

Abstract
Conventional laser scanning microscopy for multiple fluorescent stains can be a useful tool if the problems of autofluorescence and cross-talk are eliminated. The technique of spectral imaging was employed to unmix five different fluorophores – ranging in emission from 435 to 665 nm – applied to a Pseudomonas aeruginosa biofilm with overlapping spectra and which was not possible using traditional channel mode operation. Using lambda scanning and linear unmixing, the five fluorophores could be distinguished with regions of differentiation apparent.
Keywords: Biofilm, Spectral imaging, Pseudomonas aeruginosa
1. Introduction
The use of laser scanning confocal microscopy in the fields of biomedical and biological sciences has become widespread as the cost of the technology becomes more nominal and its applications more versatile. The use of multiple fluorophores to distinguish features of tissues, cells and bacteria has been facilitated by the availability of multi-track filters and commercial fluorescent dyes and proteins with high specificity and sensitivity. However, one fundamental issue is that of spectral cross-talk (also known as bleed-through or cross-bleed) where emission from one fluorophore is detected on a second filter set or photomultiplier detection channel that is designated for another fluorophores emission. One possible culprit is that single fluorophore adsorption and emission spectra on the chosen specimen can differ from the manufacturer’s specifications, which may result in overlapping spectra. This issue can lead to deceptive results and complications in image analysis if not detected. Autofluorescence can also contribute to spectral cross-talk when the specimen exhibits natural fluorescent properties that are not taken into consideration when comprehensively analyzing the fluorescence intensities of regions of interest (ROI). In addition, consistent microscope settings, such as gain, concentration of dye and similarity of laser emission intensities between fluorophores are critical and often overlooked factors when choosing fluorophores for combination staining. Inadequate evaluation and statement of the settings and controls used in published reports impedes the ability of other laboratories to interpret and reproduce data.
Laser scanning confocal microscopy has been a very useful tool in characterizing biofilm structure. Biofilms are complex communities of bacteria, either single species or polymicrobial, with distinct regions or structures. Single fluorophore analysis of biofilms grown in flow cells has been successful in detailing features such as macrocolony mushroom-like projections (Klayman, et al., 2008; Bridier, et al., 2010; Yang, et al., 2011a). Dual staining can be used for metabolic activity analysis in mature biofilms (Baum, et al., 2009; Yang, et al., 2011b). Multiple fluorophores have been used to characterize various bacterial communities. For example, biofilm floc characterization from drinking water treatment plants via independent subsequent scans of three fluorophores to differentiate DNA, alpha-polysaccharides and protein (Sun, et al., 2011); cell clustering in flow cell biofilms using two general protein and a DNA stain (Stewart, et al., 2007); biofilm flocs from wastewater treatment plants using fluorophores for polysaccharides, DNA and protein (Nosyk, et al., 2008); and biofilm granules formed in aerobic batch cultures using fluorophores for proteins, DNA, lipids and polysaccharides (Chen, et al., 2007).
In this study, a panel of five fluorophores were evaluated to label DNA, protein, lipids and two types of extracellular polysaccharides on a Pseudomonas aeruginosa biofilm grown under low shear conditions. The biofilm was first imaged using standard channel mode then spectral imaging was carried out to eliminate spectral cross-talk. Different combinations of fluorophores along with different mounting media were tested to obtain the clear images without saturation.
2. Materials and Methods
2.1 Biofilm samples
Pseudomonas aeruginosa strain 14 (PA14) was grown overnight in 5 mL of Luria-Bertani (LB) broth. The culture was then diluted in fresh LB broth to an optical density (OD) of 0.05 and placed in a high aspect ratio vessel (HARV) (Synthecon, TX, USA) with no headspace and incubated at 37°C with a rotation rate of 25 rpm for a period of 24 – 48 hours. Suspended biofilms were collected and rinsed in sterile phosphate buffered saline (PBS) three times and placed in 4% paraformaldehyde solution overnight at 4°C. After overnight fixation, the biofilm was then placed in a cryo-mould with Optimal Cutting Temperature (OCT) solution and allowed to equilibrate overnight. The specimen was then frozen at −80°C for a period of 48 hours before being sectioned. Sectioning was carried out on a Microm Cryostat (Thermo Scientific, Walldorf, Germany) in the Microscopy Imaging Center at the University of Vermont and 10 μm sections were affixed to Superfrost+ Gold slides (Fisher Scientific, Waltham, MA, USA) and stored at −20°C.
