

Abstract
Tumor-initiating cells (TICs) are a sub-population of cells that exhibit a robust ability to self-renew and contribute to the formation of primary tumors, the relapse of previously treated tumors, and the development of metastases. TICs have been identified in various tumors, including those of the breast, and are particularly enriched in the basal-like and claudin-low subtypes of breast cancer. The signaling pathways that contribute to the function and maintenance of TICs are under intense study. We explored the potential involvement of the NF-κB family of transcription factors in TICs in cell lines that are representative of basal-like and claudin-low breast cancer. NF-κB was found to be activated in breast cancer cells that form tumorspheres efficiently. Moreover, both canonical and non-canonical NF-κB signaling is required for these cells to self-renew in vitro and to form xenograft tumors efficiently in vivo using limiting dilutions of cells. Consistent with this, canonical and non-canonical NF-κB signaling is activated in TICs isolated from breast cancer cell lines. Experimental results indicate that NF-κB promotes the function of TICs by stimulating epithelial-to-mesenchymal transition (EMT) and by upregulating the expression of the inflammatory cytokines IL-1β and IL-6. The results suggest the use of NF-κB inhibitors for clinical therapy of certain breast cancers.
Keywords: NF-κB, basal-like breast cancer, tumor-initiating cells, EMT, IL-6, IL-1β
Introduction
Tumors are comprised of a heterogeneous population of cells, including bulk epithelial tumor cells, inflammatory cells, and a sub-population of cells termed cancer stem cells or tumor-initiating cells (TICs) (1). The primary characteristic of TICs is their ability to self-renew, which is measured in vitro by the formation of spheroid cellular structures termed tumorspheres (2, 3). Additionally, TICs exhibit elevated motility and invasiveness in vitro that correlates with high metastatic potential in vivo (4–6), and are frequently radio- (7, 8) and chemoresistant (9, 10). Importantly, TICs are thought to drive the progression of primary tumors, promote tumor recurrence, and stimulate the development of metastases at distance sites (4, 5). The importance of TICs in the clinical outcome of breast cancer is evidenced by the observation that an increase in their abundance following initial systemic treatment correlates with worse prognosis (11). TICs have been observed in multiple subtypes of human breast cancer (12) and are particularly enriched in the basal-like and claudin-low subtypes (12–14).
The NF-κB family of transcription factors contains five members, p65 (RelA), RelB, c-Rel, p105/p50, and p100/p52 (15, 16). In most cells, NF-κB proteins exist as hetero- and homodimers in the cytoplasm bound to a class of inhibitory proteins called IκBs. In response to a wide variety of cellular stimuli, NF-κB becomes active via one of two pathways. In the canonical pathway, NF-κB activation depends on the IκB kinase complex (IKK), which contains two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ or NEMO. Upon stimulation, IκBα is phosphorylated at Ser32/36 by IKK in a manner that requires IKKβ, which results in the degradation of IκBα and the release of the p65-p50 dimer to accumulate in the nucleus (15). Phosphorylation of p65 at Ser536 by IKK is also important for its activity (17). Separately, the non-canonical NF-κB pathway is regulated by an IKKα homodimer. In this cascade, RelB-p100 heterodimers are processed to RelB-p52 heterodimers in a manner that depends on IKKα. In the nucleus, NF-κB dimers activate genes including those involved in cell cycle regulation (e.g. cyclin D1), suppression of apoptosis (e.g. Bcl-2 and Bcl-xL), and inflammation (e.g. cytokines such as IL-6 and IL-8) (15).
Activation of NF-κB is strongly associated with oncogenesis, as it is known to promote the oncogenic phenotype through processes including cell proliferation, inflammation, cell invasion and suppression of apoptosis (18, 19). Consistent with this, both canonical and non-canonical NF-κB signaling is activated in human breast cancer cell lines and primary breast tumors (20–24). Recently, IKK/NF-κB was shown to be important in TICs isolated from HER2+ breast cancer (25, 26). Others have observed that NF-κB functions to promote proliferation in basal-like breast cancer cells (27). Here, we have explored a potential role for NF-κB in TIC function in cells derived from basal-like and claudin-low breast cancer cells. Specifically, we show that NF-κB signaling is more highly activated in breast cancer cell lines that undergo efficient self-renewal. Moreover, inhibition of either canonical or non-canonical NF-κB signaling blunts the self-renewal of human breast cancer cells in vitro. Inhibition of NF-κB also reduces the formation of xenograft tumors in the mammary fat pads of nude mice in vivo. Mechanistically, we provide evidence that NF-κB promotes the function of TICs through stimulation of epithelial-to-mesenchymal transition (EMT) and the production of inflammatory cytokines that are encoded by NF-κB target genes. Collectively, these data demonstrate that canonical and non-canonical NF-κB signaling play critical roles in the function of TICs derived from basal-like and claudin-low subtypes of breast cancer cells.
Results
NF-κB signaling is preferentially activated in tumorsphere-forming breast cancer cells
Cell lines representing the basal-like subtype (SUM149) and the claudin-low subtype (MDA-MB231) of breast cancer were utilized to investigate the role of NF-κB in the function of TICs. A hallmark of breast cancer TICs is the ability to drive the formation of spheroid structures termed tumorspheres (or mammospheres) in serum-free culture, which reflects the ability of these cells to self-renew in vitro (reviewed in (2, 28)). It was observed that both SUM149 and MDA-MB231 cells efficiently form tumorspheres over at least three cycles of culture (Figure 1a). It was then determined whether the ability of basal-like and claudin-low cancer cells to form tumorspheres correlates with the level of basal NF-κB activation in the bulk population. Importantly, both p65 and IκBα are preferentially phosphorylated in SUM149 and MDA-MB231 cells that form tumorspheres efficiently, compared to MCF10A cells which form tumorspheres less efficiently (Figure 1b) (29).
Figure 1. NF-κB signaling is preferentially activated in tumorsphere-forming breast cancer cells.

(A) Primary and tertiary tumorspheres formed by the indicated bulk populations of basal-like and claudin-low breast cancer cells in serum-free culture on low-adhesion plates. (B) Phosphorylation of p65 and IκBα as markers of NF-κB activation in the indicated bulk populations of breast cancer cells (SUM149 and MDA-MB231) or immortalized breast (MCF10A) cells.
