

Abstract
Reactive oxygen species (ROS) play an important pathogenic role in the development of many diseases, including kidney disease. Major ROS generators in the glomerulus of the kidney are the p47phox-containing NAPDH oxidases NOX1 and NOX2. The cytosolic p47phox subunit is a key regulator of the assembly and function of NOX1 and NOX2 and its expression and phosphorylation are up-regulated in the course of renal injury, and have been shown to exacerbate diabetic nephropathy. However, its role in non-diabetic-mediated glomerular injury is unclear. To address this, we subjected p47phox-null mice to either adriamycin-mediated or partial renal ablation-mediated glomerular injury. Deletion of p47phox protected the mice from albuminuria and glomerulosclerosis in both injury models. Integrin α1-null mice develop more severe glomerulosclerosis than wild type mice in response to glomerular injury mainly due to increased production of ROS. Interestingly, the protective effects of p47phox knockout were more profound in p47phox/integrin α1 double knockout mice. In vitro analysis of primary mesangial cells showed that deletion of p47phox led to reduced basal levels of superoxide and collagen IV production. Thus, p47phox-dependent NADPH oxidases are a major glomerular source of ROS, contribute to kidney injury, and are potential targets for antioxidant therapy in fibrotic disease.
Keywords: glomerulosclerosis, partial renal ablation, adriamycin, integrins, reactive oxygen species, NOX
Introduction
Glomerulosclerosis, characterized by excessive extracellular matrix (ECM) deposition in the glomeruli of the kidneys, is the common final pathway of many chronic kidney diseases irrespective of their etiologies.1, 2 The development of glomerulosclerosis is modulated by multiple factors including reactive oxygen species (ROS) and integrins.3, 4
ROS are a group of active intermediate oxygen molecules produced under both physiological and pathological conditions. Although low levels of ROS are essential for physiological processes, excessive production leads to oxidative stress, which is an important mediator of pathological conditions, including kidney injury.5-8 ROS are produced during many intracellular processes and major cellular sources of ROS are the mitochondrial respiratory chain, the NOX family NADPH oxidases, the xanthine oxidase, the uncoupled endothelial nitric oxide synthase and the cytochrome P450 systems.9, 10
The NOX family NADPH oxidases are superoxide generating enzymes that catalyze electron transfer from NADPH onto molecular oxygen. The first NADPH oxidase described was the neutrophil phagocytic NADPH oxidase that contains the membrane-bound catalytic subunit gp91phox/NOX2.11 NOX2 is expressed in many different cell types and tissues including the kidney.12, 13 The phagocytic NADPH oxidase requires the assembly of five subunits to function properly: the two membrane-bound gp91phox/NOX2 and p22phox subunits and the cytosolic regulatory subunits p40phox, p47phox, p67phox. In addition, assembly of this NADPH oxidase is regulated by the balance between inactive (GDP-bound) and active (GTP-bound) small GTPase Rac. GTP-bound Rac leads to the translocation of cytosolic subunits to the membrane, association with the membrane subunits and formation of an active NOX2 containing complex.14, 15
In addition to the gp91phox/NOX2, six other catalytic subunits have been identified, including NOX1, NOX3, NOX4, NOX5, Duox1 and Duox2.11 NOX1, like NOX2, is expressed in many different tissues including the kidney12, 16 and requires p22phox and the regulatory subunit p47phox as well as active Rac for its stabilization and function. Its dependence on p22phox, however, is less stringent than that observed for NOX2.11 NOX4, originally identified as a NADPH oxidase homolog, is also highly expressed in the kidney.17, 18 It requires interactions with p22phox, but not the cytosolic organizer subunit p47phox or Rac for its stabilization and function.11 Due to its selective expression in the kidney, NOX4 has been suggested to be the major NOX in kidney and the major source of ROS in animal models of diabetic nephropathy.18, 19 However, recent studies have cast doubt on the pathological role of NOX4 in kidney injury, since NOX4-null mice showed worse glomerular and tubular kidney injury than wild type mice.20, 21 Interestingly, deleting NOX2 did not change the severity of tubulointerstitial injury in the kidney nor did it alter the phenotype of NOX4-null mice subjected to the same injury.13, 21 In contrast to the data showing no role for NOX2 in tubulointerstitial disease, p47phox-null mice have reduced kidney hypertrophy and mesangial matrix expansion in diabetic nephropathy.22-24 As p47phox affects both NOX1 and NOX2, this finding suggests that simultaneous inactivation of both these NOXs is required to alleviate diabetic nephropathy. The role of p47phox-containing NOXs in non-diabetic glomerular injury is unknown.
Integrins can also regulate the levels of ROS in both physiological and pathological conditions. Integrins are transmembrane receptors for matrix components formed by two non-covalently associated α and β subunits that combine to form 24 different heterodimers with different ligand specificity to matrix molecules.25 Upon binding to matrix, integrins initiate multiple cell signaling pathways which regulate critical cellular functions such as survival, proliferation, and matrix homeostasis.26, 27 Integrin α1β1, a major collagen binding receptor, is highly expressed on all major cell types in the glomeruli of the kidney and it has been identified as an important negative regulator of collagen synthesis.4, 28 In this context, integrin α1-null mice develop more severe glomerulosclerosis than wild type mice in response to glomerular injury and this is mainly due to increased production of pro-fibrotic ROS.29, 30 In contrast, mice lacking another collagen receptor, integrin α2β1, are protected from ROS-mediated injury.31 These findings suggest that integrin α1β1 decreases, while integrin α2β1 increases ROS production.32 The mechanism whereby integrin α1-null glomerular cells produce increased basal levels of ROS is in part due to increased growth factor receptor-mediated Rac activation.28 Thus, integrin α1-null mice and cells are predisposed to produce excessive collagen because of increased activity of Rac-dependent NOXs (NOX1 and NOX2) and they are an excellent model to define the mechanisms whereby ROS production contributes to glomerulosclerosis.
To determine the role of p47phox-dependent NOXs in non-diabetic glomerular injury, we subjected p47phox null mice as well as p47phox null mice crossed with the integrin α1-null mice to adriamycin- and partial renal ablation-mediated glomerular injury. We provide evidence that deletion of p47phox protects mice from renal-injury mediated albuminuria and glomerulosclerosis found in both injury models and these effects are more profound in mice on the integrin α1-null background. This protection is also accompanied by decreased production of free radicals, decreased activation of the EGF receptor/GTP-Rac axis and consequent decreased collagen production. Thus, our data indicate that p47phox-containing NOXs are an important source of ROS in the kidney and point to p47phox as a potential target for amelioration of albuminuria and renal fibrosis following injury.