2.2 Staining procedure
Stains were selected using Fluorescence SpectraViewer (Invitrogen) for their compatible emission spectra. SYTO 9, SYPRO Ruby, DiD and Concanavalin A-Tetramethylrhodamine conjugate (ConA-TMR) were purchased from Invitrogen (Carlsbad, CA, USA). Calcofluor White M2R (CFW) and Fluorescein (FITC) were purchased from Sigma (St Louis, MO, USA).
Frozen sections were brought to room temperature and the OCT removed with three PBS washes. Three sections representing the top, middle and bottom of each biofilm sample were chosen and were stained simultaneously. All stains were applied in a volume of 100 μL and incubated in a dark environment. In between stains, sections were rinsed with 1 mL sterile PBS, unless stated otherwise. SYTO 9 (600 μg L−1) was applied to the section for 15 min at room temperature to stain DNA. ConA-TMR (500 mg L−1) was applied to the section and incubated for 30 min at room temperature to bind to α-mannopyranosyl and α-glucopyranosyl residues. Next, CFW (100 mg L−1) was added to the specimen and left for 30 min at room temperature to stain β-polysaccharides. Subsequently, the lipid stain DiD (263 mg L−1) was incubated with the specimen at 37°C for 30 min. For the final protein stain, the specimen was rinsed in milliQ water and incubated for 30 min at room temperature in SYPRO Ruby (ready-to-use 1× solution) or for 60 min at room temperature in FITC solution (50 mg L−1). Excess liquid was removed from the specimens and they were mounted in Vectashield (Vector Laboratories, Inc., Burlingame, CA, USA) or ProLong Gold Antifade Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
2.3 Fluorescent microscopy and image analysis
The structure of the biofilm was visualized using confocal laser scanning with a Zeiss LSM 510 META Laser Scanning Microscope (Carl Zeiss MicroImaging, LCC, Thornwood, NY, USA). Channel mode visualization was done using the 63× (1.4 NA) objective with oil immersion. Spectral imaging of the biofilm utilized a 25× (0.7 NA) objective with oil immersion and visualized with Zeiss AIM Image software. During initial image collection, confocal tiles were obtained using channel mode (4-track: 405/488/543/633 nm excitation). After cross-talk was observed during channel mode, lambda scanning settings were implemented (405/488/543 nm excitation with 405 nm at 6.4%, 488 nm at 0.9%, 543 nm 100% strength; Gain: 631; Amplifier Gain: 1; Amplifier Offset: 0.02; Pinhole 92 μm; Filters: 411–754, Step: 10.7 nm) and individual spectra were used for unmixing. The use of the 633 nm laser was not required for excitation of the DiD stain as adequate emission was detected using the 543 nm laser at full strength. Configuration settings were established from the singly stained controls and then applied to all the subsequent imaging sessions so there was comparable consistency in the imaging process.
Each stained section was initially examined using a far red filter to avoid photo-bleaching the stained section. Full spectrum lambda tile scans (an array of single images presented as one image) of the ROI and surrounding area were taken, and then the ROI would be extracted as an individual single image. A minimum of 25 single fields of views were captured for each stained section and displayed as a tiled array. Consecutive sections would also be stained and examined to confirm the ROI was present in multiple sections of the biofilm.
3. Results
3.1 Channel mode scanning resulted in cross-talk between fluorophores used on a Pseudomonas aeruginosa biofilm
The five fluorophores originally chosen for this study were spectrally unique with their peaks separated by a minimum of 20 nm according to the published spectral curves. However, during optimization of the channel mode settings it was determined that there was cross-talk occurring between SYTO 9 and SYPRO Ruby (Figure 1A). When singly stained specimens were excited using a 488 nm laser, SYTO 9 emission (approximately 500 nm) was detected in the SYPRO Ruby filter which was set to detect only an emission spectrum of 604 – 754 nm. When the same specimen was scanned using lambda mode, the detection filters after 577 nm did not have substantial intensity to attribute to the emission being displayed during a channel mode scan (Figure 1B). The high intensity of the emission being detected in the SYPRO Ruby channel mode did not alter with a reduction in the laser power from 10% to 6%. During configuration optimization using the META detector, adjustment of the dichroic beam splitter to divert all emissions above 610 nm from the SYTO 9 dedicated detector did not eliminate this cross-talk from appearing. DiD also demonstrated cross-talk with other fluorophores such as ConA-TMR and SYTO 9, even though the emission from the DiD is in the far red region of the spectrum (data not shown).
