been assembled and hence cannot be used for genome rearrangement-based phylogenetic analysis. Either rabbit or swine or both were included in preliminary analyses, but all had very long branches (Supplementary Fig. 1), perhaps because of errors in assembly.
2.3. Breakpoint reuse rate measurement
We further analyzed the breakpoint reuse rate among the placental mammals. Breakpoints are not exact positions in the genome. Rather, they represent genomic regions whose resolution is dictated by synteny blocks. In rearrangement analysis, genomes are basically represented by sequences of synteny blocks interspaced with breakpoint regions that are typically much shorter than synteny blocks. Synteny blocks hide effects of much more frequent and intractable evolutionary events like mutations and micro-rearrangements and allow one to focus on analysis of large-scale genome rearrangements. Large-scale genome rearrangements are detected from different orderings of synteny blocks in different genomes. The span of such rearrangements is therefore defined in terms of synteny blocks and their actual breakpoints (i.e., positions where rearrangements break the genome) are known only to belong to certain breakpoint regions. So there is no way to determine exact locations of breakpoints which are therefore treated as genomic regions.
In fact, exact locations of breakpoints are not that important for large-scale rearrangement analysis. It is now known that mammalian genomes are formed by mosaic of “solid” and “fragile” regions where the latter are prone to rearrangements (Alekseyev and Pevzner 2007). From this perspective, it does not matter much whether two breakpoints exactly coincide or just colocated within a short breakpoint region. Both cases are treated as breakpoint reuse and simply indicate that the corresponding breakpoint region is “fragile”.
Using the principle of parsimony, the breakpoints reuse rate was computed among genomes (Alekseyev, 2008). Since each gene rearrangement causes two breakpoints, the breakpoint reuse rate is measured by multiplying two by the number of rearrangements which is then divided by the total number of breakpoints (for a 2-genome comparison). The breakpoint reuse rate measurement ranges in between the interval of 1 and 2, where the value 1 implies no breakpoint reuse. One caveat is that a high breakpoint reuse rate may indicate that the parsimony assumption is not valid.
2.4. Synteny block length analysis
The gene order sequence data were used to compute synteny block lengths of all pairwise comparisons among the mammalian genomes. The frequency distribution of the synteny block lengths was fitted to the expected exponential Probability Density Function (Zdobnov and Bork, 2007) and a power law function using MatLab.
2.5. Gene order rate calculation
Each of the 1000 jackknife random samples resulted in a DCJ distance-based phylogenetic tree. Trees with different topologies from the consensus tree were discarded. We then collected the branch lengths from the remaining jackknifing trees. To test that any two branches were of different lengths, the mean values of jackknife-generated branch lengths were compared by a z-test. All statistical analyses were performed using the R statistical software package (R Develop Core Team, 2008).
2.6. Sequence-based phylogenetic tree construction
The shared orthologous amino acid sequences were aligned separately using PRANK software (Loytynoja and Goldman, 2005, 2008). PRANK models insertions and deletions as distinct evolutionary events and thus dramatically reduces bias in sequence alignment (Loytynoja and Goldman, 2005, 2008). In each genome, the orthologous sequences were concatenated together and transformed as a Phylip format. All ambiguous amino acid sites were removed which left 3,873,035 sites per genome.
Phylogenetic trees were constructed using Neighbor-Joining (Saitou and Nei, 1987), Maximum-Likelihood (Schmidt et al., 2002), Maximum-Parsimony (Eck and Dayhoff, 1966), and Bayesian (Huelsenbeck and Ronquist, 2001) methods. To establish the confidence of internal branches of the resulting trees, 1000 bootstrapped replicates were resampled in the NJ and MP trees separately. Likewise, 1000 puzzling steps were applied in the ML tree. The puzzling steps are akin to the bootstrapped random samples in testing the tree topology. In the Bayesian tree, posterior probabilities were calculated by using a Metropolis-coupled Markow chain Monte Carlo approach with sampling according to the Metropolis–Hastings algorithm.
The NJ and MP methods were implemented in MEGA4 software (Tamura et al., 2007). In the NJ tree, the pairwise genomic distances were computed using Jones-Taylor-Thornton (JTT) model (Jones et al., 1992) with gamma correction. The gamma distribution parameter alpha (alpha=0.32) was estimated from the concatenated dataset by TREE-PUZZLE software (Schmidt et al., 2002) implemented by the Mobyle online server (Neron et al., 2009). The ML tree was constructed using TREE-PUZZLE software on the Mobyle online server with JTT model and gamma correction. The Bayesian analysis was implemented in the MPI version of MrBayes software (Altekar et al., 2004). One cold and three heated Markov chain Monte Carlo (MCMC) chains with default chain temperatures were run for a total of 10,000 generations with trees sampled every 10 generations. The first 25% of all runs were discarded as “burn-in”. A majority-rule consensus tree was constructed from the post-burn-in trees. To assess whether rate variation across the tree has an impact on Bayesian tree, the covarion model was invoked in some Bayesian tree reconstructions. Different protein rate variation models, including the gamma-distributed rate model (rates=gamma) and the proportion of invariable sites model combined with the gamma model (rates=invgamma), were used. Average standard deviation of split frequencies = 0.00000 is reached at the end of the calculations.
3. Results
3.1. Gene order trees
We obtained 9,212 orthologs shared by the 10 organisms (Supplementary Table 1), accounting for 49.4% of the smallest proteome (i.e. opossum; Table 1). These shared orthologous amino acid sequences were used in gene order and sequence-based phylogeny construction. The Double Cut and Join (DCJ, Fig. 4A) and breakpoint (Fig. 4B) distance-based gene order trees of the five mammalian orders were consistent. The distance-based gene order trees resolved the evolutionary relationships among hominid and non-hominid primates with high statistical support (Fig. 4). In addition, the gene order trees supported a Primates-Carnivora-Perissodactyla clade excluding Rodentia. This relationship received high statistical support in the breakpoint tree (Fig. 4B). Moreover, the gene order trees showed that Artiodactyla branched before Perissodactyla and Carnivora (Fig. 4). However, the distance-based gene order trees did not resolve the phylogenetic position of Artiodactyla (Fig. 4).
Figure 4.