Results
Loss of p47phox ameliorates kidney injury
To identify the role of NADPH oxidase-mediated ROS production in glomerular injury, we used p47phox-null (p47phoxKO) mice crossed onto the integrin α1-null (α1KO) mouse. This choice is dictated by the fact that integrin α1KO mice develop exacerbated glomerulosclerosis following injury due in part to increased baseline and injury-mediated ROS production.29 Glomerular injury was induced either by a single intravenous injection of adriamycin29 or by partial renal ablation.31
Mice received a single intraperitoneal injection of adriamycin (10mg/kg) and their weight was assessed at 0, 1, 2, 3 and 4 weeks after injection. As previously reported, integrin α1KO mice lost ∼ 20% of body weight at 2 and 3 weeks after adriamycin injection.29 In contrast, no body weight loss was observed in injured wild type control, p47phoxKO or p47phoxKO/integrin α1KO mice (DKO) (Fig. 1A). At 4 weeks, ∼ 50% mortality was observed in integrin α1KO mice and ∼ 20% in wild type mice, while no deaths were observed in either p47phoxKO or DKO mice. To determine whether there were differences in renal function among the various groups, we analyzed albuminuria by measuring urine albumin-to-creatinine ratio at 1 and 4 weeks after adriamycin injection. One and four weeks were chosen because at 1 week albuminuria is observed in both wild type and integrin α1KO mice, but glomerular matrix deposition and injury are evident primarily in the latter group, and at 4 weeks albuminuria decreases in both groups, but it stays more elevated (together with overall kidney damage) in the integrin α1KO group.29 One week after adriamycin injection, albuminuria was evident in both wild type and integrin α1KO mice, although it is was more prominent in the latter group (Figs. 1B, C). In contrast, significantly lower albuminuria was evident in p47phoxKO and DKO compared to injured wild type and integrin α1KO mice, respectively. Albuminuria decreased in all groups at 4 weeks after injury, although it was significantly lower in p47phoxKO and DKO mice (Figs. 1B, C). Similar results for the urine albumin-to-creatinine ratio were seen in mice post-partial renal ablation, with albuminuria in 12 weeks injured p47phoxKO or DKO mice significantly lower than that measured in injured WT and integrin α1KO mice (Suppl. Fig. 1A). Thus, p47phox deletion ameliorates both adriamycin- and partial renal ablation-induced albuminuria. To characterize and quantify the degree of adriamycin-mediated injury, the kidneys were subjected to pathological examination. Four weeks after adriamycin injection, integrin α1KO mice showed more severe glomerular and tubular injury than injured WT mice. Injury was characterized by exacerbated mesangial matrix expansion, glomerulosclerosis, tubular accumulation of proteinacious casts, and interstitial fibrosis (Figs. 2A-D). By contrast, the kidneys of injured p47phoxKO and DKO mice looked relatively normal with minimal matrix mesangial expansion, glomerulosclerosis and tubulointerstitial fibrosis (Figs. 2A-D). Similar differences in glomerular injury were seen among the different strains of mice 12 weeks after partial renal ablation (Suppl. Fig. 1B). These results show that loss of p47phox in mice that either do or do not express integrin α1β1 ameliorates adriamycin- and renal ablation-mediated glomerular injury.
Figure 1. Loss of p47phox improves adriamycin-mediated albuminuria.
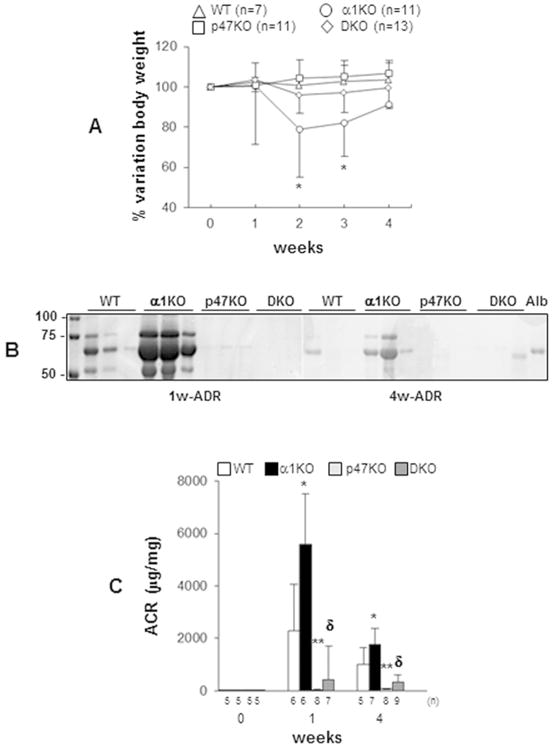
Kidney injury was induced by a single tail vein injection of adriamycin (10 mg/Kg b.w.) in wild type (WT), integrin α1KO, p47phoxKO, integrin α1KO/p47phoxKO (DKO) mice and changes in body weight (A) and urine albuminuria (B, C) were examined over time. (*) indicates significant differences (p≤0.05) between α1KO and WT, or α1KO and p47phoxKO, or α1KO and DKO. Values are the mean ± SD of the number of mice indicated. (B) SimplyBlue staining of 1 μl urine showing elevated albuminuria in adriamycin-treated integrin α1KO mice compared to WT, p47phoxKO or DKO mice. Alb = mouse serum albumin (1.25 μg/lane). (C) Urine albumin excretion expressed as albumin-to-creatinine ratio (ACR) in untreated or adriamycin-treated WT, α1KO, p47phoxKO, and DKO mice. Values represent the mean ± SD of the mice indicated. Difference between injured WT and α1KO (*) or injured WT and p47phoxKO (**) or injured α1KO and DKO (δ) mice were significant (p≤0.05).
Figure 2. Loss of p47phox improves adriamycin-induced kidney injury.