Fig 1.
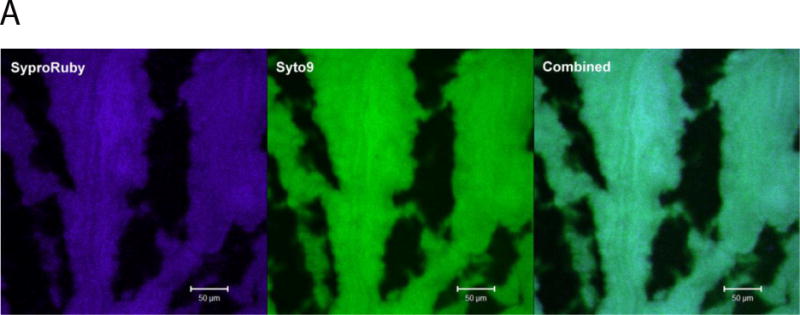

Autofluorescence was investigated as a possible cause as pseudomonads produce pigments that have natural fluorescent properties such as pyoverdin (reviewed in (Meyer, 2000)). PA14 fluorescence was quenched by using paraformaldehyde solution to fix the biofilms overnight. Unstained specimens that were mounted in either Vectashield or ProLong Antifade Reagent, showed no significant autofluorescence in any channels when excited by all 4 lasers (405/488/543/633nm). The minimal autofluorescence that was detected during this scan was then used as a reference spectrum that was subtracted from the lambda spectra during linear unmixing.
3.2 Assessing the importance of calibration and optimization of staining and mounting procedures
The Zeiss META detector is regularly calibrated using FocalCheck fluorescence microscope test slides (Invitrogen, F36913) which evaluate the sensitivity of the 10.7 nm bandwidth detection and its ability to differentiate overlapping spectra. This is done by focusing on spectral check beads, 6 μm in diameter, which has a mixture of eight reference beads ranging in emission from 511 – 692 nm and four ring/core beads ranging in emission from 511 – 676 nm. The routine maintenance and calibration eliminated the META detector as a source of the cross-talk.
Each individual fluorophore was optimized so that minimal saturation could be achieved under identical settings even though the extinction coefficient varies between the selected fluorophores (Table 1). CFW proved to be difficult to optimize even though its low extinction coefficient allowed higher concentrations to be used for staining. Due to the nature of the specimen, there were large amounts of beta-polysaccharides in a few localized areas which resulted in some oversaturation, however use of 100 mg L−1 of CFW resulted in the entire specimen being stained adequately. The protein stain SYPRO Ruby was limited by its ready-to-use formulation, consequently impeding its optimization. Increasing the incubation time for SYPRO Ruby did result in higher emission signals from the specimen, but it was not sufficient to continue using it for spectral imaging. Other protein-specific fluorophores such as Pacific Orange (Invitrogen, Carlsbad, CA, USA) and Dapoxyl (Invitrogen, Carlsbad, CA, USA) were both tested as replacements; however their emission spectra were not compatible with the existing panel of fluorophores. Due to the chosen fluorophores spectra, FITC was chosen and its optimization led to a concentration of 50 mg L−1 being used for all specimens.
TABLE 1.