Canonical NF-κB signaling is required for basal-like breast cancer cells to efficiently self-renew in vitro
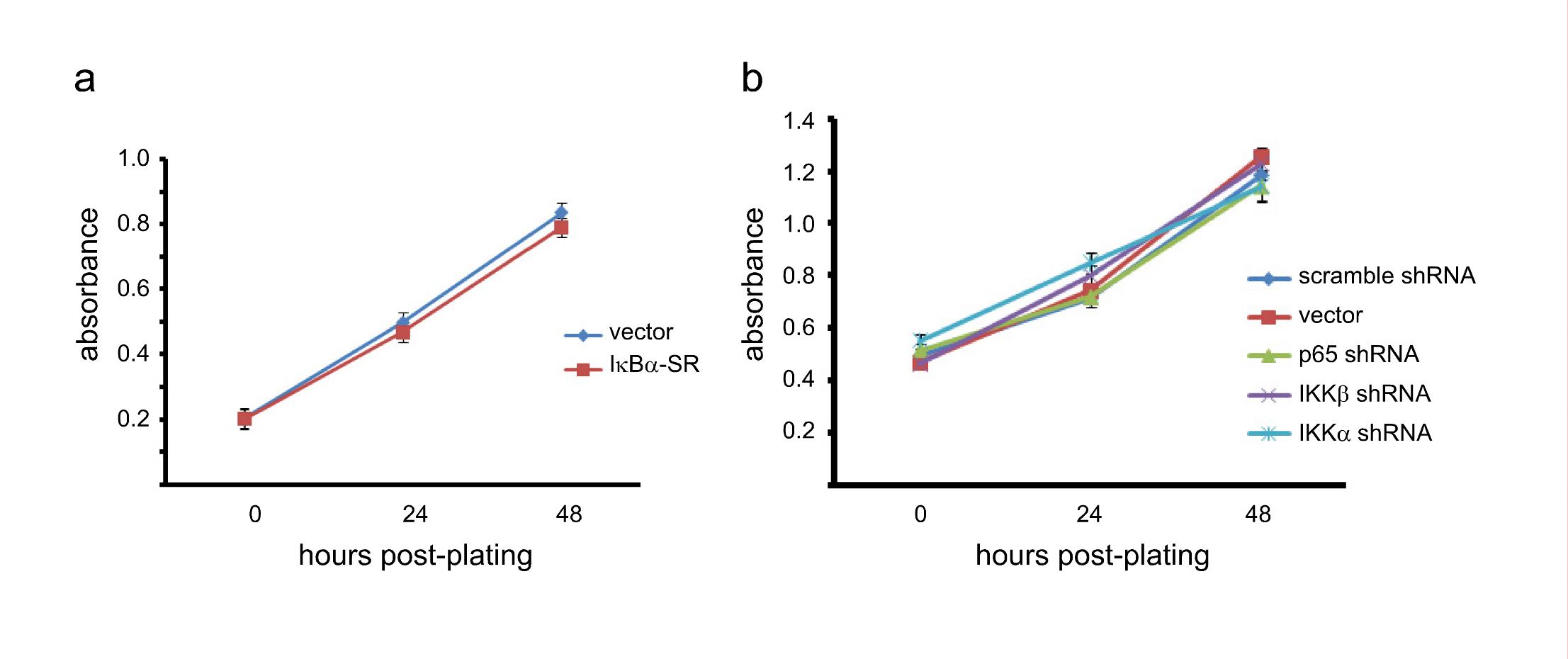
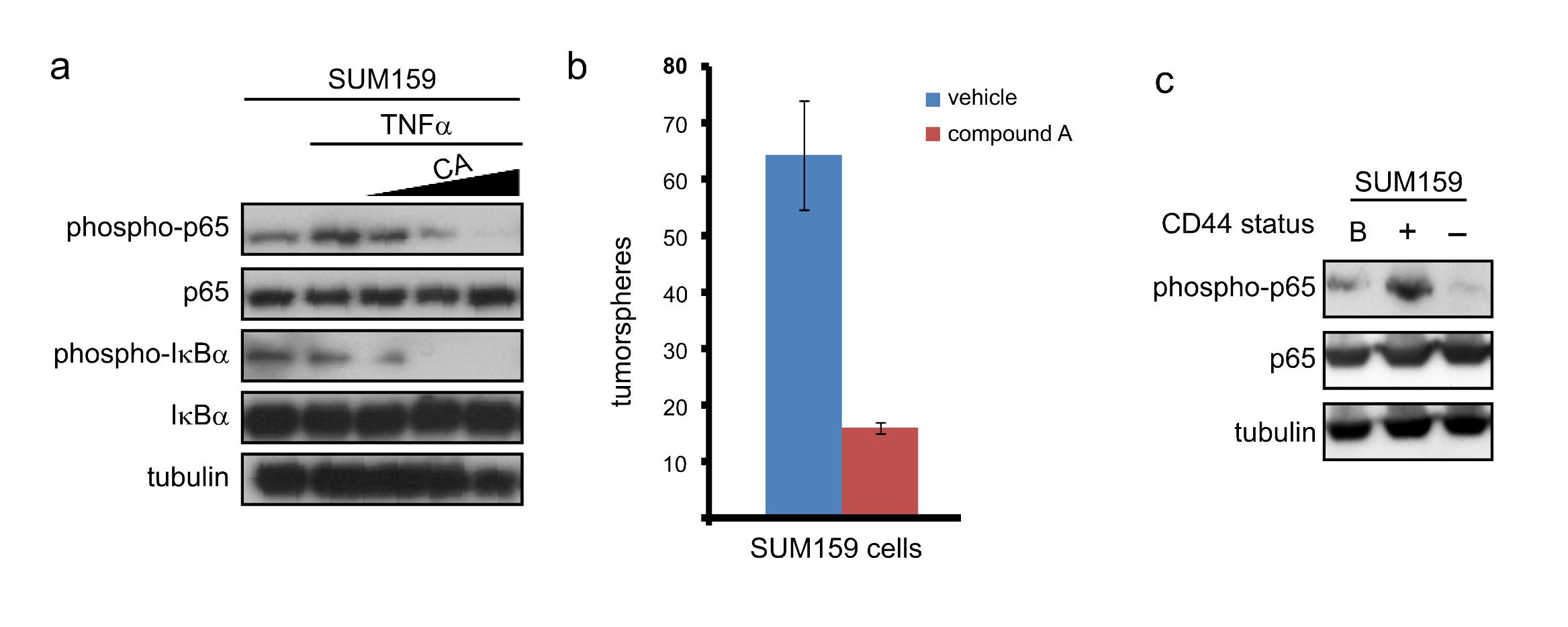
To inhibit NF-κB, SUM149 cells were stably infected with a retrovirus expressing either an empty vector or IκBα-SR (a modified form of IκBα that cannot be phosphorylated) and selected with puromycin (Figure 2a). To assay for self-renewal, 100 SUM149 cells expressing empty vector or IκBα-SR were plated in serum-free media on low-adhesion plates and the number of tumorspheres was determined five days later. Importantly, cells in which NF-κB was inhibited by expression of IκBα-SR formed three-fold fewer tumorspheres than control cells expressing an empty vector (Figure 2b). To confirm these results, an RNAi knockdown approach was utilized. SUM149 cells were stably infected with an empty lentiviral vector, a lentivirus encoding a scrambled shRNA, or a lentivirus encoding an shRNA construct targeting p65 or IKKβ (Figure 2c). Subsequently, self-renewal of these cells was assayed through tumorsphere formation as described above. Importantly, knockdown of either p65 or IKKβ resulted in a statistically significant, approximately five-fold reduction in the ability of SUM149 cells to form tumorspheres (Figure 2d). Notably, expression of IκBα-SR or knockdown of IKKβ or p65 does not alter the growth of SUM149 cells, as assessed by MTT assay (Supplemental Figures 1a and 1b). Additionally, further analysis demonstrated that tumorspheres were arising from single cells (data not shown). This indicates that any variation in the formation of tumorspheres is due to reduced self-renewal, rather than an inhibition of cell growth or cytotoxicity. Interestingly, shRNA-based knockdown of IKKα (Figure 2c) induced a statistically significant, approximately five-fold reduction in the self-renewal of SUM149 cells (Figure 2d) without altering overall cell growth (Supplemental Figure 1b). Treatment with an IKKβ inhibitor (compound A, (30)) blocked both basal and TNFα-induced phosphorylation of p65 (Figure 2e) and led to a reduction in tumorsphere formation that was very similar to the results obtained with shRNA knockdown of different IKK and NF-κB subunits (Figure 2f). These data demonstrate that canonical NF-κB signaling, driven by IKK, promotes self-renewal in basal-like breast cancer cells (also see below).
Figure 2. Canonical NF-κB signaling is required for basal-like breast cancer cells to efficiently self-renew.