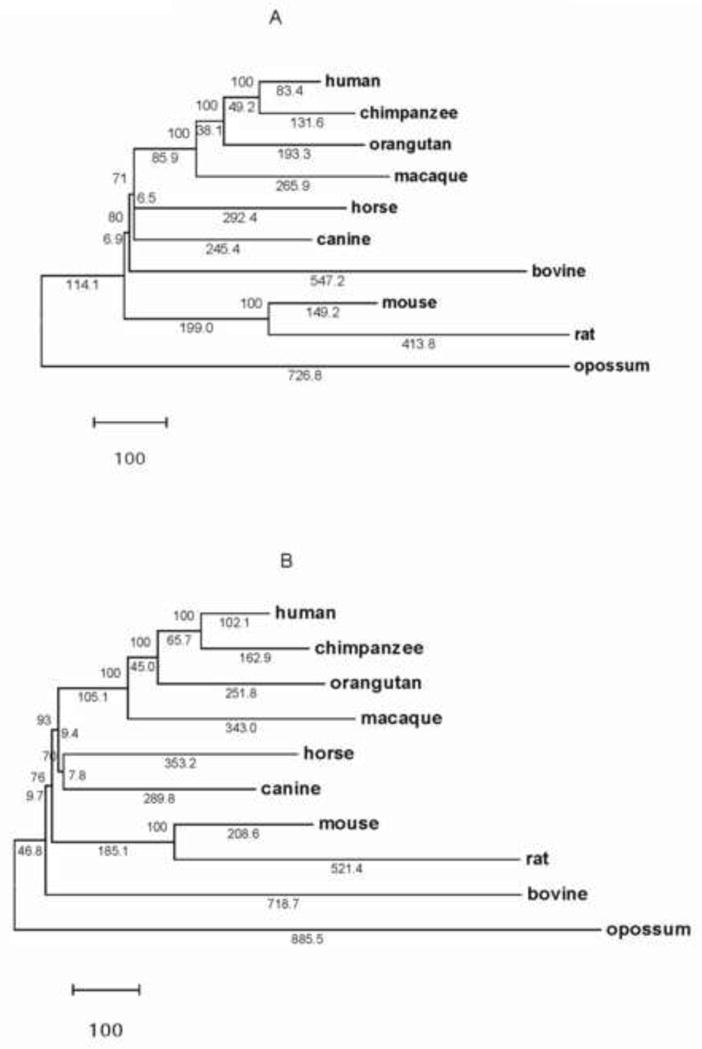
Phylogeny of Primates (human, chimpanzee, orangutan, macaque), Laurasiatheria (canine, horse, bovine) and Rodentia (mouse, rat) inferred from (A) a DCJ distance and (B) a breakpoint distance matrix tree. Values above branches show the number of times that the clade defined by that node was supported by 1000 jackknife trees. Values below branches show number of genome rearrangement events. Opossum is the outgroup.
3.2. Sequence-based trees
The Neighbor-Joining (NJ, Fig. 5A), Maximum-Likelihood (ML, Fig. 5B) and Maximum-Parsimony (MP, Fig. 5C) trees using concatenated amino acid sequences were congruent. With high statistical support, these sequence-based trees unanimously supported Primates, Carnivora, Perissodactyla and Artiodactyla as a monophyletic group and Rodentia as the outgroup. We also analyzed the same concatenated sequences with Bayesian approaches in MrBayes software which implements the covarion model to take heterotachy into account. In contrast to other molecular sequence phylogenies, the Bayesian trees (Fig. 6) consistently supported a Primates-Rodentia clade excluding Carnivora, Perissodactyla and Artiodactyla with high posterior probability, regardless of whether the gamma-distributed model or a combination of a proportion of invariable sites model and the gamma-distributed model was used, and regardless of whether rate variation across the tree (i.e. heterotachy) was considered.
Figure 5.
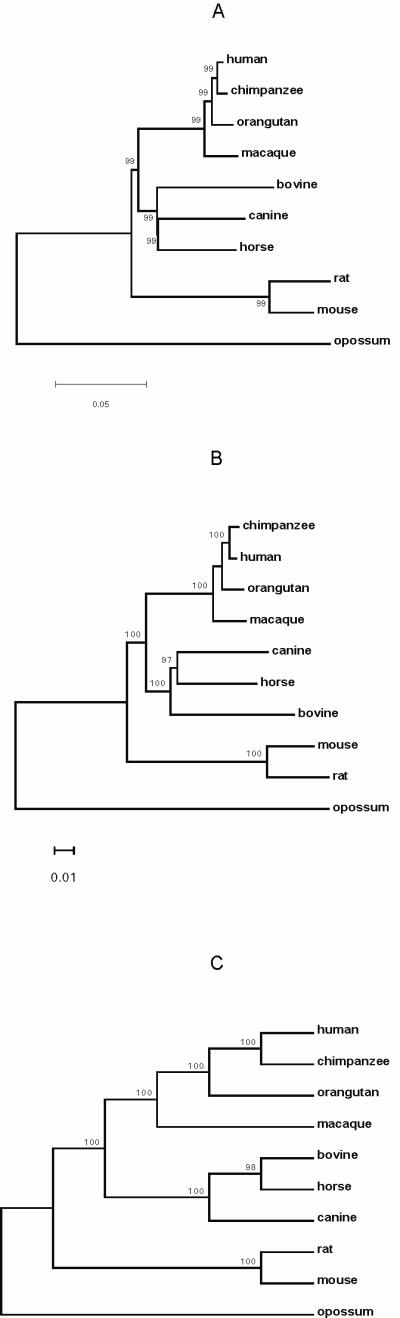
Phylogeny of Primates (human, chimpanzee, orangutan, macaque), Laurasiatheria (canine, horse, bovine) and Rodentia (mouse, rat) inferred from concatenated amino acid sequences: (A) Neighbor-Joining (NJ) method with JTT model and gamma correction, (B) Maximum-Likelihood (ML) with JTT model and gamma correction, and (C) Maximum-Parsimony (MP). Values above branches show the number of times that the clade defined by that node were supported by 1000 bootstrapped pseudoreplicates (NJ, MP) or 1000 puzzling steps (ML).
Figure 6.

Bayesian phylogeny of Primates (human, chimpanzee, orangutan, macaque), Laurasiatheria (canine, horse, bovine) and Rodentia (mouse, rat) inferred from concatenated amino acid sequences, assuming (A) gamma-distributed rate variation across sites (rates=gamma) and rate variation across tree (covarion=yes); (B) a proportion of invariable sites combined with the gamma model (rates=invgamma) and rate variation across tree (covarion=yes); (C) gamma-distributed rate variation across sites (rates=gamma, covarion=no); (D) a proportion of invariable sites combined with the gamma model (rates=invgamma, covarion=no).
The sequence-based trees were also inconsistent regarding the evolutionary relationship among Carnivora, Perissodactyla and Artiodactyla. The ML and NJ tree supported Artiodactyla as an outgroup of the Carnivora-Perissodactyla clade, whereas the MP and Bayesian trees supported Carnivora as an outgroup of the Perissodactyla-Artiodactyla clade.
3.3. Evolutionary rates
A breakpoint reuse rate analysis showed that there is limited breakpoint reuse in the placental mammals (Table 2). The synteny block lengths in the placental mammals did not follow the expected exponential distribution (Fig. 7A), but it was fit to a power function (Fig. 7B).
Table 2.
Breakpoint re-use rate 1 in mammalian genomes
| canine | bovine | horse | human | opossum | macaque | mouse | orangutan | chimpanzee | rat | |
|---|---|---|---|---|---|---|---|---|---|---|
| canine | 1.15 | 1.15 | 1.21 | 1.6 | 1.29 | 1.46 | 1.3 | 1.31 | 1.46 | |
| bovine | 1.13 | 1.16 | 1.53 | 1.25 | 1.35 | 1.25 | 1.27 | 1.39 | ||
| horse | 1.19 | 1.6 | 1.22 | 1.41 | 1.25 | 1.29 | 1.46 | |||
| human | 1.61 | 1.07 | 1.42 | 1.2 | 1.14 | 1.43 | ||||
| opossum | 1.61 | 1.66 | 1.61 | 1.61 | 1.66 | |||||
| macaque | 1.42 | 1.21 | 1.24 | 1.43 | ||||||
| mouse | 1.46 | 1.5 | 1.19 | |||||||
| orangutan | 1.07 | 1.47 | ||||||||
| chimpanzee | 1.5 | |||||||||
| rat |
Breakpoint re-use rate ranges from 1 without reuse to 2 with extensive reuse.
Figure 7.