Fluorophores used in multiple staining protocol
| Fluorophore | Target molecule | Excitation (nm) | Emission (nm) | Extinction coefficient (cm−1 M−1) | References |
|---|---|---|---|---|---|
| SYTO 9 | DNA | 485 | 498 | >50,000a | (Strathmann, et al., 2002; Stocks, 2004; Hangland, 2005) |
| FITC | Protein | 492 | 518 | 88,000 | (Wilderspin and Green, 1983; Hangland, 2005) |
| SYPRO Ruby | Protein | 450 | 610 | Not knownb | (Berggren, et al., 2000; Hangland, 2005) |
| Concanavalin A with Tetramethylrhodamine | Alpha-polysaccharides | 555 | 580 | 99,000 | (Strathmann, et al., 2002; Hangland, 2005) |
| Calcofluor White M2R | Beta-polysaccharides | 365 | 435 | 19,200 | (Chen, et al., 2007) |
| DiD | Cell membranes and lipids | 644 | 665 | 16,250 | (Hangland, 2005) |
Range given in manufacturer’s product information
Manufacturer’s proprietary knowledge
The mounting media Vectashield was replaced with ProLong Gold Antifade Reagent as the delicate specimens would collapse during imaging. This resulted in a more consistent imaging of the green and far-red fluorophores, more stable emissions and a great reduction in photo-bleaching. It also changed one of the spectral curves of ConA-TMR which influences the linear unmixing. The spectral peak shifted from 577 nm in the Vectashield mounting media to 588 nm in the ProLong Gold Antifade Reagent both of which are within 10 nm of the manufacturer’s published peak of 580 nm.
3.4 Lambda scanning and spectral unmixing differentiated regions of the biofilm
The main benefit of spectral unmixing is that each pixel is analyzed and compared to pre-determined spectral emission curves from similar specimens stained singly with each fluorophore which leads to enhanced interpretation of the image. For this study, background fluorescence was subtracted during the linear unmixing. The application of spectral imaging successfully separated the five fluorophores and removed the minimal autofluorescence from the specimen. In the biofilm specimens, the production of alpha and beta polysaccharides is shown to be more concentrated around the edges of the extensions of the biofilm (Figure 2) with the production of beta polysaccharides (Figure 2A) being more concentrated in the outer edges. The alpha polysaccharides (Figure 2B) were found in similar areas but were not as extensive as the beta polysaccharides. Spectral imaging was sensitive enough to also detect low levels of accumulated lipids (Figure 2C) amongst the abundant levels of DNA (Figure 2D) and protein (Figure 2E). Accumulation of each of the staining targets could be found in multiple regions of the stained specimen and subsequent specimens from different biofilm samples.
Fig 2.
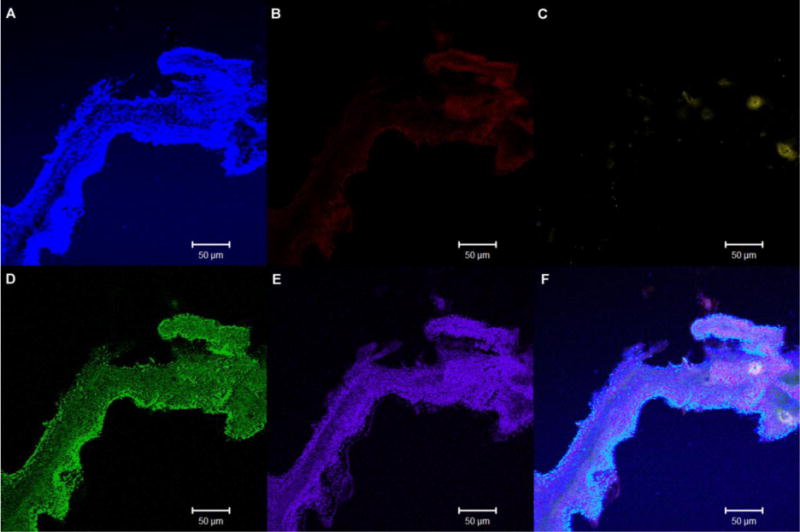
4. Conclusions
Conventional laser scanning microscopy uses filters with a relatively broad bandwidth to detect fluorescent emissions. This is particularly useful when the specimen has been stained with a single fluorophore and autofluorescence is minimal. However, when applied to a complex specimen that has been stained with a panel of fluorophores, image interpretation can be challenging. Using multiple fluorophores to highlight specific targets has the advantage of showing all the features in one scan of the specimen and has been used for aerobic granules (Chen, et al., 2007). When using a similar staining procedure on a Pseudomonas aeruginosa biofilm, there was cross-talk between channels for two of the five fluorophores when using channel mode; cross-talk was absent in lambda scanning mode (Figure 1A).