(A) Immunoblot of the indicated proteins in SUM149 cells stably expressing an empty vector or IκBα-SR. (B) Quantification of tumorspheres formed by 100 SUM149 cells expressing empty vector or IκBα-SR. (C) Immunoblot of the indicated proteins by 100 SUM149 cells stably infected with the indicated shRNA constructs. (D) Quantification of tumorspheres formed by 100 SUM149 cells stably expressing the indicated shRNA constructs. (E) Phosphorylation of p65 and IκBα as markers of activation of canonical NF-κB signaling in SUM149 cells pre-treated with increasing doses of compound A and then treated with TNFα. (F) Quantification of tumorspheres formed by 100 SUM149 cells treated daily with 5 µM compound A.
In addition to the breast cancer cells analyzed above, we analyzed the role of NF-κB in the claudin-low representative cell line SUM159 (reviewed in (31, 32)). Notably, similar results were obtained in SUM159 cells compared to SUM149 cells. Specifically, inhibition of IKK with compound A blocked NF-κB activity as measured by inhibition of phosphorylation of p65 and IκBα (Supplemental Figure 2a). Additionally, and consistent with the findings using basal-like breast cancer cells, compound A blocked the formation of tumorspheres derived from SUM159 cells (Supplemental Figure 2b).
Non-canonical NF-κB activity is required for breast cancer cells to self-renew in vitro
As shown above, IKKα contributes to the self-renewal of basal-like breast cancer cells. IKKα typically acts in non-canonical NF-κB signaling (reviewed in (15, 16)), although it has recently been reported to promote canonical NF-κB signaling in breast cancer cells of the HER2+ subtype (33). Knockdown with p100/p52 siRNA efficiently reduced the precursor 100 kD component and almost completely eliminated the processed p52 subunit (Figure 3a). Consistent with the IKKα knockdown results (Figures 2c and 2d), a statistically significant reduction in tumorsphere formation was observed in cells in which p100/p52 was inhibited by siRNA (Figure 3b). To further address a role for non-canonical NF-κB in promoting breast cancer cell tumorsphere formation, RelB was knocked down with siRNA (Figure 3a). As shown in Figure 3b, knockdown of this component of the non-canonical NF-κB pathway suppressed tumorsphere formation in both SUM149 and MDA-MB231 cells. These data, along with those shown in Figure 2, demonstrate that both canonical and non-canonical NF-κB are two signaling pathways that promote the self-renewal of breast cancer cells.
Figure 3. Non-canonical NF-κB signaling is required for basal-like breast cancer cells to self-renew.