Frequency of synteny block lengths. (A) The Probability Density Function (PDF) fits an exponential distribution (f(x)= 0.318*e−0.318x, R2= 0.6812); (B) The PDF fits a power function (f(x)= 0.381* x−1.381, R2= 0.9682
We tested whether the evolutionary rates of gene order change were consistent with a molecular clock model. Among the placental mammals analyzed in this study, the bovine and rat lineages had an accelerated gene rearrangement rate as compared to other placental mammals (z-test, P<0.001, in both cases). Although the rat rearranged significantly faster than all Primates (z-test, P<0.001), the mouse had a significantly greater gene rearrangement rate than human (z-test, P<0.001), chimpanzee (z-test, P<0.001), and orangutan (z-test, P<0.05) but no difference from macaque (z-test, N.S.). In addition, significant within-order differences in the gene rearrangement rates were observed in Rodentia separately (z-test, P<0.001 in both cases). In the NJ and ML trees, we also observed the intraordinal differences in the amino acid substitution rates in Rodentia (z-test, P<0.001 in both cases).
Discussion
The effect of choice of outgroup on phylogenetic reconstruction using sequence data was discussed previously (Janke et al., 1994). Since amphibian and bird diverged from the mammalian ancestor about 350 and 300 million years ago, respectively, (Graur and Martin, 2004), multiple substitutions may lead to saturation of nucleotide changes, which will degrade the phylogenetic signal (Janke et al., 1994). However, marsupials diverged from placentals about 130 million years ago based on paleontological evidence, making it suitable as an outgroup for the placental radiation (Janke et al., 1994). In the case of the gene order character, excessive gene rearrangement events lead to reuse of chromosomal breakpoints, making it difficult to resolve the species relationship.
In addition, the chicken (Gallus gallus) genome is reduced in size along with many absent gene families as compared to other vertebrates (Hughes and Friedman, 2008). Inclusion of the chicken genome reduced the dataset size of common orthologous genes, which are the data source for gene order phylogeny construction (Luo et al., 2009). Therefore, a marsupial mammal (i.e. opossum) is a better choice of an outgroup in reconstructing the basal relationship among placental mammals.
The distance-based gene order phylogenetic approach yielded well-resolved evolutionary relationships among some placental mammals. For instance, the relationship among hominid and non-hominid Primates revealed by gene order trees (Fig. 4) were congruent with that shown in the nucleotide substitution trees (Tocheri et al., 2008) and amino acid concatenated sequence-based trees (Fig. 5). In addition, the gene order trees provided an alternative cladistic character to investigate basal interordinal mammalian relationships which have been unresolved by the nucleotide substitution process. For instance, many studies have investigated but not resolved the evolutionary relationship of Primates, Rodentia, and some orders of Laurasiatheria (e.g. Carnivora, Artiodactyla and Perissodactyla) (Hallstrom et al., 2007; Madsen et al., 2001; Misawa and Nei, 2003; Murphy et al., 2001a; Murphy et al., 2001b). Likewise, the relationship among Carnivora, Perissodactyla and Artiodactyla is unclear (Arnason and Janke, 2002; Kullberg et al., 2006; Murphy et al., 2001a). The gene order phylogeny supported that Rodentia is an outgroup of Primates, Carnivora and Perissodactyla, and also supported that Artiodactyla as an outgroup of Carnivora and Perissodactyla. However, Artiodactyla, Carnivora and Perissodactyla were not a monophyletic group in the distance-based gene order phylogeny, and the DCJ tree differed from the breakpoint tree in the phylogenetic position of Artiodactyla.
Analysis of breakpoint reuse is important for reconstruction of ancestral genomes and rearrangement history (Alekseyev and Pevzner, 2009). Unambiguous reconstruction is possible when breakpoint reuse is limited. In contrast, extensive breakpoint reuse eliminates tracks of earlier rearrangements and thus represents a major obstacle to reconstruction of the rearrangement history. The low breakpoint reuse rate obtained in the present study (Table 2) is consistent with a previous report of 20% reuse of breakpoints during mammalian evolution (Murphy et al., 2005), suggesting that chromosomal rearrangements occurred randomly and the gene order sequence is reliable for reconstructing the placental mammalian phylogeny.
The short but well-supported basal branches leading to the Primate-Artiodactyla-Carnivora-Perissodactyla clade (Fig. 4) suggested that only a few synapomorphies joined the Primates-Carnivora-Perissodactyla clade, whereas the long leaf branches (Fig. 4) indicated that most autapomorphies occurred after the divergence of these placental orders. Short basal branches were also revealed in trees using mitochondrial genomic sequences (Corneli, 2002). Note that the branch lengths shown (Fig. 4) were related to the number of the orthologous genes shared by all of the genomes in the phylogenetic tree; with less genomes included in the phylogenetic tree, a greater number of orthologous genes were shared among the genomes in comparison and could be used to compute the gene rearrangements, which would result in longer branches. Preliminary analyses with a smaller number of genomes were consistent with this hypothesis (not shown).
The amino acid sequence-based trees constructed by NJ, ML, and MP methods were generally consistent with the distance-based gene order trees. The former supported a monophyly of Primates-Artiodactyla-Carnivora-Perissodactyla (Fig. 5), which is consistent with the DCJ tree. However, a Bayesian tree from the concatenated amino acid sequences supported the Euarchontoglires hypothesis (i.e. monophyly of Primates and Rodentia). One advantage of the Bayesian method implemented in MrBayes software over NJ, ML, and MP methods is that it provides options to model heterotachy in phylogeny construction. However, because inclusion of heterotachy in our Bayesian analyses did not affect the results, it seems unclear whether heterotachy is responsible for the differences between trees constructed by the Bayesian method and by the NJ, ML, and MP methods.
Another notable difference between the molecular sequence and gene order based trees is whether the Artiodactyla-Carnivora-Perissodactyla forms a monophyletic clade. Whatever method was used, the amino acid sequence trees supported a monophyly of these three orders (Fig. 5 and 6), whereas the gene order trees did not (Fig. 4).
Ohno proposed the random breakage model (Ohno, 1973) which described that chromosomal rearrangements occur randomly and breaks of the ancestral genome are uniformly distributed along the chromosomal length (Zdobnov and Bork, 2007). Several follow-up studies provided evidence supporting the random breakage of chromosomes (International Human Genome Sequencing Consortium, 2001; Mural et al., 2002; Nadeau and Taylor, 1984). It was claimed that an exponential distribution of synteny blocks of a certain length should be expected, provided the random breakage occurred (Zdobnov and Bork, 2007). However, data available in the literature from pairwise comparisons among sequenced genomes suggested that synteny blocks of a certain length fit better to a power function than to an exponential distribution (Zdobnov and Bork, 2007). Other studies suggested that the random breakage model cannot explain closely located breakpoints (i.e. synteny block length < 1 Mbp), and proposed the fragile breakage model could account for a significant number of short synteny blocks observed from genomic sequences (Pevzner and Tesler, 2003). Here, we showed that the distribution of synteny block lengths cannot fit to an exponential function (Fig. 7A), but the power function seems a better fit (Fig. 7B), which is consistent with previous observations (Zdobnov and Bork, 2007). The approximate power law distribution of synteny block lengths is consistent with the finding of a low breakpoint reuse rate, indicating random breakages among chromosomes.
Phylogenomic analysis dated the basal placental mammal divergences at 95–100 million years ago, but many of the interordinal divergences occurred only a few million years later (Hallstrom and Janke, 2008). Paleontological evidence also supported a sudden radiation of placental mammals in the late Cretaceous period (Archibald, 2003; Cannarozzi et al., 2007). The narrow time interval within which basal placental divergences took place renders it problematic in constructing the sequence-based (Hallstrom and Janke, 2008) and short interspersed repetitive elements (SINEs) based phylogenetic relationships (Miyamoto 1999). Although using a large amount of data and improving taxon sampling might have enhanced the resolution of narrow divergences, such efforts did not resolve the deep mammalian relationships (Hallstrom and Janke, 2008).