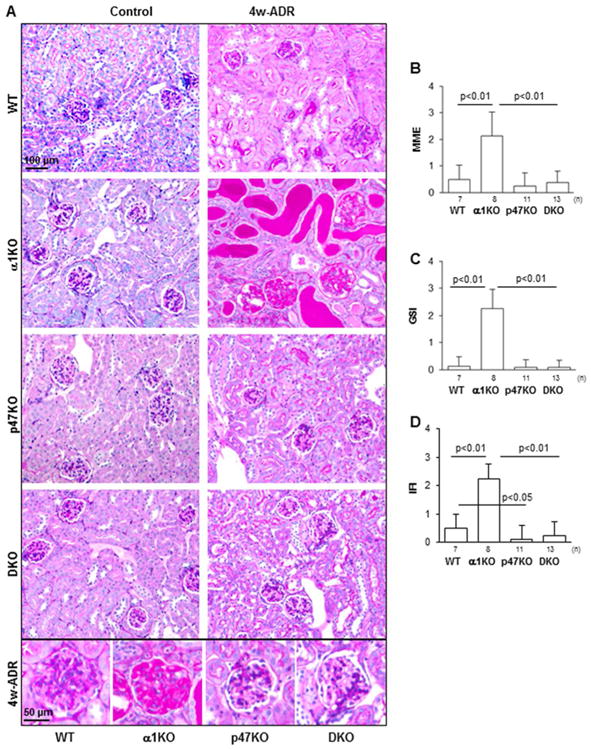
(A) Representative light micrographs of periodic-acid-Schiff (PAS) stained kidneys from uninjured (Control) or adriamycin-treated (4w-ADR) WT, α1KO, p47phoxKO, and DKO mice. Loss of p47phox rescues the severe glomerular and tubular damage observed in injured integrin α1KO mice. (B-D) Matrix mesangial expansion (MME) (B), global glomerulosclerosis index (GSI) (C), and interstitial fibrosis index (IFI) (D) were evaluated 4 weeks after adriamycin injection and scored as described in the Methods. Values represent the mean ± SD of the number of mice indicated.
Loss of p47phox ameliorates kidney fibrosis
Increased collagen deposition is a key pathological feature of glomerular injury leading to glomerulosclerosis. Trichrome staining of kidney sections from 4 week adriamycin-injured mice revealed increased accumulation of fibrillar collagen in both glomeruli and tubules of integrin α1KO mice compared to injured WT mice (Fig. 3A). In contrast, kidneys of injured p47phoxKO and DKO showed low levels of fibrillar collagen (Fig. 3A). Fibrillar collagen staining was not evident in uninjured mice independently of their genotype (Fig. 3A).
Figure 3. Loss of p47phox improves adriamycin-induced matrix deposition.
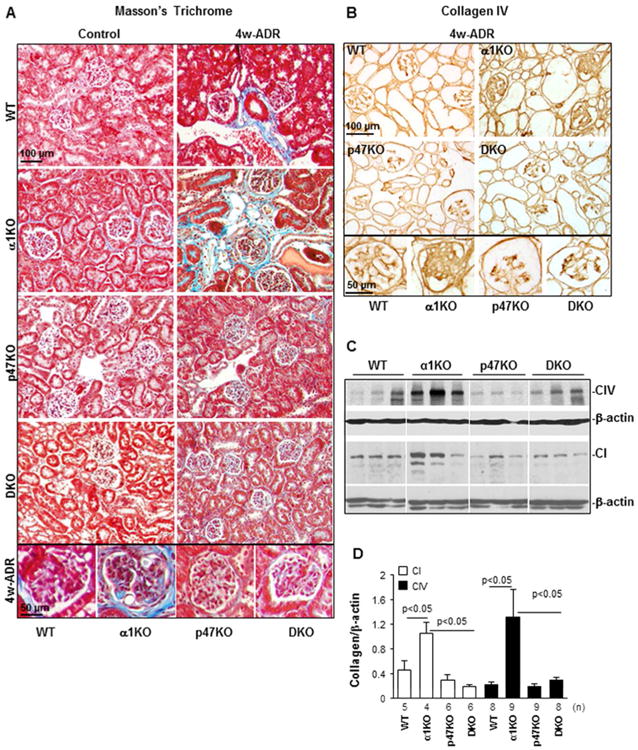
(A) Masson's Trichrome staining of kidneys from uninjured (Control) or adriamycin-treated (4w-ADR) WT, α1KO, p47phoxKO, and DKO mice. Note the presence of fibrillar collagen (blue) in both glomeruli and tubules of integrin α1KO mice which was reduced in kidneys of p47phoxKO and DKO mice. (B) Collagen IV staining of kidney sections from 4 weeks injured WT, α1KO, p47phoxKO and DKO mice. Note the high levels of glomerular collagen IV in integrin α1KO mice which were reduced in kidneys of p47phoxKO and DKO mice. (C) Equal amount of kidney lysates (20 μg/lane) from WT, α1KO, p47phoxKO, and DKO mice (n=3 shown) 4 weeks after adriamycin injection were analyzed by Western blot for levels of collagen IV and collagen I. (D) The collagen I, collagen IV and β-actin bands were analyzed by densitometry analysis and the levels of collagen IV are expressed as collagen I/β-actin or collagen IV/β-actin ratio. The values represent the mean ± SD of the number of kidneys indicated.
As collagens I and IV are the principal fibrillar and non-fibrillar collagens induced in glomerulosclerosis, we examined their expression levels in kidney either by immunohistochemistry or Western blot analysis. Increased levels of collagen IV were evident in injured integrin α1KO mice compared to wild type mice and this was independent of the type of renal injury (Figs. 3B-D, Suppl. Figs. 1C, 1D). Moreover, decreased collagen IV deposition (especially in glomeruli) was detected in both p47phoxKO and DKO mice (Figs. 3B-D, Suppl. Figs. 1C, 1D). Similar to Trichrome staining, increased levels of collagen I were evident in injured integrin α1KO mice compared to WT mice and these levels were significantly decreased in the kidneys of DKO mice (Figs. 3C, 3D). These results suggest that p47phox, at least in part, accounts for the increased injury and collagen deposition observed in the injured α1KO mice.
Loss of p47phox ameliorates adriamycin-induced oxidative stress
p47phox is a critical regulatory subunit of the NOX1 and NOX2 containing NADPH oxidases and deleting it reduces in vivo ROS production and therefore oxidative stress.33, 34 Thus, we examined markers of oxidative stress in vivo to determine whether reduced ROS production correlates with the decreased renal damage observed in the p47phoxKO background. Urinary levels of F2-isoprostane and nitrotyrosine in kidney tissues were analyzed as they are recognized markers of in vivo oxidative stress. The levels and/or expression of both markers peaked at 1 week after adriamycin injury in all groups analyzed (Figs. 4A-D). The nitrotyrosine staining in the kidneys of injured mice localized primarily to podocytes, mesangial cells, proximal tubule cells, and infiltrating cells (Fig. 4B) which have been all shown to express p47phox. Both oxidative stress markers were significantly reduced in DKO mice compared to injured integrin α1KO mice. Similarly, p47phoxKO mice showed significantly decreased oxidative stress markers in both urine and kidney tissues compared to injured WT mice (Figs. 4A-D). At 4 weeks, the overall levels of F2-isoprostane and nitrotyrosine were decreased in all four groups, although the levels of nitrotyrosine remained significantly higher in the kidneys of injured integrin α1KO mice (Figs. 4A-D).