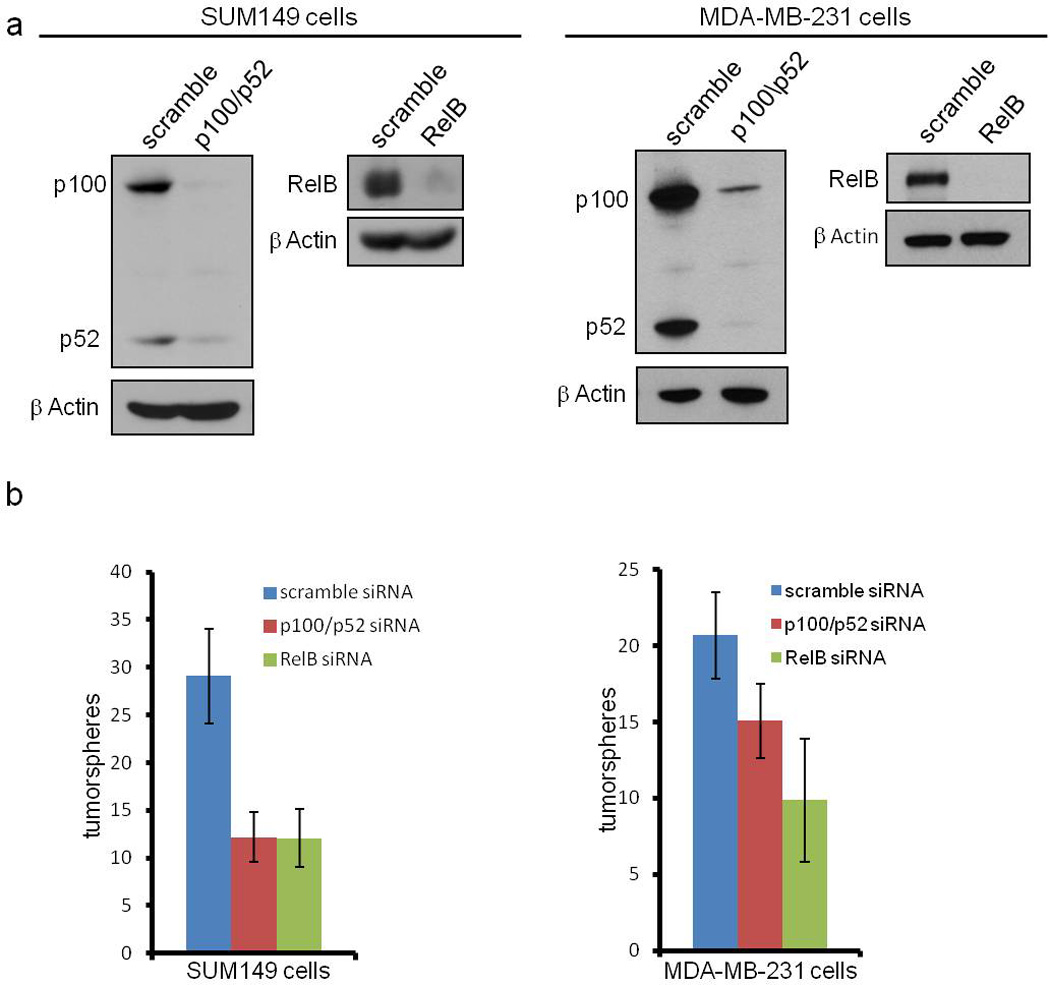
(A) Immunoblot of the indicated proteins in SUM149 and MDA-MB231 cells expressing scrambled siRNA or siRNA targeting p100/p52 or RelB using the indicated antibodies. (B) Quantification of tumorspheres formed by 100 SUM149 or MDA-MB231 cells expressing the indicated siRNA constructs. p values for the right panel of (B) are: p = 0.0413 for scramble siRNA compared to p100/p52 siRNA and p = 0.0011 for scramble siRNA compared to RelB siRNA.
NF-κB promotes the self-renewal of breast cancer cells in vivo
In vivo, self-renewal is assayed by measuring the ability of cells to establish primary tumors when injected at limiting dilutions, which is specifically associated with the function of TICs (2, 34, 35). To directly test the role of NF-κB in the self-renewal of basal-like breast cancer cells in vivo, SUM149 cells expressing empty vector or IκBα-SR at low (102 cells/100 µL) or high (106 cells/100 µL) density were prepared. These cells were injected into the mammary fat pad of nude mice and monitored as described in the methods. In these experiments, cells in which NF-κB signaling was deficient exhibited both delayed tumor onset and reduced overall tumor size (Table 1). Specifically, while the high density of SUM149 cells expressing empty vector formed palpable tumors at six weeks and reached an average tumor volume of 138 mm3 by ten weeks, the high density of SUM149 cells expressing IκBα-SR did not form palpable tumors until week 8 and these tumors maintained a significantly smaller size (11 mm3) at week 10 (Table 1). Importantly, the low density of SUM149 cells expressing empty vector formed palpable tumors at week 9 and these tumors continued to increase in size at a significant rate (Table 1). Conversely, the low density of SUM149 cells expressing IκBα-SR did not form tumors (Table 1). These data demonstrate that NF-κB is required for xenograft-generated tumorigenesis in a context (limiting dilutions of cells, reviewed in (2)) that depends significantly on self-renewal.
Table 1. Inhibition of NF-κB signaling blunts tumorigenesis of limiting dilutions of basal-like breast cancer cells.
Quantification of the average tumor volume of xenograft tumors formed in the mammary fat pad of nude mice following injection of limiting dilutions of SUM149 cells expressing an empty vector or IκBα-SR.
| average tumor volume (mm3) |
|||||
|---|---|---|---|---|---|
| week 6 | week 7 | week 8 | week 9 | week 10 | |
| SUM149 vector | |||||
| 102 cells | — | — | — | 1.33 | 8.25 |
| 106 cells | 8.33 | 4.67 | 24.67 | 83.25 | 137.58 |
| SUM149 IκBα-SR | |||||
| 102 cells | — | — | — | — | — |
| 106 cells | — | — | 10.67 | 10.67 | 10.67 |
Canonical and non-canonical NF-κB are activated in breast cancer TICs and required for maintenance of TICs in the bulk population
The cell surface profile most commonly associated with breast cancer TICs is CD44+CD24- (7, 13, 34, 36–38), although CD44+, EpCAM+, or ALDEFLUOR-positivity (which depends on the activity of the enzyme ALDH1) are also indicative of this sub-population of cells (12, 26, 34, (39–43). After examining the various cell surface profiles previously reported to enrich for TICs (data not shown), we chose to analyze CD44+ cells, which could be identified and isolated efficiently and, more importantly, efficiently self-renew (see below). By calculating the percentage yield from the isolation protocol, bulk populations of both the SUM149 (Figures 4g and 4h) and MDA-MB231 cell lines (data not shown) were found to contain approximately 10% CD44+ cells. This percentage is lower than earlier reports (2, 12, 44), although different groups have reported a range of the proportion of TICs even within the same cell line, particularly when different cell surface markers are utilized (12, 44).