Many possible causes have been proposed to account for the difficulty in resolving narrow divergences, as in the case of ordinal and higher level divergences among placental mammals (Hallstrom and Janke, 2008). One possible explanation is introgression from species hybridization, a process known as the incorporation of genes from one species into the gene pool of another (Mallet, 2005). This happens where closely related populations hybridize with each other before they become genetically isolated (Hallstrom and Janke, 2008). Another possible cause is lineage sorting which results from fixation of different alleles in the descendant lineages from a speciation event. Lineage sorting jumbles the genomes of species which radiated over a short period of evolutionary time and also gives rise to the inconsistency between gene history and species phylogeny (Hallstrom and Janke, 2008).
Short branches suggest few changes of character states during a rapid taxon radiation (Boore, 2006), indicating that an extremely conserved character is not able to resolve such a rapid radiation and instead that a relatively fast evolving character is required (Corneli, 2002). On the other hand, an ancient relationship suggests that the fast evolving character may accumulate backward mutations, resulting in homoplasy and in turn obscuring the phylogenetic signal (Boore, 2006; Corneli, 2002). Although traditional sequence substitution models have resolved many controversies, resolving short branches remains a notoriously difficult problem (Murphy et al., 2004). For instance, mitochondrial DNA sequences are highly saturated at many positions with respect to ancient interordinal divergences (Corneli, 2002).
However, many properties of gene rearrangements are favorable to resolving rapid radiations. One important characteristic is the lower rate of change of gene order in contrast to nucleotide substitution. Likewise, in a deeper evolutionary relationship, multiple nucleotide substitutions may obscure the phylogenetic signal (Boore, 2006), whereas relatively few chromosomal breakpoints are reused. In addition, the molecular sequence character has only four (nucleotides) or 20 (amino acids) possible character states, hence convergent and parallel substitutions might have occurred among different lineages over a long period of time. In contrast, there are a large number of states in a gene order character, each of which represents a permutation of genes in the genome, so that identical changes are unlikely to occur in two separate lineages (Boore, 2006). The potential usage of genome rearrangement as a powerful evolutionary character to resolve short branches and contested relationships in mammalian phylogeny has already been addressed by Murphy et al. (2004) and Boore (2006). Our results suggest that the genome rearrangement character is useful and complementary to nucleotide substitution in resolving the deeper mammalian relationships.
In mammalian genomes, the order of genes in some genomic regions is more conserved than other regions (Murphy et al., 2004), implying that the various genes and blocks of genes resulting from rearrangement events are not entirely independent of each other. However, by applying a jackknifing technique, we assume that the sequential ordering of genes is arranged by a rearrangement process which occurs uniformly across the genome. A similar assumption is made in a bootstrapping test of a sequence-based phylogeny, in which aligned sites (nucleotides or amino acids) are assumed independent of each other. However, in many cases they are correlated (Felsenstein, 1985). For instance, in a protein-coding gene, a single codon consists of three adjacent sites. In most cases, replacement of the first or second position of a codon results in an amino acid substitution. In another example, when a slightly deleterious mutation is fixed during a bottleneck, afterwards another advantageous mutation that compensates for the deleterious mutation may occur. This compensatory mutation may then be fixed by positive selection. Such compensatory changes usually occurred near to the site of the deleterious mutation (Hughes, 2008). Both examples show that the aligned sites, like that of gene order, are not independent of each other.
Therefore, both bootstrapping and jackknifing techniques generate an approximate statistic rather than a true statistic. The essence of the two techniques is to add perturbation to the data, and to assess whether the original data contain sufficient phylogenetic signal to resist the perturbation. A high jackknifing or bootstrap support value on an internal branch indicates the branch is reliable (Felsenstein, 1985). In fact, the jackknifing test has been used and verified in the gene order phylogenies of prokaryotic species (Belda et al., 2005; Luo et al., 2008; Luo et al., 2009). A recent simulation study demonstrated the jackknifing test is useful in gene order data (Shi et al., 2010).
However, in some cases high bootstrap support in a sequence-based tree may indicate the reinforcement of certain systematic errors contained in the data, which results in an incorrect tree (Gadagkar et al., 2005). Wildman and others (2007) showed that Maximum Parsimony (MP), Maximum Likelihood (ML), and Bayesian methods supported the Euarchontoglires clade, whereas Neighbor Joining (NJ) yielded a basal position of murid rodents from their sampling of Primates, artiodactyls and carnivores. Both of these mutually exclusive topologies were well supported by the bootstrap test (Wildman et al., 2007). The possible systematic errors in molecular sequence data include nucleotide or amino acid compositional bias, long-branch attraction, and heterotachy (Wildman et al., 2007). In gene order data, there is no compositional bias issue, because its character states are not analogous to those of nucleotide and amino acid sites. Long-branch attraction due to parallel changes is less severe in the gene order data than in nucleotide (or amino acid) sequence data, because gene order has many more character states and hence less parallel changes than the other (Boore, 2006). Furthermore, currently there is little evidence for heterotachy in the genome rearrangement process.
Computer simulation suggested that, at the 85% as the threshold of confidence value, only 20% of the jackknife trees contained false positive branches (Shi et al., 2010). The jackknifing test is able to identify 95% of the incorrect branches under the 85% threshold confidence value (Shi et al., 2010). One shortcoming of the jackknifing technique is that a low confidence value could be assigned to correct branches, suggesting that interpretation of branches with low confidence values should be carefully examined (Shi et al., 2010). In the case of the distance-based mammalian gene order phylogeny, the DCJ tree (Fig. 4A) and breakpoint tree (Fig. 4B) have the same topology, but the DCJ tree has lower confidence values than the breakpoint tree for the branches leading to swine, bovine, canine and horse (Fig. 4). The high confidence values of these branches in the breakpoint tree lend additional support for these branches in the DCJ tree.
The question of whether a phylogenetic analysis is reliable based on a limited number of taxa has been much debated (DeBry, 2005; Hillis, 1998; Hillis et al., 2003; Hughes and Friedman, 2007; Mitchell et al., 2000; Poe, 1998; Pollock et al., 2002; Rannala et al., 1998; Rosenberg and Kumar, 2001, 2003; Zwickl and Hillis, 2002). Increased taxon sampling serves to break up long branches and thereby avoids erroneous clustering of fast evolving taxa known as the long-branch attraction problem (Hughes and Friedman, 2007; Pollock et al., 2002). Although there are only 10 taxa included in the gene order phylogenetic analysis (Fig. 4), there is no evidence that supports long-branch attraction as the cause of the basal position of Rodentia in relationship to Carnivora and Perissodactyla. Mouse had a much lower rate than rat in gene rearrangements, and inclusion of mouse avoided the potential long-branch attraction problem. However, it is not clear whether the basal position of Artiodactyla in the breakpoint tree was due to long-branch attraction.
An early study by Wu and Li (1985) showed that the rodent lineages evolved significantly faster than human; they made comparisons of protein-coding regions of 11 nuclear genes, the 5′ and 3′ untranslated regions of five different mRNAs, and the beta-globin gene family of rodents and human. Later, Janke and others (1994) argued the use of Artiodactyla or Carnivora as an outgroup by Wu and Li (1985) was not appropriate, which may account for the observed acceleration of molecular rates in the rodent lineage. They further showed that the majority of mitochondrial genes conformed to a molecular clock model in human and rodents (Janke et al., 1994). In fact, before the publication of Janke et al., other studies using chicken as an outgroup reached the same conclusion that rodent evolved significantly faster than Primates (Bulmer et al., 1991; Li et al., 1990). This conclusion was confirmed by recent studies using large datasets (Huttley et al., 2007).