Using individually-stained specimens assisted in determining which filters were detecting phantom emission that was not being detected in lambda mode (Figure 1B). Exposing a set of individually-stained specimens to the full scanning parameters for the entire fluorophore panel may be time-consuming; however, it provides validation that each fluorophores spectral curve on the specimen is valid, accurate and its emission unique to its target. Each microscope is unique, which may result in the published spectra being slightly altered; hence, optimization and calibration controls need to be carried out with each new specimen and panel of fluorophores so that the results are interpreted correctly. We could not determine the cause of cross-talk in channel mode as scanning the singly stained SYTO 9 specimen in lambda mode (Figure 1B) did not detect any significant emission in the same wavelengths of SYPRO Ruby emission. The effect of changing mounting media on the fluorophores spectral curves was also originally unknown in this system of fluorophores. Only one spectral peak, ConA-TMR, was shifted. Published studies have, however, reported on the anti-fade properties of such mounting media and their compatibility with the commonly used fluorophores (Krenik, et al., 1989; Longin, et al., 1993; Florijn, et al., 1995; Ono, et al., 2001; Farshid, et al., 2010; Malkani and Schmid, 2011).
A key factor to the success of an imaging procedure is to fully characterize the components involved. In an ideal world, the fluorophores would all have similar extinction coefficients, the specimen would be uniformly stained and the microscope would have a single set of parameters that would image the specimen. However, each microscope, fluorophore and specimen is unique and the interaction between all three is complex (reviewed in (Berg, 2004)). Areas of misinterpretation and technique modifications have been reported in the literature for other complex specimens, such as brain tissue (Ito, et al., 2003), plant cells (Gray, et al., 1999; Berg, 2004) and embryonic tissue (Germroth, et al., 1995). In this study, we have shown that through the careful selection of multiple fluorophores, optimization of the staining protocol, use of appropriate controls, and utilization of spectral imaging with linear unmixing, that multiple fluorophore staining of a mature P. aeruginosa biofilm, can generate reproducible and reliable image data.
Highlights.
Five fluorophores on a biofilm specimen can be successfully unmixed.
We examine spectral effects of different mounting media.
Thorough examination of settings and controls are required for confocal images.
Acknowledgments
The study was supported by a NASA grant NNX09AO60A and NIH grant P20 RR021905-06. The project described in this article was supported by Award Number 1S10RR019246 from the National Center for Research Resources for purchase of the Zeiss 510 META confocal scanning laser microscope.
We thank Dr. Douglas Taatjes, Director of the Microscopy Imaging Center, University of Vermont for his critical comments on the manuscript. We thank Janet Schwarz, Microscopy Imaging Center, University of Vermont and Robert Almstrand at the University of Gothenburg, Gothenburg, Sweden for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baum MM, et al. Characterization of structures in biofilms formed by a Pseudomonas fluorescens isolated from soil. BMC Microbiol. 2009;9:103. doi: 10.1186/1471-2180-9-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg RH. Evaluation of spectral imaging for plant cell analysis. J Microsc. 2004;214:174–81. doi: 10.1111/j.0022-2720.2004.01347.x. [DOI] [PubMed] [Google Scholar]
- Berggren K, et al. Background-free, high sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gels using a luminescent ruthenium complex. Electrophoresis. 2000;21:2509–2521. doi: 10.1002/1522-2683(20000701)21:12<2509::AID-ELPS2509>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Bridier A, et al. The biofilm architecture of sixty opportunistic pathogens deciphered using a high throughput CLSM method. J Microbiol Methods. 2010;82:64–70. doi: 10.1016/j.mimet.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chen MY, et al. Staining of extracellular polymeric substances and cells in bioaggregates. Appl Microbiol Biotechnol. 2007;75:467–74. doi: 10.1007/s00253-006-0816-5. [DOI] [PubMed] [Google Scholar]