Figure 4. Canonical and non-canonical NF-κB signaling is preferentially activated in TICs and required for the maintenance of TICs in breast cancer cells.

(A) FACS analysis of the indicated populations of SUM149 cells stained with CD44-APC. (B) Quantification of tumorspheres formed by 100 cells from the indicated cell populations of SUM149 or MDA-MB231 cells. (C) Phosphorylation of p65 as a marker of activation of canonical NF-κB signaling in the indicated populations of SUM149 and MDA-MB231 cells. (D) Cleavage of p100 to p52 as a marker of activation of non-canonical NF-κB signaling in the indicated populations of SUM149 and MDA-MB231 cells. (E) Phosphorylation of IKKα and IKKβ in the indicated populations of SUM149 cells. (F) Phosphorylation of TAK1 in the indicated populations of SUM149 cells. (G, H) Percentage of TICs isolated from SUM149 cells stably expressing IκBα-SR (G) or the indicated shRNA constructs (H).
While the bulk population and CD44− cells each contain only a small proportion of cells that are positive for CD44, nearly all the cells in the CD44-isolated population robustly express CD44 (Figure 4a). This confirms that the isolation protocol successfully enriches for CD44+ cells. Notably, CD44+ cells form tumorspheres significantly more efficiently than CD44− cells (Figure 4b), which demonstrates that the CD44+ isolation protocol enriches for TIC function. Given that NF-κB is required for the self-renewal of TICs (Figures 2 and 3), the activation of NF-κB was assessed in lysates from breast cancer TICs (CD44+ cells) compared to lysates isolated from the bulk population of cells and non-TICs (CD44− cells). Importantly, phosphorylation of p65 (Figure 4c) and levels of p52 (Figure 4d) are detected in CD44+ cells in both SUM149 and MDA-MB231 cells, indicating that both canonical and non-canonical NF-κB signaling is activated in breast cancer TICs. As before, the role of NF-κB in TICs of the claudin-low SUM159 cells was also assessed. Specifically, CD44+ cells isolated from SUM159 cells also exhibit elevated NF-κB activity, as measured by phosphorylation of p65 (Supplemental Figure 2c).
To determine if pathways upstream of NF-κB are also activated in breast cancer TICs, phosphorylation of IKKα and IKKβ was assessed by immunoblot of cellular lysates prepared from the bulk population of SUM149 cells and its TIC and non-TIC counterparts. In support of the concept that NF-κB activation is IKK-dependent in these cells, an increased signal for phosphorylated IKKα/β was observed in TICs isolated from SUM149 cells (Figure 4e). This is also consistent with evidence that canonical and non-canonical NF-κB pathways are important in TICs (Figures 2, 3, 4c, and 4d). Finally, the activation of TAK1, a kinase that is an upstream activator of IKK (45), was examined. Similar to IKK, phosphorylation of TAK1, which is indicative of its activation (46), was enriched in CD44+ cells (Figure 4f).
Given that NF-κB is preferentially activated in breast cancer TICs (Figures 4a-4f) and required for their self-renewal in vitro and in vivo (Figures 2 and 3 and Table 1), it was determined whether NF-κB is important in the maintenance of TICs in the bulk population of basal-like breast cancer cells. To this end, the percentage of TICs in the bulk population of SUM149 cells was determined in the presence or absence of overexpression of IκBα-SR. The resulting data showed that inhibition of NF-κB reduced the percentage of CD44+ cells by approximately 50% (Figure 4g). Similarly, stable knockdown of p65, IKKβ, or IKKα each reduced the percentage of CD44+ cells by approximately 50% (Figure 4h). Taken together, these data demonstrate that both canonical and non-canonical NF-κB signaling is preferentially activated in breast cancer TICs, consistent with observations from TICs in the bulk population of cells (Figures 2 and 3), and that NF-κB is important for the maintenance of the TIC population.
NF-κB promotes expression of markers of EMT in TICs and TGFβ-induced self-renewal
EMT is a process by which an epithelial cell releases from the basement membrane and transforms into a spindle-like, mesenchymal cell expressing vimentin and fibronectin (reviewed in (47)). EMT has been demonstrated to promote the self-renewal of immortalized breast cells (36, 37), but this has not been examined in specific subtypes of breast cancer cells. NF-κB is one of multiple signaling pathways implicated in the regulation of EMT, as activation of NF-κB is required for EMT that occurs during Ras-driven transformation (48). Thus, we hypothesized that NF-κB promotes the function of breast cancer TICs by stimulating EMT. Expression of mesenchymal markers was analyzed in the bulk population of SUM149 or MDA-MB231 cells, and in CD44+ and CD44− cells. Both vimentin and fibronectin were detected in the bulk population as well as CD44+ and CD44− cells (data not shown). Importantly, expression of mesenchymal markers depends on NF-κB, as vimentin expression is decreased in the bulk population of SUM149 and MDA-MB231 cells expressing IκBα-SR (Figure 5a). To extend these results, knockdown of the RelA/p65 NF-κB subunit reduced vimentin and fibronectin expression in SUM149 cells (Supplementary Figure 3).
Figure 5. NF-κB activation promotes expression of markers of EMT in TICs and TGFβ-induced self-renewal of basal-like breast cancer cells.

(A) Immunoblot of vimentin in SUM149 or MDA-MB231 cells stably expressing an empty vector or IκBα-SR. (B) Quantification of tumorspheres formed by 100 SUM149 cells stably expressing empty vector or IκBα-SR, followed by treatment with vehicle control or TGFβ.