These conflicting findings may result from the distinctive evolutionary patterns between nuclear and mitochondrial genes. For instance, recombination is common in nuclear genes but not in mitochondrial genes in mammals (Innan and Nordborg, 2002). In mammalian mitochondria, transitions predominate over transversions, while this is not the case for nuclear genes (Belle et al., 2005). These differences indicate that nuclear and mitochondrial genes are not directly comparable in the evolutionary rate analysis, and the rate of the mitochondrial DNA change may not represent that of nuclear genomic change. Although there was evidence that the evolutionary rates of the several nuclear genes are constant across the placental mammals (Easteal, 1988, 1990), a small number of nuclear genes is not sufficient to draw certain conclusions on lineage-specific rates of evolution (Easteal, 1992). The concatenated nuclear amino acid sequence-based phylogenetic analysis using Maximum-Likelihood and Neighbor-Joining methods in the present study supported that Rodentia evolved significantly faster than Primates (z-test, P<0.001).
Our gene rearrangement rate analysis supported that the rat lineage evolved significantly faster than Primates, and the mouse lineage evolved significantly faster than human, chimpanzee, and orangutan, but not macaque. This is consistent with the previous findings that the rearrangement rate of mouse is in-between various Primates (Coghlan et al., 2005). In addition, the rat lineage showed a significantly increased rate of rearrangement as compared to mouse. Other studies detected 180 conserved elements (genomic segments of preserved gene order) shared between human and mouse and 109 shared between human and rat (O’Brien et al., 1999), suggesting that the rearrangement rate of rat is greater than that of mouse. This is also consistent with the concatenated amino acid sequence-based analysis in the present study, but different from several previous reports, which showed that rat had either slightly more neutral substitutions in protein-coding gene regions than in mouse (Rat Genome Sequencing Project Consortium, 2004) or almost identical rates with mouse (Huttley et al., 2007).
A non-clock-like manner of gene order changes has been observed in animal mitochondrial genomes (Boore, 1999; Boore and Brown, 1998), and is a common property of genome-level cladistic characters (Boore, 2006). Clock-like characters (e.g. nucleotide substitution) perform poorly in resolving short basal branches because the signal to noise ratio in the data closely matches the ratio of the time periods of the internal branches to the leaf branches (Boore, 2006). However, non-clocklike characters (e.g. genome rearrangement) may have had some occasional and abrupt changes occurring and being fixed during rapid radiation (Boore, 2006). Although non-clocklike character is not desirable to estimate the time of lineage splitting, it is the non-clocklike behavior that makes a genome-level character such as genome rearrangement especially useful in resolving narrow and ancient divergences (Boore, 2006).
Supplementary Material
Supplementary Figure 1. Phylogeny of Primates (human, chimpanzee, orangutan, macaque), Laurasiatheria (canine, horse, bovine) and Rodentia (mouse, rat) inferred from (A, C, E) a DCJ distance (swine or rabbit may or may not be included); (B, D, F) a breakpoint distance matrix tree (swine or rabbit may or may not be included). Values above branches show the number of times that the clade defined by that node was supported by 1000 jackknife trees. Values below branches show number of genome rearrangement events. Opossum is the outgroup.
Supplementary Table 1. The 9212 orthologous genes shared by 10 mammalian genomes.
Supplementary Table 2. The 6740 orthologous genes shared by 11 mammalian genomes (with rabbit genome included)
Gene order phylogeny placed rodents outside other orders of placental mammals.
Rapid radiation in distant past makes phylogenetic reconstruction difficult.
Gene order may be a good character in mammals because breakpoint re-use is low.
Acknowledgments
This research was supported by grant GM43940 from the National Institute of Health to A.L.H and grant GM078991 from the National Institute of Health to J.T. Most computations were performed on a 128-core shared memory computer of the High Performance Computing Group at the University of South Carolina. We also thank the generous support from Research Computing Center at the University of Georgia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alekseyev M, Pevzner P. Are there rearrangement hotspots in the human genome? PLoS Comput Biol. 2007;3:e209. doi: 10.1371/journal.pcbi.0030209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyev M, Pevzner P. Breakpoint graphs and ancestral genome reconstructions. Genome Res. 2009;19:943–957. doi: 10.1101/gr.082784.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyev MA. Multi-break rearrangements and breakpoint re-uses: from circular to linear genomes. J Comput Biol. 2008;15:1117–1131. doi: 10.1089/cmb.2008.0080. [DOI] [PubMed] [Google Scholar]
- Altekar G, Dwarkadas S, Huelsenbeck JP, Ronquist F. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics. 2004;20:407–415. doi: 10.1093/bioinformatics/btg427. [DOI] [PubMed] [Google Scholar]
- Archibald JD. Timing and biogeography of the eutherian radiation: fossils and molecules compared. Mol Phylogenet Evol. 2003;28:350–359. doi: 10.1016/s1055-7903(03)00034-4. [DOI] [PubMed] [Google Scholar]
- Arnason U, Adegoke JA, Bodin K, Born EW, Esa YB, Gullberg A, Nilsson M, Short RV, Xu X, Janke A. Mammalian mitogenomic relationships and the root of the eutherian tree. Proc Natl Acad Sci USA. 2002;99:8151–8156. doi: 10.1073/pnas.102164299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnason U, Adegoke JA, Gullberg A, Harley EH, Janke A, Kullberg M. Mitogenomic relationships of placental mammals and molecular estimates of their divergences. Gene. 2008;421:37–51. doi: 10.1016/j.gene.2008.05.024. [DOI] [PubMed] [Google Scholar]
- Arnason U, Gullberg A, Janke A. Phylogenetic analyses of mitochondrial DNA suggest a sister group relationship between Xenarthra (Edentata) and Ferungulates. Mol Biol Evol. 1997;14:762–768. doi: 10.1093/oxfordjournals.molbev.a025816. [DOI] [PubMed] [Google Scholar]
- Arnason U, Gullberg A, Janke A. The mitochondrial DNA molecule of the aardvark, Orycteropus afer, and the position of the Tubulidentata in the eutherian tree. Philos Trans R Soc Lond, B, Biol Sci. 1999;266:339–345. doi: 10.1098/rspb.1999.0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnason U, Janke A. Mitogenomic analyses of eutherian relationships. Cytogenet Genome Res. 2002;96:20–32. doi: 10.1159/000063023. [DOI] [PubMed] [Google Scholar]
- Belda E, Moya A, Silva FJ. Genome rearrangement distances and gene order phylogeny in {gamma}-Proteobacteria. Mol Biol Evol. 2005;22:1456–1467. doi: 10.1093/molbev/msi134. [DOI] [PubMed] [Google Scholar]