- Farshid G, et al. Some mounting media can lead to rapid fading of SISH signals in breast cancer. Pathology. 2010;42:481–3. doi: 10.3109/00313025.2010.494294. [DOI] [PubMed] [Google Scholar]
- Florijn RJ, et al. Analysis of antifading reagents for fluorescence microscopy. Cytometry. 1995;19:177–82. doi: 10.1002/cyto.990190213. [DOI] [PubMed] [Google Scholar]
- Germroth PG, et al. Confocal microscopy of thick sections from acrylamide gel embedded embryos. Microsc Res Tech. 1995;30:513–20. doi: 10.1002/jemt.1070300608. [DOI] [PubMed] [Google Scholar]
- Gray JD, et al. Technical Advance: Confocal measurement of the three-dimensional size and shape of plant parenchyma cells in a developing fruit tissue. Plant J. 1999;19:229–236. doi: 10.1046/j.1365-313x.1999.00512.x. [DOI] [PubMed] [Google Scholar]
- Hangland RP. The Molecular Probes® Handbook—A Guide to Fluorescent Probes and Labeling Technologies. Carlsbad, USA: Invitrogen Corp; 2005. [Google Scholar]
- Ito K, et al. Cautionary observations on preparing and interpreting brain images using molecular biology-based staining techniques. Microsc Res Tech. 2003;62:170–86. doi: 10.1002/jemt.10369. [DOI] [PubMed] [Google Scholar]
- Klayman BJ, et al. Measurements of accumulation and displacement at the single cell cluster level in Pseudomonas aeruginosa biofilms. Environ Microbiol. 2008;10:2344–54. doi: 10.1111/j.1462-2920.2008.01660.x. [DOI] [PubMed] [Google Scholar]
- Krenik KD, et al. Comparison of antifading agents used in immunofluorescence. J Immunol Methods. 1989;117:91–7. doi: 10.1016/0022-1759(89)90122-1. [DOI] [PubMed] [Google Scholar]
- Longin A, et al. Comparison of anti-fading agents used in fluorescence microscopy: image analysis and laser confocal microscopy study. J Histochem Cytochem. 1993;41:1833–40. doi: 10.1177/41.12.8245431. [DOI] [PubMed] [Google Scholar]
- Malkani N, Schmid JA. Some secrets of fluorescent proteins: distinct bleaching in various mounting fluids and photoactivation of cyan fluorescent proteins at YFP-excitation. PLoS One. 2011;6:e18586. doi: 10.1371/journal.pone.0018586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JM. Pyoverdines: pigments, siderophores and potential taxonomic markers of fluorescent Pseudomonas species. Arch Microbiol. 2000;174:135–42. doi: 10.1007/s002030000188. [DOI] [PubMed] [Google Scholar]
- Nosyk O, et al. A standardized pre-treatment method of biofilm flocs for fluorescence microscopic characterization. J Microbiol Methods. 2008;75:449–456. doi: 10.1016/j.mimet.2008.07.024. [DOI] [PubMed] [Google Scholar]
- Ono M, et al. Quantitative comparison of anti-fading mounting media for confocal laser scanning microscopy. J Histochem Cytochem. 2001;49:305–12. doi: 10.1177/002215540104900304. [DOI] [PubMed] [Google Scholar]
- Stewart PS, et al. Observations of cell cluster hollowing in Staphylococcus epidermidis biofilms. Lett Appl Microbiol. 2007;44:454–7. doi: 10.1111/j.1472-765X.2007.02112.x. [DOI] [PubMed] [Google Scholar]
- Stocks SM. Mechanism and use of the commercially available viability stain, BacLight. Cytometry A. 2004;61:189–95. doi: 10.1002/cyto.a.20069. [DOI] [PubMed] [Google Scholar]
- Strathmann M, et al. Application of fluorescently labelled lectins for the visualization and biochemical characterization of polysaccharides in biofilms of Pseudomonas aeruginosa. J Microbiol Methods. 2002;50:237–48. doi: 10.1016/s0167-7012(02)00032-5. [DOI] [PubMed] [Google Scholar]
- Sun C, et al. Characterization of membrane biofouling at different operating conditions (flux) in drinking water treatment using confocal laser scanning microscopy (CLSM) and image analysis. J Membr Sci. 2011;382:194–201. [Google Scholar]
- Wilderspin AF, Green NM. The reaction of fluorescein isothiocyanate with thiols: a method for assay of isothiocyanates. Anal Biochem. 1983;132:449–55. doi: 10.1016/0003-2697(83)90032-5. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. Distinct roles of extracellular polymeric substances in Pseudomonas aeruginosa biofilm development. Environ Microbiol. 2011a;13:1705–17. doi: 10.1111/j.1462-2920.2011.02503.x. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. Pattern differentiation in co-culture biofilms formed by Staphylococcus aureus and Pseudomonas aeruginosa. FEMS Immunol Med Microbiol. 2011b;62:339–347. doi: 10.1111/j.1574-695X.2011.00820.x. [DOI] [PubMed] [Google Scholar]