TGFβ is a well-characterized inducer of EMT and has been shown to promote the self-renewal of at least immortalized breast cancer cells (36). To first confirm that TGFβ promotes self-renewal of basal-like breast cancer cells, 100 SUM149 cells expressing empty vector were plated on low-adhesion plates in serum-free media and treated every other day for seven days with 10 ng/mL TGFβ or vehicle control. Such treatment induced a statistically significant, three-fold increase in the number of tumorspheres formed by these cells (Figure 5b), suggesting that EMT is important for the function of TICs in basal-like breast cancer cells. Importantly, it was investigated whether the ability of TGFβ-induced EMT to stimulate self-renewal of basal-like breast cancer cells depends on NF-κB. Specifically, SUM149 cells expressing IκBα-SR to inhibit NF-κB were also included in the above TGFβ experiment. As previously observed (Figure 2b), expression of IκBα-SR reduced the self-renewal of SUM149 cells (Figure 5b). TGFβ treatment of SUM149 cells expressing IκBα-SR resulted in the formation of a significantly smaller number of tumorspheres than TGFβ treatment of cells with proficient NF-κB signaling (Figure 5b). These data suggest that NF-κB promotes the self-renewal of basal-like breast cancer cells at least in part by stimulating EMT.
IL-1β and IL-6 stimulate the self-renewal of basal-like breast cancer cells downstream of NF-κB
Inflammatory cytokines that are NF-κB target genes have been demonstrated to be involved in self-renewal (29, 49, 50), but the role of NF-κB in this process has not been thoroughly examined. We hypothesized that a subset of cytokine NF-κB target genes may be important to promote breast cancer TICs. To test this hypothesis, SUM149 cells were examined for expression of three inflammatory cytokines (IL-1β, IL-6, and IL-8) and for whether NF-κB is involved in regulating their expression in these cells. Analysis of mRNA expression by real-time PCR demonstrated that the IκBα-SR-expressing cells exhibit reduced levels of IL-1β, IL-6, and IL-8 mRNAs compared to vector control cells (Figure 6a). Additionally, ELISA analysis revealed significantly decreased levels of secreted IL-1β, IL-6, and IL-8 in the media of SUM149 cells in which NF-κB is inhibited compared to the vector control cells (Figure 6b). These data confirm that NF-κB is critical for the expression and secretion of IL-1β, IL-6, and IL-8 in basal-like breast cancer cells.
Figure 6. IL-1β and IL-6 stimulate the self-renewal of basal-like breast cancer cells downstream of NF-κB.

(A) Real-time PCR showing expression of IL-1β, IL-6, or IL-8 in the bulk population of SUM149 cells stably expressing an empty vector or IκBα-SR. (B) ELISA analysis showing the abundance of IL-1β, IL-6, or IL-8 in the media of SUM149 cells stably expressing empty vector or IκBα-SR. (C) Quantification of tumorspheres formed by 100 SUM149 cells expressing empty vector or IκBα-SR, followed by treatment with IL-1β, IL-6, IL-8 or vehicle control.
To determine whether IL-1β, IL-6, or IL-8 promote the self-renewal of basal-like breast cancer cells, 100 SUM149 cells expressing an empty vector were plated on low-adhesion plates in serum-free media and treated every other day for seven days with either IL-1β, IL-6, IL-8 or vehicle control. Tumorspheres were enumerated following the completion of this treatment schedule. Importantly, treatment of SUM149 cells expressing empty vector with either IL-1β or IL-6 potently increased the number of tumorspheres by four-fold and two-fold, respectively (Figure 6c). These data suggest that IL-1β and IL-6 are important modulators of the ability of basal-like breast cancer cells to self-renew. Treatment of SUM149 cells with CXCL7, the product of an NF-κB target gene, produced small increases in the ability of these cells to form tumorspheres (data not shown). Conversely, treatment with exogenous IL-8 failed to promote the formation of tumorspheres in control SUM149 cells (Figure 6c). Consistent with the results described above, addition of an IL-6 receptor antagonistic antibody suppressed tumorsphere formation approximately 25% and addition of recombinant IL-1β receptor antagonist suppressed tumorsphere formation approximately 30% (Supplementary Figure 4).
Since IL-1β and IL-6 are known NF-κB target genes (51) and were upregulated by NF-κB in SUM149 cells (Figures 6a and 6b), we investigated whether NF-κB promotes self-renewal by inducing their expression and secretion. Specifically, we tested whether treatment with IL-1β or IL-6 can rescue the ability to self-renew in SUM149 cells in which NF-κB is inhibited. To do so, the above experiment was also performed using SUM149 cells expressing IκBα-SR. As before (Figure 2B), expression of IκBα-SR reduces the ability of untreated SUM149 to form tumorspheres (Figure 6C). Notably, treatment with either IL-1β or IL-6, but not IL-8, partially rescued the ability of IκBα-SR-expressing cells to form tumorspheres (Figure 6c). These data indicate that IL-1β and IL-6 promote self-renewal of basal-like breast cancer cells downstream of NF-κB.
Discussion
Most solid tumors, including those of the breast (52), are characterized by a hierarchy of cells including a sub-population of cells that can self-renew and give rise to the differentiated cells that comprise the bulk of the tumor. These TICs promote tumor initiation, cellular motility and invasiveness, tumor recurrence, and are typically radio- and chemoresistant. As such, characterizing the functional and phenotypic differences between the bulk population of cancer cells and TICs is critical to understanding tumorigenesis and to gain insight into new approaches for cancer therapy. The transcription factor NF-κB is widely implicated in a variety of oncogenic mechanisms in both hematologic malignancies as well as solid tumors, including cancer cell proliferation, survival, and metastasis (18, 19). Here, we have shown the involvement of NF-κB in promoting TICs in a basal-like breast cancer cell line, SUM149, and two claudin-low lines, MDA-MB231 and SUM159. Interestingly, both canonical and non-canonical NF-κB appear to be important in promoting TICs in these cancer cell lines. It will be interesting to determine if distinct, or overlapping functions of these two pathways are operative in the maintenance of TICs. Work by others has indicated the association of NF-κB activity with other TICs. For example, prostate cancer TICs exhibit increased canonical NF-κB activity (53). Also, published work indicates that canonical NF-κB signaling is important in TICs in the HER2+ breast tumor subtype (25, 26, 54). One study utilized inhibitors which are not specific to NF-κB to suggest an involvement of NF-κB in promoting MCF7 breast cancer tumorspheres (55).