Figure 4. Loss of p47phox improves adriamycin-induced oxidative stress.
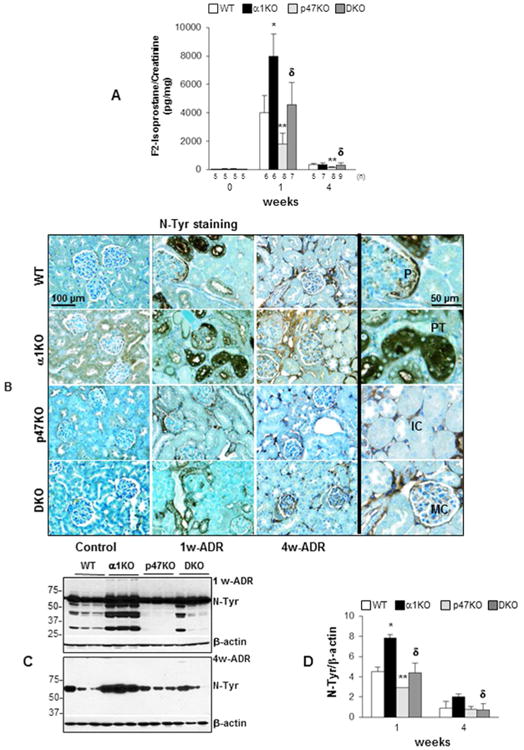
(A) The levels of urinary F2-isoprostane in the mice indicated were analyzed by ELISA before or after adriamycin injection and were expressed F2-isoprostane/urine creatinine ratio. Values are the mean ± SD of the number of mice indicated. (*), (**) and (δ) are as in Fig. 1. (B) Paraffin sections of kidneys from uninjured (Control) or adriamycin-treated (1w-ADR and 4w-ADR) WT, α1KO, p47phoxKO, or DKO mice were stained with anti-nitrotyrosine (N-Tyr) antibodies to evaluate the degree of oxidative stress-induced tyrosine nitration (brown staining). Slides were counterstained with toluidine blue (blue staining). The high-magnification images on the right show nitrotyrosine staining in infiltrating cells (IC), mesangial cells (MC), podocytes (P), and proximal tubules (PT). (C) Equal amount of kidney lysates (20 μg/lane) from WT, α1KO, p47phoxKO, and DKO mice (n=3) 1 and 4 weeks after adriamycin injection were analyzed by Western blot for levels of nitro-tyrosine. (D) The nitro-tyrosine and β-actin bands were analyzed by densitometry analysis and the levels of tyrosine nitration are expressed as N-Tyr/β-actin ratio. The values represent the mean ± SD of 3 kidneys/genotype. (*), (**) and (δ) are as in Fig. 1.
As the expression of p47phox increases in the course of renal injury and correlates with the degree of injury, we analyzed the levels of p47phox in the kidneys of uninjured (Control), 1 week and 4 weeks after adriamycin-injured mice. While no basal expression of p47phox was detected in uninjured mice, increased expression of this NADPH regulatory subunit was evident primarily in 1 week injured integrin α1KO mice and remained elevated at 4 weeks (Fig. 5A, 5B). The translocation of the p47phox subunit to the plasma membrane is a key step for the formation of an active NOX2 containing complex.14,15 Thus, we analyzed the membrane localization of the p47phox by performing Western blot analysis of membrane enriched fractions of kidneys from uninjured (control) and 4 week-adriamycin injured wild type and integrin α1KO mice. Increased levels of membrane-associated p47phox were detected in kidneys of injured integrin α1KO mice (Fig. 5C).
Figure 5. Analysis of p47phox levels and localization in kidneys from uninjured and adriamycin-injured mice.
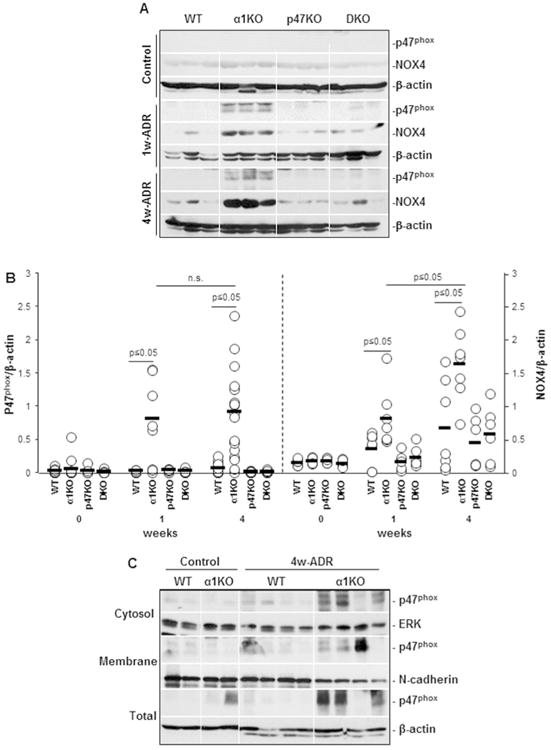
(A) Equal amount of kidney lysates (20 μg/lane) from uninjured (Control) or adriamycin-treated (1w-ADR and 4w-ADR) WT, α1KO, p47phoxKO, and DKO mice (n=3 shown) were analyzed by Western blot for levels of p47phox and NOX4. (B) The p47phox, NOX4 and β-actin bands were analyzed by densitometry and the levels of p47phox and NOX4 are expressed as p47phox/β-actin or NOX4/β-actin ratio. Open circles represent values of individual kidneys, while the bar represents the mean value. (C) Equal amount of kidney lysates (Total) membrane enriched (Membrane) and membrane deprived (Cytosol) fractions (40 μg/lane) from uninjured (Control) or adriamycin-treated (4w-ADR) WT and integrin α1KO mice (n=3 shown) were analyzed by Western blot for p47phox localization. Membranes were re-blotted with anti-ERK, anti-N-cadherin, and anti β-actin antibodies to verify the purity of various fractions.