- Belle EMS, Piganeau G, Gardner M, Eyre-Walker A. An investigation of the variation in the transition bias among various animal mitochondrial DNA. Gene. 2005;355:58–66. doi: 10.1016/j.gene.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Bergeron A, Mixtacki J, Stoye J. A unifying view of genome rearrangements. Algorithms in Bioinformatics. 2006:163–173. [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL. The use of genome-level characters for phylogenetic reconstruction. Trends Ecol Evol. 2006;21:439–446. doi: 10.1016/j.tree.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Boore JL, Brown WM. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev. 1998;8:668–674. doi: 10.1016/s0959-437x(98)80035-x. [DOI] [PubMed] [Google Scholar]
- Bulmer M, Wolfe KH, Sharp PM. Synonymous nucleotide substitution rates in mammalian genes: implications for the molecular clock and the relationship of mammalian orders. Proc Natl Acad Sci USA. 1991;88:5974–5978. doi: 10.1073/pnas.88.14.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarozzi G, Schneider A, Gonnet G. A phylogenomic study of human, dog, and mouse. PLoS Comput Biol. 2007;3:1e2. doi: 10.1371/journal.pcbi.0030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Adachi J, Janke A, Pääbo S, Hasegawa M. Phylogenetic relationships among eutherian orders estimated from inferred sequences of mitochondrial proteins: Instability of a tree based on a single gene. J Mol Evol. 1994;39:519–527. doi: 10.1007/BF00173421. [DOI] [PubMed] [Google Scholar]
- Cao Y, Janke A, Waddell PJ, Westerman M, Takenaka O, Murata S, Okada N, Pääbo S, Hasegawa M. Conflict Among Individual Mitochondrial Proteins in Resolving the Phylogeny of Eutherian Orders. J Mol Evol. 1998;47:307–322. doi: 10.1007/pl00006389. [DOI] [PubMed] [Google Scholar]
- Chen X, Zheng J, Fu Z, Nan P, Zhong Y, Lonardi S, Jiang T. Assignment of orthologous genes via genome rearrangement. IEEE/ACM Trans Comput Biol Bioinform. 2005;2:302–315. doi: 10.1109/TCBB.2005.48. [DOI] [PubMed] [Google Scholar]
- Coghlan A, Eichler EE, Oliver SG, Paterson AH, Stein L. Chromosome evolution in eukaryotes: a multi-kingdom perspective. Trends Genet. 2005;21:673–682. doi: 10.1016/j.tig.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Corneli PS. Complete Mitochondrial Genomes and Eutherian Evolution. J Mammal Evol. 2002;9:281–305. [Google Scholar]
- DeBry RW. The systematic component of phylogenetic error as a function of taxonomic sampling under parsimony. Syst Biol. 2005;54:432–440. doi: 10.1080/10635150590946745. [DOI] [PubMed] [Google Scholar]
- Desper R, Gascuel O. Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J Comput Biol. 2002;9:687–705. doi: 10.1089/106652702761034136. [DOI] [PubMed] [Google Scholar]
- Easteal S. Rate constancy of globin gene evolution in placental mammals. Proc Natl Acad Sci USA. 1988;85:7622–7626. doi: 10.1073/pnas.85.20.7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easteal S. The Pattern of Mammalian Evolution and the Relative Rate of Molecular Evolution. Genetics. 1990;124:165–173. doi: 10.1093/genetics/124.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easteal S. A mammalian molecular clock? Bioessays. 1992;14:415–419. doi: 10.1002/bies.950140613. [DOI] [PubMed] [Google Scholar]
- Eck RV, Dayhoff MO. Atlas of Protein Sequence and Structure. National Biomedical Research Foundation; Silver Springs, Maryland: 1966. [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP - Phylogeny Inference Package. Cladistics. 1989;5:164–166. [Google Scholar]
- Gadagkar SR, Rosenberg MS, Kumar S. Inferring species phylogenies from multiple genes: Concatenated sequence tree versus consensus gene tree. J Exp Zool B Mol Dev Evol. 2005;304B:64–74. doi: 10.1002/jez.b.21026. [DOI] [PubMed] [Google Scholar]
- Goodman M, Czelusniak J, Beeber JE. Phylogeny of primates and other eutherian orders: a cladistic analysis using amino acid and nucleotide sequence data. Cladistics. 1985;1:171–185. doi: 10.1111/j.1096-0031.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Graur D, Martin W. Reading the entrails of chickens: molecular timescales of evolution and the illusion of precision. Trends Genet. 2004;20:80–86. doi: 10.1016/j.tig.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Hallstrom B, Janke A. Resolution among major placental mammal interordinal relationships with genome data imply that speciation influenced their earliest radiations. BMC Evol Biol. 2008;8:162. doi: 10.1186/1471-2148-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom BM, Kullberg M, Nilsson MA, Janke A. Phylogenomic data analyses provide evidence that Xenarthra and Afrotheria are sister groups. Mol Biol Evol. 2007;24:2059–2068. doi: 10.1093/molbev/msm136. [DOI] [PubMed] [Google Scholar]
- Hillis DM. Taxonomic sampling, phylogenetic accuracy, and investigator bias. Syst Biol. 1998;47:3–8. doi: 10.1080/106351598260987. [DOI] [PubMed] [Google Scholar]
- Hillis DM, Pollock DD, McGuire JA, Zwickl DJ. Is sparse taxon sampling a problem for phylogenetic inference? Syst. Biol. 2003;52:124–126. doi: 10.1080/10635150390132911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Near Neutrality: Leading Edge of the Neutral Theory of Molecular Evolution. Ann N Y Acad Sci. 2008;1133:162–179. doi: 10.1196/annals.1438.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. The effect of branch lengths on phylogeny: An empirical study using highly conserved orthologs from mammalian genomes. Mol Phylogenet Evol. 2007;45:81–88. doi: 10.1016/j.ympev.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Genome size reduction in the chicken has involved massive loss of ancestral protein-coding genes. Mol Biol Evol. 2008;25:2681–2688. doi: 10.1093/molbev/msn207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttley GA, Wakefield MJ, Easteal S. Rates of genome evolution and branching order from whole genome analysis. Mol Biol Evol. 2007;24:1722–1730. doi: 10.1093/molbev/msm094. [DOI] [PubMed] [Google Scholar]
- Innan H, Nordborg M. Recombination or Mutational Hot Spots in Human mtDNA? Mol. Biol Evol. 2002;19:1122–1127. doi: 10.1093/oxfordjournals.molbev.a004170. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Janke A, Feldmaier-Fuchs G, Thomas WK, von-Haeseler A, Paabo S. The Marsupial Mitochondrial Genome and the Evolution of Placental Mammals. Genetics. 1994;137:243–256. doi: 10.1093/genetics/137.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke A, Xu X, Arnason U. The complete mitochondrial genome of the wallaroo (Macropus robustus) and the phylogenetic relationship among Monotremata, Marsupialia, and Eutheria. Proc Natl Acad Sci USA. 1997;94:1276–1281. doi: 10.1073/pnas.94.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T. A combinatorial approach to genome-wide ortholog assignment: beyond sequence similarity search. Proceedings of the 18th annual symposium on Combinatorial Pattern Matching; London, Canada: Springer-Verlag; 2007. [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Bioinformatics. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Karere GM, Froenicke L, Millon L, Womack JE, Lyons LA. A high-resolution radiation hybrid map of rhesus macaque chromosome 5 identifies rearrangements in the genome assembly. 2008;92:210–218. doi: 10.1016/j.ygeno.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowski B, Thornton JW. Performance of maximum parsimony and likelihood phylogenetics when evolution is heterogeneous. Nature. 2004;431:980–984. doi: 10.1038/nature02917. [DOI] [PubMed] [Google Scholar]