We propose that NF-κB promotes the function of TICs through several mechanisms. First, NF-κB may promote TIC self-renewal by stimulating EMT. Second, NF-κB promotes TICs by stimulating the expression of cytokines, such as IL-1β and IL-6. Notably, IL-6 has been implicated in the induction of EMT in breast cancer cells (56). Several reports support the finding that IL-6 promotes the function of TICs downstream of NF-κB. MCF10A cells transformed by Src gain the ability to form tumorspheres in a manner that depends on expression of IL-6 (29). Additionally, it was found that treatment of a claudin-low breast cancer cell line with IL-6 increased the proportion of TICs (50). That NF-κB upregulates the expression of many cytokines, and correspondingly can be activated downstream of these cytokines, indicates an important mechanism for sustaining NF-κB activation and promoting its TIC self-renewal properties in certain cancers. Additional roles for NF-κB in TICs likely include the upregulation of other key genes. Given the potential benefit of targeting TICs in breast cancer patients, the identification of NF-κB as a key regulator of TICs in basal-like and claudin-low breast cancer cells, along with previous work studying NF-κB in Her2+ TICs, represents a significant opportunity for the development of more effective chemotherapeutics for breast cancer.
Materials and Methods
Cell culture and reagents
SUM149, MDA-MB231, SUM159, and MCF10A cells were maintained as described (Supplementary Materials). Details regarding shRNA constructs and antibodies are also found in Supplementary Materials.
Tumorsphere formation assay
Cells growing adherently in serum-containing media were trypsinized with TrypLE Select (Invitrogen) to generate a single cell solution and then enumerated using a hemocytometer. Subsequently, 100 cells per well were plated in 3 mL of Mammocult© media (Stem Cell Technologies) on 6-well low-adhesion plates (Corning). Cells were treated with compound A, TGFβ, cytokines, or vehicle controls as described in the text. The number of tumorspheres formed per well were counted visually. Cell density is a critical parameter in the tumorsphere formation assay and cells may aggregate if cell density is too high (28). As such, we ensured that a disperse, low density (100 cells in 3 mL of media) solution of cells was prepared for each tumorsphere formation assay to avoid cell aggregation. Furthermore, plates were not moved during the growth period, to avoid cell aggregation. These precautions were taken to ensure the clonality of tumorspheres formed during this assay (28).
Immunoblotting
FACS
Cell sorting was performed using a Beckman-Coulter (Dako) CyAn and FlowJo software. See Supplementary Materials.
Isolation of TICs
Cells were trypsinized using TrypLE Select (Invitrogen) and dissociated by incubation in Accutase (Invitrogen) for 15 minutes at 37°C. The resulting cell solution was passed through a pre-separation filter (Miltenyi Biotec) to generate a single cell suspension. Cells were then incubated with 100 µL of Dead Cell Removal microbeads (Miltenyi Biotec) per 107 cells for 15 minutes at room temperature. Subsequently, the cell and microbead solution was resuspended in 20 mL MACS buffer (Miltenyi Biotec) passed through an LS column (Miltenyi Biotec) that was pre-moistened with MACS buffer and placed in a magnetized field. Live cells from the eluate were collected. Next, the resulting live cells were resuspended in 100 µL of MACS buffer per 107 cells and incubated with 75 µL CD44 microbeads (Miltenyi Biotec) and 75 µL FcR blocking reagent (Miltenyi Biotec) per 107 cells for 30 minutes at 4°C. Subsequently, the cell and microbead solution was resuspended in 20 mL MACS buffer (Miltenyi Biotec) passed through a LS column (Miltenyi Biotec) that was pre-moistened with MACS buffer and placed in a magnetic field. The CD44− cells in the eluate were collected, then the LS column was removed from the magnetized field, and the CD44+ cells were collected from the column in 5 mL MACS buffer.
MTT
The MTT assays were performed as previously described (57) using CellTiter cell viability reagent (Promega). See Supplementary Materials.
Enzyme-linked immunosorbent assay (ELISA)
ELISA kits (BD Biosciences) were utilized according to the manufacturer’s instructions.
Quantitative real-time PCR
Real-time PCR was performed and analyzed as previously described (58) using Taqman Gene Expression Assay primer-probe sets from Applied Biosystems.
Xenograft tumor formation
Cells were trypsinized with TrypLE Select (Invitrogen), a single cell solution generated, and the cells were enumerated using a hemocytometer. Cell solutions were generated in 50:50 media:Matrigel (BD Biosciences) at concentrations of 106 or 102 cells/100 µL. 100 µL of the resulting solutions were orthotopically injected into the mammary fat pad of athymic nude-Foxn1nu mice (Harlan Laboratories). Injection of each cell solution was repeated in triplicate. All murine studies were conducted in accordance with guidelines from the UNC Institutional Animal Care and Use Committee on approved protocol 08-266.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Funding We would like to thank members of the Baldwin lab for helpful discussions and comments. Drs. Megan Kendellen and Jennifer Bradford were supported by a T32 National Research Service Award (5-T32-CA009156-37) awarded to the Lineberger Comprehensive Cancer Center at the University of North Carolina at Chapel Hill. Dr. Megan Kendellen was also supported by an F32 National Research Service Award (1F32CA153439-01) from the National Institutes of Health and a Post-doctoral Fellowship from the American Cancer Society (120296-PF-11-093-01-DDC). Dr. Jennifer Bradford was also supported by an F32 National Research Service Award (1F32CA162628-01) from the National Institutes of Health. Research support was provided by NCI grants CA138937 and CA73756 and by Department of Defense grant BC073048. Additional research support was provided by the Samuel Waxman Cancer Research Foundation.
Abbreviations
- NF-κB
nuclear factor kappa B
- IκB
inhibitor of kappa B
- IKK
IκB kinase
- EMT
epithelial-mesenchymal transition
- TIC
tumor-initiating cell
- IL-6
interleukin 6
- IL-8
interleukin 8
- IL-1β
interleukin 1 beta
- TGFβ
transforming growth factor beta
Footnotes
Competing Interests/Conflicts: The authors declare that they have no competing or conflicting interests.
Author Contributions: Dr. Megan Kendellen performed experiments and wrote the manuscript. Dr. Jennifer Bradford performed key experiments and edited the manuscript. Dr. Cortney Lawrence performed key experiments. Kelly Clark performed the tumor xenograft studies. Dr. Albert Baldwin edited the manuscript.