Oxidative stress was still detected in the kidneys of adriamycin-treated DKO mice compared to p47KO mice (Fig. 4B), suggesting that in addition to p47phox other ROS generating enzymes might be responsible for the generation of nitrotyrosine in the kidneys of integrin α1KO/p47phox double KO mice. We therefore analyzed the levels of NOX4, a ROS-producing enzyme highly expressed in the kidneys and upregulated in kidney injury,35, 36 in the kidneys of control and adriamycin-treated mice. While no difference in the basal levels of NOX4 were observed in kidneys of uninjured mice, increased expression of this enzyme was observed in the kidneys of 1 and 4 week injured mice, although it was more significant in the kidneys of integrin α1KO mice (Figs. 5A, 5B). Thus p47phox and NOX4 contribute to the renal damage observed in the integrin α1KO mice.
Loss of p47phox attenuates podocyte injury and macrophage infiltration in integrin α1KO mice
Podocytes are a major target of adriamycin-induced kidney injury.37, 38 Because these cells express p47phox and NOX4 (reviewed in39) they could contribute directly to podocyte and consequent glomerular injury by generating ROS in response to injury. To evaluate whether podocyte injury/loss directly correlates with the degree of renal damage observed in injured mice, we stained kidneys sections of control and injured mice with anti-WT1 antibodies. While no difference in the number of WT1 positive cells were observed in kidneys of uninjured mice, significantly less WT1 positive cells was observed primarily in the kidneys of 1 and 4 week injured wild type and integrin α1KO mice, although it was more significant in the latter group (Figs. 6A, 6B).
Figure 6. Loss of p47phox improves adriamycin-induced podocyte loss.
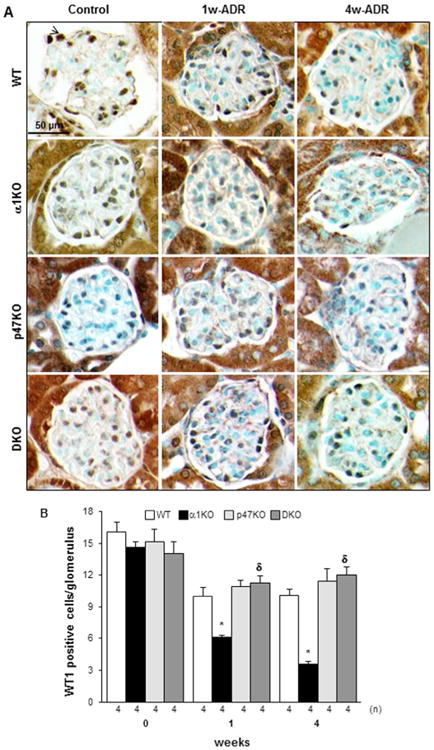
(A) WT-1 staining in kidneys from uninjured (Control) or adriamycin-treated (1w-ADR and 4w-ADR) WT, α1KO, p47phoxKO, and DKO mice. Slides were counterstained with toluidine blue (blue staining). (B) The number of WT1 positive cells (arrowhead) was evaluated in 20 randomly chosen glomeruli per kidney. The values represent the mean ± SD of the kidneys indicated. (*) and (δ) are as in Fig. 1. Note the decreased number of podocytes in injured integrin α1KO mice which was reduced in kidneys of p47phoxKO and DKO mice.
Another feature associated to adriamycin-mediated injury is macrophage infiltration starting at 2 weeks after adriamycin treatment.38 As interstitial fibrosis and collagen production are still evident in adriamycin-injured integrin α1KO mice (despite decline in albuminuria and urinary ROS) at 4 weeks, we investigated whether infiltrating macrophages could account for these fibrotic features. No difference in macrophage number was observed among the 4 groups at baseline or 1 week after ADR treatment (Figs. 7A, 7B). This is consistent with the observation that infiltrating cells become evident at 2 weeks after adriamycin treatment and the primary cells affected by adriamycin are resident cells (e.g., podocytes and mesangial cells).29, 38 In contrast, at 4 weeks a significant increase in the number of F4/80 positive cells was detected only in the kidneys of integrin α1KO mice (Figs. 7A, 7B), suggesting that, in addition to ROS and resident cells, macrophages might contribute to the late fibrotic phenotype observed in this group.
Figure 7. Loss of p47phox improves adriamycin-induced macrophage infiltration.
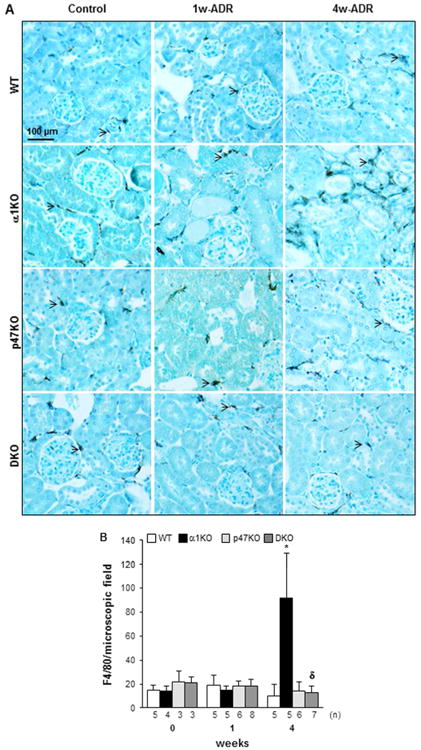
(A) F4/80 staining in kidneys from uninjured (Control) or adriamycin-treated (1w-ADR and 4w-ADR) WT, α1KO, p47phoxKO, and DKO mice. Slides were counterstained with toluidine blue (blue staining). (B) The number of macrophages (arrowhead) was evaluated in 20 randomly chosen field per kidney. The values represent the mean ± SD of the kidneys indicated. (*) and (δ) are as in Fig. 1. Note the increased number of macrophages in 4 week injured integrin α1KO mice which was reduced in kidneys of p47phoxKO and DKO mice.
Loss of p47phox leads to decreased glomerular activation of EGF receptor
We next determined the mechanism whereby loss of p47phox protects against renal injury. The EGF receptor (EGFR) contributes to the development of kidney injury through multiple mechanisms.40, 41 We showed that increased basal activation of EGFR in integrin α1KO mice contributes to kidney injury and matrix synthesis by promoting Rac-mediated activation of NADPH oxidase complex and in turn ROS production.28, 29 As ROS can potentiate the activation status of receptor tyrosine kinases42, we analyzed the levels of activated EGFR in glomeruli isolated from WT, α1KO, p47phoxKO, or DKO mice 1 week after adriamycin injection. As expected, glomeruli from injured integrin α1KO mice showed significantly higher levels of activated EGFR compared to injured WT mice, and this activation was significantly reduced in glomeruli of injured p47phoxKO and DKO mice (Figs. 8A, 8B). Thus, the p47phox/ROS axis is required for activation of EGFR in vivo.