- Kothari M, Moret BME. An experimental evaluation of inversion- and transposition-based genomic distances. Proc. 3rd IEEE Symp. on Comput. Intelligence in Bioinformatics and Comput. Biol. (CIBCB’07); 2007. pp. 151–158. [Google Scholar]
- Kullberg M, Hallstrom B, Arnason U, Janke A. Expressed sequence tags as a tool for phylogenetic analysis of placental mammal evolution. PLoS ONE. 2007;2:e775. doi: 10.1371/journal.pone.0000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullberg M, Nilsson MA, Arnason U, Harley EH, Janke A. Housekeeping Genes for Phylogenetic Analysis of Eutherian Relationships. Mol Biol Evol. 2006;23:1493–1503. doi: 10.1093/molbev/msl027. [DOI] [PubMed] [Google Scholar]
- Li WH, Gouy M, Sharp PM, O’h Uigin C, Yang YW. Molecular phylogeny of Rodentia, Lagomorpha, Primates, Artiodactyla, and Carnivora and molecular clocks. Proc Natl Acad Sci USA. 1990;87:6703–6707. doi: 10.1073/pnas.87.17.6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Moret BME. Estimating true evolutionary distances under the DCJ model. Bioinformatics. 2008;24:i114–122. doi: 10.1093/bioinformatics/btn148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Rajan V, Moret BME. Bootstrapping phylogenies inferred from rearrangement data. Proc. 11th Workshop on Algorithms in Bioinformatics WABI’11, Lecture Notes in Computer Science. 2011;6833:175–187. [Google Scholar]
- Lin Y, Rajan V, Moret BME. Fast and accurate phylogenetic reconstruction from high-resolution whole-genome data and a novel robustness estimator. J Comput Biol. 2011;18:1131–1139. doi: 10.1089/cmb.2011.0114. [DOI] [PubMed] [Google Scholar]
- Loytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci USA. 2005;102:10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loytynoja A, Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008;320:1632–1635. doi: 10.1126/science.1158395. [DOI] [PubMed] [Google Scholar]
- Luo H, Shi J, Arndt W, Tang J, Friedman R. Gene order phylogeny of the genus Prochlorococcus. PLoS ONE. 2008;3:e3837. doi: 10.1371/journal.pone.0003837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Sun Z, Arndt W, Shi J, Friedman R, Tang J. Gene order phylogeny and the evolution of methanogens. PLoS ONE. 2009;4:e6069. doi: 10.1371/journal.pone.0006069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen O, Scally M, Douady CJ, Kao DJ, DeBry RW, Adkins R, Amrine HM, Stanhope MJ, de Jong WW, Springer MS. Parallel adaptive radiations in two major clades of placental mammals. Nature. 2001;409:610–614. doi: 10.1038/35054544. [DOI] [PubMed] [Google Scholar]
- Mallet J. Hybridization as an invasion of the genome. Trends Ecol Evol. 2005;20:229–237. doi: 10.1016/j.tree.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Misawa K, Nei M. Reanalysis of Murphy et al.’s Data Gives Various Mammalian Phylogenies and Suggests Overcredibility of Bayesian Trees. J Mol Evol. 2003;57:S290–S296. doi: 10.1007/s00239-003-0039-7. [DOI] [PubMed] [Google Scholar]
- Mitchell A, Mitter C, Regier JC. More taxa or more characters revisited: combining data from nuclear protein-encoding genes for phylogenetic analyses of noctuoidea (Insecta: Lepidoptera) Syst Biol. 2000;49:202–224. [PubMed] [Google Scholar]
- Miyamoto MM. Molecular systematics: Perfect SINEs of evolutionary history? Curr Biol. 1999;9:R816–R819. doi: 10.1016/s0960-9822(99)80498-9. [DOI] [PubMed] [Google Scholar]
- Moret BME, Tang J, Wang LS, Warnow T. Steps toward accurate reconstruction of phylogenies from gene-order data. Journal of Computer and System Sciences. 2002;65:508–525. [Google Scholar]
- Mouchaty SK, Gullberg A, Janke A, Arnason U. The Phylogenetic Position of the Talpidae Within Eutheria Based on Analysis of Complete Mitochondrial Sequences. Mol Biol Evol. 2000;17:60–67. doi: 10.1093/oxfordjournals.molbev.a026238. [DOI] [PubMed] [Google Scholar]
- Mural RJ, Adams MD, Myers EW, Smith HO, Miklos GLG, Wides R, Halpern A, Li PW, Sutton GG, Nadeau J, Salzberg SL, Holt RA, Kodira CD, Lu F, Chen L, Deng Z, Evangelista CC, Gan W, Heiman TJ, Li J, Li Z, Merkulov GV, Milshina NV, Naik AK, Qi R, Shue BC, Wang A, Wang J, Wang X, Yan X, Ye J, Yooseph S, Zhao Q, Zheng L, Zhu SC, Biddick K, Bolanos R, Delcher AL, Dew IM, Fasulo D, Flanigan MJ, Huson DH, Kravitz SA, Miller JR, Mobarry CM, Reinert K, Remington KA, Zhang Q, Zheng XH, Nusskern DR, Lai Z, Lei Y, Zhong W, Yao A, Guan P, Ji RR, Gu Z, Wang ZY, Zhong F, Xiao C, Chiang CC, Yandell M, Wortman JR, Amanatides PG, Hladun SL, Pratts EC, Johnson JE, Dodson KL, Woodford KJ, Evans CA, Gropman B, Rusch DB, Venter E, Wang M, Smith TJ, Houck JT, Tompkins DE, Haynes C, Jacob D, Chin SH, Allen DR, Dahlke CE, Sanders R, Li K, Liu X, Levitsky AA, Majoros WH, Chen Q, Xia AC, Lopez JR, Donnelly MT, Newman MH, Glodek A, Kraft CL, Nodell M, Ali F, An HJ, Baldwin-Pitts D, Beeson KY, Cai S, Carnes M, Carver A, Caulk PM, Center A, Chen YH, Cheng ML, Coyne MD, Crowder M, Danaher S, Davenport LB, Desilets R, Dietz SM, Doup L, Dullaghan P, Ferriera S, Fosler CR, Gire HC, Gluecksmann A, Gocayne JD, Gray J, Hart B, Haynes J, Hoover J, Howland T, Ibegwam C, Jalali M, Johns D, Kline L, Ma DS, MacCawley S, Magoon A, Mann F, May D, McIntosh TC, Mehta S, Moy L, Moy MC, Murphy BJ, Murphy SD, Nelson KA, Nuri Z, Parker KA, Prudhomme AC, Puri VN, Qureshi H, Raley JC, Reardon MS, Regier MA, Rogers YHC, Romblad DL, Schutz J, Scott JL, Scott R, Sitter CD, Smallwood M, Sprague AC, Stewart E, Strong RV, Suh E, Sylvester K, Thomas R, Tint NN, Tsonis C, Wang G, Wang G, Williams MS, Williams SM, Windsor SM, Wolfe K, Wu MM, Zaveri J, Chaturvedi K, Gabrielian AE, Ke Z, Sun J, Subramanian G, Venter JC. A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science. 2002;296:1661–1671. doi: 10.1126/science.1069193. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, O’Brien SJ. Molecular phylogenetics and the origins of placental mammals. Nature. 2001a;409:614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Eizirik E, O’Brien SJ, Madsen O, Scally M, Douady CJ, Teeling E, Ryder OA, Stanhope MJ, de Jong WW, Springer MS. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science. 2001b;294:2348–2351. doi: 10.1126/science.1067179. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Larkin DM, der Wind AE-v, Bourque G, Tesler G, Auvil L, Beever JE, Chowdhary BP, Galibert F, Gatzke L, Hitte C, Meyers SN, Milan D, Ostrander EA, Pape G, Parker HG, Raudsepp T, Rogatcheva MB, Schook LB, Skow LC, Welge M, Womack JE, O’Brien SJ, Pevzner PA, Lewin HA. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science. 2005;309:613–617. doi: 10.1126/science.1111387. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Pevzner PA, O’Brien SJ. Mammalian phylogenomics comes of age. Trends Genet. 2004;20:631–639. doi: 10.1016/j.tig.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Taylor BA. Lengths of chromosomal segments conserved since divergence of man and mouse. Proc Natl Acad Sci USA. 1984;81:814–818. doi: 10.1073/pnas.81.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neron B, Menager H, Maufrais C, Joly N, Maupetit J, Letort S, Carrere S, Tuffery P, Letondal C. Mobyle: a new full web bioinformatics framework. Bioinformatics. 2009;25:3005–3011. doi: 10.1093/bioinformatics/btp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara H, Okada N, Hasgawa M. Rooting the eutherian tree: the power and pitfalls of phylogenomics. Genome Biol. 2007;8:R199. doi: 10.1186/gb-2007-8-9-r199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien SJ, Menotti-Raymond M, Murphy WJ, Nash WG, Wienberg J, Stanyon R, Copeland NG, Jenkins NA, Womack JE, Marshall Graves JA. The promise of comparative genomics in mammals. Science. 1999;286:458–481. doi: 10.1126/science.286.5439.458. [DOI] [PubMed] [Google Scholar]
- Ohno S. Ancient linkage groups and frozen accidents. Nature. 1973;244:259–262. doi: 10.1038/244259a0. [DOI] [PubMed] [Google Scholar]
- Penny D, Hasegawa M. Molecular systematics: The platypus put in its place. Nature. 1997;387:549–550. doi: 10.1038/42352. [DOI] [PubMed] [Google Scholar]
- Pevzner P, Tesler G. Human and mouse genomic sequences reveal extensive breakpoint reuse in mammalian evolution. Proc Natl Acad Sci USA. 2003;100:7672–7677. doi: 10.1073/pnas.1330369100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poe S. Sensitivity of phylogeny estimation to taxonomic sampling. Syst Biol. 1998;47:18–31. doi: 10.1080/106351598261003. [DOI] [PubMed] [Google Scholar]
- Pollock DD, Zwickl DJ, McGuire JA, Hillis DM. Increased taxon sampling is advantageous for phylogenetic inference. Syst Biol. 2002;51:664–671. doi: 10.1080/10635150290102357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumo DE, Finamore PS, Franek WR, Phillips CJ, Tarzami S, Balzarano D. Complete mitochondrial genome of a neotropical fruit bat, Artibeus jamaicensis, and a new hypothesis of the relationships of bats to other eutherian mammals. J Mol Evol. 1998;47:709–717. doi: 10.1007/pl00006430. [DOI] [PubMed] [Google Scholar]
- Rannala B, Huelsenbeck JP, Yang Z, Nielsen R. Taxon sampling and the accuracy of large phylogenies. Syst Biol. 1998;47:702–710. doi: 10.1080/106351598260680. [DOI] [PubMed] [Google Scholar]
- Rat Genome Sequencing Project Consortium. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Reyes A, Gissi C, Catzeflis F, Nevo E, Pesole G, Saccone C. Congruent Mammalian Trees from Mitochondrial and Nuclear Genes Using Bayesian Methods. Mol Biol Evol. 2004;21:397–403. doi: 10.1093/molbev/msh033. [DOI] [PubMed] [Google Scholar]
- Reyes A, Gissi C, Pesole G, Catzeflis FM, Saccone C. Where Do Rodents Fit? Evidence from the Complete Mitochondrial Genome of Sciurus vulgaris. Mol Biol Evol. 2000;17:979–983. doi: 10.1093/oxfordjournals.molbev.a026379. [DOI] [PubMed] [Google Scholar]
- Rokas A, Holland PWH. Rare genomic changes as a tool for phylogenetics. Trends Ecol Evol. 2000;15:454–459. doi: 10.1016/s0169-5347(00)01967-4. [DOI] [PubMed] [Google Scholar]
- Rosenberg MS, Kumar S. Incomplete taxon sampling is not a problem for phylogenetic inference. Proc Natl Acad Sci USA. 2001;98:10751–10756. doi: 10.1073/pnas.191248498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg MS, Kumar S. Taxon sampling, bioinformatics, and phylogenomics. Syst Biol. 2003;52:119–124. doi: 10.1080/10635150390132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Shi J, Tang J. An experimental evaluation of corrected inversion and DCJ distance metric through simulation. Proceeding of the 4th International Conference on Bioinformatics and Biomedical Engineering (iCBBE); 2010. pp. 1–4. [Google Scholar]
- Shi J, Zhang Y, Luo H, Tang J. Using jackknife to assess the quality of gene order phylogenies. BMC Bioinformatics. 2010;11:168. doi: 10.1186/1471-2105-11-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer MS, Cleven GC, Madsen O, de Jong WW, Waddell VG, Amrine HM, Stanhope MJ. Endemic African mammals shake the phylogenetic tree. Nature. 1997;388:61–64. doi: 10.1038/40386. [DOI] [PubMed] [Google Scholar]
- Springer MS, de Jong WW. Which Mammalian Supertree to Bark Up? Science. 2001;291:1709–1711. doi: 10.1126/science.1059434. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Glazko GV, Nei M. Overcredibility of molecular phylogenies obtained by Bayesian phylogenetics. Proc Natl Acad Sci USA. 2002;99:16138–16143. doi: 10.1073/pnas.212646199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- R Develop Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- Tocheri MW, Orr CM, Jacofsky MC, Marzke MW. The evolutionary history of the hominin hand since the last common ancestor of Pan and Homo. J Anat. 2008;212:544–562. doi: 10.1111/j.1469-7580.2008.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildman DE, Uddin M, Opazo JC, Liu G, Lefort V, Guindon S, Gascuel O, Grossman LI, Romero R, Goodman M. Genomics, biogeography, and the diversification of placental mammals. Proc Natl Acad Sci USA. 2007;104:14395–14400. doi: 10.1073/pnas.0704342104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CI, Li WH. Evidence for higher rates of nucleotide substitution in rodents than in man. Proc Natl Acad Sci USA. 1985;82:1741–1745. doi: 10.1073/pnas.82.6.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos S, Attie O, Friedberg R. Efficient sorting of genomic permutations by translocation, inversion and block interchange. Bioinformatics. 2005;21:3340–3346. doi: 10.1093/bioinformatics/bti535. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, Bork P. Quantification of insect genome divergence. Trends Genet. 2007;23:16–20. doi: 10.1016/j.tig.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Zhang M, Arndt W, Tang J. An Exact Solver for the DCJ Median Problem. Pac Symp Biocomput. 2009;14:138–149. [PMC free article] [PubMed] [Google Scholar]
- Zwickl DJ, Hillis DM. Increased taxon sampling greatly reduces phylogenetic error. Syst Biol. 2002;51:588–598. doi: 10.1080/10635150290102339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Phylogeny of Primates (human, chimpanzee, orangutan, macaque), Laurasiatheria (canine, horse, bovine) and Rodentia (mouse, rat) inferred from (A, C, E) a DCJ distance (swine or rabbit may or may not be included); (B, D, F) a breakpoint distance matrix tree (swine or rabbit may or may not be included). Values above branches show the number of times that the clade defined by that node was supported by 1000 jackknife trees. Values below branches show number of genome rearrangement events. Opossum is the outgroup.
Supplementary Table 1. The 9212 orthologous genes shared by 10 mammalian genomes.
Supplementary Table 2. The 6740 orthologous genes shared by 11 mammalian genomes (with rabbit genome included)