Contributor Information
Megan F. Kendellen, Email: megankendellen@gmail.com.
Jennifer W. Bradford, Email: jennifer_bradford@med.unc.edu.
Cortney L. Lawrence, Email: cllawren@email.unc.edu.
Kelly S. Clark, Email: clarkks@med.unc.edu.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Charafe-Jauffret E, Ginestier C, Birnbaum D. Breast cancer stem cells: tools and models to rely on. BMC Cancer. 2009;9:202. doi: 10.1186/1471-2407-9-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 4.Croker AK, Allan AL. Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med. 2008;2:374–390. doi: 10.1111/j.1582-4934.2007.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawson JC, Blatch GL, Edkins AL. Cancer stem cells in breast cancer and metastasis. Breast Cancer Res Treat. 2009;2:241–254. doi: 10.1007/s10549-009-0524-9. [DOI] [PubMed] [Google Scholar]
- 6.Abraham BK, Fritz P, McClellan M, Hauptvogel P, Athelogou M, Brauch H. Prevalence of CD44+/CD24-/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin Cancer Res. 2005;3:1154–1159. [PubMed] [Google Scholar]
- 7.Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;24:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 8.Hambardzumyan D, Squatrito M, Holland EC. Radiation resistance and stem-like cells in brain tumors. Cancer Cell. 2006;6:454–456. doi: 10.1016/j.ccr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;9:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 10.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;33:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee HE, Kim JH, Kim YJ, Choi SY, Kim SW, Kang E, et al. An increase in cancer stem cell population after primary systemic therapy is a poor prognostic factor in breast cancer. Br J Cancer. 2011;11:1730–1738. doi: 10.1038/bjc.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;4:1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K, et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;3 doi: 10.1186/bcr2108. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakshatri H, Srour EF, Badve S. Breast cancer stem cells and intrinsic subtypes: controversies rag on. Curr Stem Cell Res Ther. 2009;1:50–60. doi: 10.2174/157488809787169110. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109:81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 16.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 17.Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, et al. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J Immunol. 2004;10:6336–6344. doi: 10.4049/jimmunol.172.10.6336. [DOI] [PubMed] [Google Scholar]
- 18.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;51:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 19.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;7092:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 20.Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS., Jr. Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene. 2000;9:1123–1131. doi: 10.1038/sj.onc.1203412. [DOI] [PubMed] [Google Scholar]
- 21.Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, et al. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;12:2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr, Sledge GW., Jr. Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol. 1997;7:3629–3639. doi: 10.1128/mcb.17.7.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakshatri H, Goulet RJ., Jr. NF-kappaB and breast cancer. Curr Probl Cancer. 2002;5:282–309. doi: 10.1067/mcn.2002.129977. [DOI] [PubMed] [Google Scholar]
- 24.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, et al. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;27:10137–10142. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci U S A. 2007;40:15852–15857. doi: 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu M, Sakamaki T, Casimiro MC, Willmarth NE, Quong AA, Ju X, et al. The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. 2010;24:10464–10473. doi: 10.1158/0008-5472.CAN-10-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi N, Ito T, Azuma S, Ito E, Honma R, Yanagisawa Y, et al. Constitutive activation of nuclear factor-kappaB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 2009;9:1668–1674. doi: 10.1111/j.1349-7006.2009.01228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pastrana E, Silva-Vargas V, Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011;5:486–498. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;4:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziegelbauer K, Gantner F, Lukacs NW, Berlin A, Fuchikami K, Niki T, et al. A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br J Pharmacol. 2005;2:178–192. doi: 10.1038/sj.bjp.0706176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;1:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;5 doi: 10.1186/bcr2635. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merkhofer EC, Cogswell P, Baldwin AS. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene. 2010;8:1238–1248. doi: 10.1038/onc.2009.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;7:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A. 2004;14:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;4:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;8 doi: 10.1371/journal.pone.0002888. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, et al. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006;5 doi: 10.1186/bcr1610. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009;4 doi: 10.1186/bcr2333. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;5:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;47:6120–6130. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;2 doi: 10.1186/bcr1982. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storci G, Sansone P, Mari S, D'Uva G, Tavolari S, Guarnieri T, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol. 2010;3:682–691. doi: 10.1002/jcp.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;2 doi: 10.1186/bcr1982. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;6844:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 46.Yu Y, Ge N, Xie M, Sun W, Burlingame S, Pass AK, et al. Phosphorylation of Thr-178 and Thr-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NFkappaB and AP-1 activation as well as IL-6 gene expression. J Biol Chem. 2008;36:24497–24505. doi: 10.1074/jbc.M802825200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;6:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;4:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;2:485–497. doi: 10.1172/JCI39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast Cancer Stem Cells Are Regulated by Mesenchymal Stem Cells through Cytokine Networks. Cancer Res. 2011;2:614–624. doi: 10.1158/0008-5472.CAN-10-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 52.Fillmore C, Kuperwasser C. Human breast cancer stem cell markers CD44 and CD24: enriching for cells with functional properties in mice or in man? Breast Cancer Res. 2007;3 doi: 10.1186/bcr1673. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun. 2011;2 doi: 10.1038/ncomms1159. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhat-Nakshatri P, Appaiah H, Ballas C, Pick-Franke P, Goulet R, Jr, Badve S, et al. SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24- phenotype. BMC Cancer. 2010;10 doi: 10.1186/1471-2407-10-411. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou J, Zhang H, Gu P, Bai J, Margolick JB, Zhang Y. NF-kappaB pathway inhibitors preferentially inhibit breast cancer stem-like cells. Breast Cancer Res Treat. 2008;3:419–427. doi: 10.1007/s10549-007-9798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;33:2940–2947. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilson W, 3rd, Baldwin AS. Maintenance of constitutive IkappaB kinase activity by glycogen synthase kinase-3alpha/beta in pancreatic cancer. Cancer Res. 2008;19:8156–8163. doi: 10.1158/0008-5472.CAN-08-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steinbrecher KA, Wilson W, 3rd, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol. 2005;19:8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.