Figure 8. Loss of p47phox improves adriamycin-mediated activation of pro-fibrotic pathways.
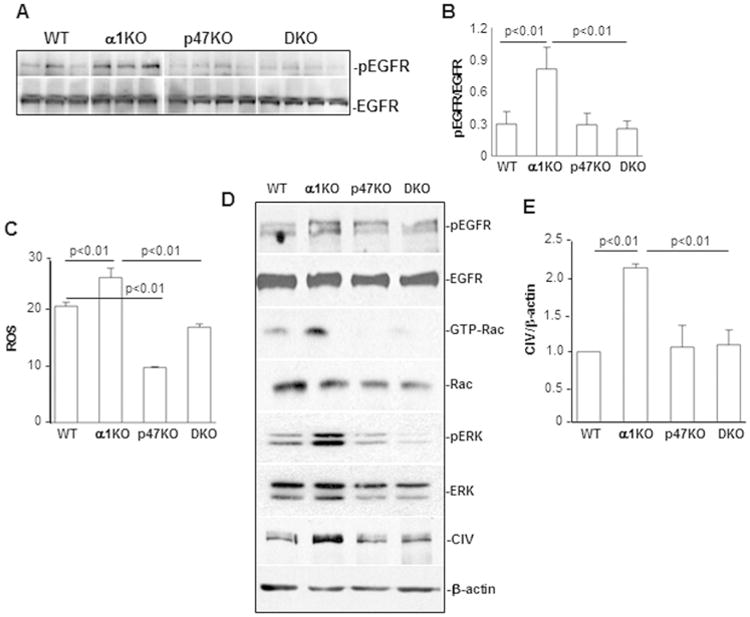
(A) Equal amount of glomerular lysates (20 μg/lane) from 1 week adriamycin-treated WT, α1KO, p47phoxKO, and DKO mice were analyzed by Western blot for levels of phosphorylated and total EGF receptor (EGFR). (B) pEGFR and EGFR bands were quantified by densitometry analysis and the levels of activated EGFR are expressed as pEGFR/EGFR ratio. Values are the mean ± SD of 3-4 mice/genotype. (C) Primary mesangial cells isolated from the mice indicated were cultured in serum free medium. Twenty-four hours later, cells were incubated with 2 μM dihydro-rhodamine and 2 hours later ROS generation was determined by FACS as described in the Methods. The level of ROS was expressed as a ratio of fluorescence intensity of cells with dihydro-rhodamine vs. that of cells without dihydro-rhodamine. Data represent the mean ± SD of 3 samples/genotypes. The same experiment was repeated twice with similar result. (D) Equal amount of cell lysates (20 μg/lane) from the serum starved cells indicated were analyzed by Western blot for the phosphorylated and total EGFR, activated and total Rac, phosphorylated and total ERK, as well as collagen IV (CIV) levels. (E) The collagen IV and β-actin bands were analyzed and expressed as described in Fig. 3. The values represent the mean ± SD of 3 independent experiments.
Crosstalk between the EGFR-Rac pathway and p47phox regulates ROS and collagen IV production in vitro
In addition to podocytes, mesangial cells are a major glomerular cell type responsible for production of ROS and collagen IV. To further dissect the mechanisms whereby p47phox regulates ROS and collagen IV production, the levels of activated EGFR, Rac, as well as the levels of ROS and collagen IV production were analyzed in mesangial cells isolated from WT, α1KO, p47phoxKO, and DKO mice. Consistent with the in vivo data, integrin α1KO mesangial cells showed significantly higher levels of activated EGFR, activated Rac (GTP-Rac), ROS and collagen IV (Figs. 8C-E) than the other cell types analyzed. In contrast, deletion of p47phox profoundly decreased activation of EGFR, GTP-Rac, ROS and collagen IV production (Figs. 8C-E).
We previously showed that integrin α1KO mesangial cells have increased basal level of activated ERK 43. This MAPK kinase is not only downstream EGFR,44-46 but also promotes ROS production by regulating the expression and/or phosphorylation of p47phox.47, 48 Therefore, we analyzed the levels of activated ERK in mesangial cells and found that, in contrast to the activated levels observed in integrin α1KO mesangial cells, deletion of p47phox resulted in decreased levels of activated ERK (Fig. 8D). These results suggest that integrin α1β1-mediated downregulation of ROS production is mainly mediated by the p47phox-dependent NADPH oxidase complex, and this complex also plays a role in regulating the activation of EGFR, Rac, ERK, and collagen IV production (Figs. 9A, 9B).
Figure 9.
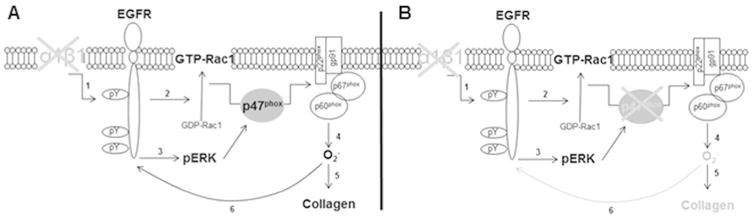
Schematic reorientation of how loss of p47phox ameliorates the fibrotic response in integrin α1KO cells or mice. (A) Loss of integrin α1β1 leads to increased activation of EGFR (1) which in turn promotes p47phox membrane translocation and the assembly of the NADPH oxidase system by activating Rac1 (2) and/or ERK (3). NADPH-generated superoxide (4) can then exert a pro-fibrotic action by promoting collagen synthesis (5) and prolonging EGFR activation (6). (B) Loss of p47phox reduces this fibrotic response by dampening ROS production (4), collagen synthesis (5), and EGFR activation (6).
Discussion
Increased ROS production or oxidative stress has been documented in various human diseases, and studies using animal models have demonstrated that increased ROS levels are an important pathogenic mechanism in the development of fibrotic injuries. The overall goal of this study was to evaluate the role of p47phox, the regulatory subunit of NOX1 and NOX2, in the development and progression of glomerular kidney disease. To do this, we used p47phox-null mice crossed to integrin α1-null mice, a model of increased ROS production and exacerbated glomerulosclerosis following injury.28, 29 Using both ROS-mediated and non-ROS mediated glomerular injury models; we provide evidence that deletion of p47phox protects mice from albuminuria and kidney fibrosis which is consistent with reduced in vivo oxidative stress in the p47phox-null background. In addition, we provide evidence that deletion of p47phox in mesangial cells leads to significantly reduced ROS and collagen IV production. Thus, our study indicates that p47phox-dependent NOXs are an important source of ROS in the kidney and suggests that p47phox can be viewed as a potential target for amelioration of albuminuria and renal fibrosis following injury.
NOX1, NOX2, and NOX4 are major sources of ROS in various organs, including the kidneys. The finding that the expression of these oxidases is upregulated in the course of fibrosis has initiated a series of studies aimed to determine their contribution to fibrotic disease. NOX4, for example, has been implicated in the initiation and progression of diabetic nephropathy.18 The finding that glucose induces upregulation of NOX4 and oxidative stress in kidney tubular cells, and this effect is prevented by inhibition of the protease ADAM17, suggests that the ADAM17/NOX4 axis is a key mediator of glucose-promoted kidney tubular injury.49 Moreover, the recent finding that inhibition of mTOR prevents NOX-4-mediated ROS generation and glomerular podocyte apoptosis in a model of type 1 diabetes, suggests that NOX4 mediates mTOR-induced podocyte instability in the course of glomerular injury.19 Consistent with a deleterious role of NOX4 protein in glomerular injury, we show that the kidneys of adriamycin-injured integrin α1KO mice have increased levels of NOX4 and p47phox levels, both of which could contribute to the severe glomerular injury observed in these mice. At present, it is unclear how loss of integrin α1β1 might lead to increased NOX4 production, although MAPK kinases have been implicated in the regulation of NOXs under physiological and pathological conditions (reviewed in50). Thus, further studies are needed to determine whether the increased ERK activation we detected in integrin α1KO mesangial cells might contribute directly to the regulation of NOX4.
Although studies indicate that activation of NOX4 plays a deleterious role in fibrotic diseases, recent studies have cast a shadow on the pathological role of NOX4 in kidney injury. In this context, NOX4-null mice show worse pathology than wild type mice following glomerular and/or tubular injury.20 Moreover, NOX4-null mice develop exaggerated cardiac contractile dysfunction, hypertrophy, and dilatation during exposure to chronic overload.51 Collectively, these results not only suggest that NOX4 might paradoxically play a protective role in stress-induced kidney injury, but also indicate that other sources of ROS-generating enzymes might play a role in disease, especially NOX1 and NOX2 complexes. p47phox-null mice have been used to investigate the role of these complexes, as well as factors able to regulate their assembly, to disease. In this regard, p47phox-null mice are protected from angiotensin-2 mediated liver fibrosis due to the inability of angiotensin-2 to promote phosphorylation and consequent activation of p47phox.52 Consistent with this finding, loss of p47phox ameliorates the susceptibility to heart disease in mice lacking the angiotensin converting enzyme 2, a negative regulator of the renin-angiotensin system, clearly indicating a positive correlation between angiotensin-2 and p47phox in fibrosis and disease.53 Despite these promising findings, it has been recently shown that loss of p47phox leads to cytoskeleton destabilization and decreased activation of kinases found in focal adhesions, thus making the heart more susceptible to overload-induced biomechanical stress.54 In addition, mice lacking p47phox show increased neutrophil alveolitis and exacerbated lung injury following LPS treatment due to enhanced NF-κB activity.34 All together these results indicate that the beneficial and/or deleterious role of p47phox is highly dependent on the type of injury and the cell type affected.
In renal disease, the contribution of p47phox has been investigated primarily in diabetes-mediated injury. Its genetic deletion reduces kidney hypertrophy, oxidative stress and mesangial matrix expansion in a spontaneous model of type 1 diabetes.24 However, no protection from albuminuria was observed in this study.24 In contrast to this finding, we show that mice lacking p47phox expression show decreased oxidative stress which is accompanied by decreased albuminuria and kidney fibrosis compared to wild type mice in two different models of glomerular injury. The beneficial effects on albuminuria are more evident when p47phox-null mice are crossed onto the integrin α1-null mouse, a model of increased NADPH-mediated oxidative stress and kidney injury.29, 30 Our results suggest that p47phox and NOX4 contribute primarily to the early phase of adriamycin-mediated renal injury. In this context, at 1 week, proteinuria, matrix accumulation, podocyte loss and glomerular injury directly correlated to the levels of renal p47phox and NOX4 expression and oxidative stress markers. This was particularly evident in 1 week injured integrin α1-null mice which showed the highest levels of p47phox and NOX4 proteins, F2 isoprostanes, and nitro-tyrosine. At 4 weeks, however, we observed an overall decrease in albuminuria and oxidative stress markers in all groups, although the p47phox and NOX4 protein levels remained elevated in the kidney of injured α1-null mice. Despite an overall decrease in oxidative stress, significantly more glomerular injury (as determined by podocyte loss) as well as glomerular and tubular interstitial sclerosis were detected in integrin α1-null mice at 4 weeks. This suggests that factors independent of ROS might contribute to the severity of injury in these mice at late phases of adriamycin-induced renal injury. A plausible explanation is that the high albuminuria detected in 1 week injured integrin α1-null mice could have triggered tubule cell death with consequent generation of pro-fibrotic and pro-inflammatory mediators.55 This possibility is supported by the significantly increase in collagen deposition and numbers of infiltrating macrophage particularly evident in the kidneys of 4 weeks injured integrin α1-null mice. Another feature of 4 week injured integrin α1-null mice was an overall increase in podocyte loss despite decreased albuminuria compared to 1 week injured integrin α1-null mice. Although this seems to be a counter intuitive result, our finding agrees with studies in humans and rodents showing that proteinuria is not uniformly associated with podocyte injury. To this end, podocyte injury does not directly correlate to the levels of proteinuria in human glomerulopathies56 and injection of antibodies to nephrin leads to rapid proteinuria in the presence of intact podocyte foot processes.57, 58
p47phox is required for both NOX1- and NOX2-mediated ROS production, suggesting that deletion of p47phox might affect both NOX pathways. Surprisingly, the NOX2-null mice show no overall renal protection upon induction of type 1 diabetes.13 On the other hand, inhibition of the NOX1/NOX4 axis with the dual inhibitor GKT136901 protects mice from renal injury induced by type 2 diabetes.59 Thus, it is conceivable that the p47phox/NOX1 axis might be a major generator of ROS in kidney injury. Although more studies are needed in order to determine which of the two NOXs is activated by p47phox in the course or renal injury, our study shows that p47phox-dependent NADPH oxidase plays a key role in the progression of kidney injury and suggests that targeting p47phox is sufficient to ameliorate both fibrotic responses and albuminuria.
In addition to p47phox, the activity and assembly of NOX1 and NOX2 containing NADPH oxidase are mediated by the small GTPase Rac.11, 32, 60-62 We previously showed that integrin α1β1 acts as a negative regulator of ROS production by preventing EGFR phosphorylation and consequent Rac activation.28 Consistent with this finding, transfection of integrin α1KO cells with dominant negative Rac significantly decreases basal levels of ROS.28 In addition, we here show that injured integrin α1-null mice crossed onto the p47phox-null background produces less in vivo markers of oxidative stress than integrin α1-null mice. However, the levels of oxidative stress in the integrin α1/p47phox-null mice were still higher than those detected in injured p47phox- mice mice. This difference suggests that integrin α1β1 might also negatively regulate ROS productions in a Rac/p47phox independent manner. In this context, mitochondria and 5-lipoxygenase have been found to be involved in integrin-mediated ROS production.63 Moreover, the presence of integrin αvβ3 at focal adhesion sites inhibits local ROS production,64 suggesting that integrin-mediated adhesion is important in regulating the local redox environment.
In conclusion, our study suggests that p47phox contributes to the generation of ROS in kidney glomeruli and contributes to kidney injuries. This, together with the finding that the assembly of Rac-dependent NOXs is negatively regulated by integrin α1β1, points to integrins and p47phox as potential targets for fibrotic diseases.
Methods
In vivo Injury Models
All in vivo experiments were performed according to institutional animal care guidelines and conducted in Association for Assessment and Accreditation of Laboratory Animal Care–accredited facilities. p47phox-null (p47phoxKO) mice on C57/B6 background were backcrossed onto the BALB/c background for 8 generations and then were crossed with integrin α1-null (α1KO) mice onto the BALB/c background29 to generate integrin α1het/p47phoxhet. The integrin α1het/p47phoxhet mice were then crossed among themselves in order to generate wild type (WT), integrin α1KO, p47phoxKO, and integrin α1KO/p47phoxKO (DKO) mice, all on the BALB/c background. For adriamycin-mediated glomerulosclerosis, male mice (6 weeks old, 24 g body weight) received a single intravenous injection of adriamycin (10 mg/kg; Sigma Aldrich), and mice were sacrificed 0, 1 or 4 weeks after injection. One and 4 weeks were chosen because at these time points most of the integrin α1KO mice develop severe albuminuria and glomerular injury.29 Six to 18 mice/genotype were used for this set of experiments. For the remnant kidney model (5/6 nephrectomy), WT, integrin α1KO, p47phoxKO, and DKO BALB/c male mice (6–7 weeks old, 20–23 g body weight) underwent 5/6 nephrectomy or sham operation as described.31 Mice were then sacrificed 12 weeks after injury. Twelve mice/genotype were used for this set of experiments.
Clinical Parameters and Morphological Analysis
Body weight was measured weekly and expressed in grams. For the analysis of albuminuria, spot urine was collected at 0, 1, and 4 weeks after adriamycin injection or at 0 and 12 weeks after partial renal ablation and the concentration of urine albumin and creatinine was measured using the ELISA Albuwell M kit (Exocel Inc., Philadelphia, PA). The albumin-to-creatinine ratio (ACR) was expressed as micrograms per milligram.
Kidneys, removed immediately at sacrifice, were fixed in 4% formaldehyde for paraffin section for both morphological and immunohistochemistry analysis. Paraffin tissue sections were stained Masson's Trichrome Blue for the evaluation of kidney fibrosis. Paraffin sections were stained with periodic acid Schiff (PAS) to assess glomerular mesangial expansion and sclerosis, acute tubular injury, and interstitial fibrosis. The total number of glomeruli per each kidney section was counted with a total of ∼ 150 glomeruli/kidneys and 7-13 mice per each group analyzed. Global sclerosis and segmental sclerosis are presented as a percentage. A semi-quantitative index was used to evaluate the degree of glomerular mesangial expansion. Each glomerulus on a single section was graded from 0 to 4, where 0 represents no lesion, and 1, 2, 3 and 4 represent mesangial matrix expansion involving <25, 25-50, 51-75, and >75% of the glomerular tuft area, respectively. Tubular injury was scored according to the percentage of damaged tubules (loss of brush border, shedding of both necrotic and viable epithelial cells into the tubular lumen, tubular dilation, cast formation, and cell lysis): 1) <25% damaged; 2) 25–50% damaged; 3) 51–75% damaged; and 4) >75% damaged, respectively. A semi-quantitative index was used to evaluate the degree of interstitial fibrosis: 0: no interstitial fibrosis; 1) <25%; 2) 25– 50%; 3) 51–75%; and 4) >75% interstitial fibrosis, respectively. Histologic analysis was performed in a blind fashion by a renal pathologist (PP).
F2-isoprostane Assay
The levels of urinary F2-isoprostane were determined using an EIA kit following the manufacturer's instruction (Cayman Chemical Company, USA). Briefly, spot urines were diluted 100 times with water. 50 μl of standard or urine samples were placed in 96-well plate pre-coated with mouse anti-rabbit IgG. Then, 50 μl F2-isoprostane tracer and F2-isoprostane antiserum were added into each well and incubated for 18 h at 4 °C. The plates were then washed to remove all unbound reagents and captured F2-isoprostane was detected with Ellman's reagent followed by absorbance at 420 nm using multiskanascent analyzer (Thermo Labsystems). All samples were assayed in duplicate. The values of F2-isoprostane were calculated using a computer spreadsheet available from Cayman Chemical Company and expressed as a ratio to creatinine (pg/μg).
Cell Culture
Primary mesangial cells from WT, α1KO, p47phoxKO, and DKO BALB/c mice were isolated, cultured, and immortalized by retroviral-mediated expression of T-antigen as previously described.65
Immunohistochemistry
Immunohistochemistry staining for the evaluation of collagen IV and Nitro-tyrosine (N-Tyr) levels, as well as macrophage and podocyte numbers in uninjured and injured kidneys was performed on paraffin kidney sections as reported.29, 66-68 Briefly, kidney sections were incubated with rabbit anti-mouse collagen IV (1:500, AbD Serotec), rabbit anti-N-Tyr antibody (1:400, Santa Cruz), mouse anti-WT1 antibody (1:100, Santa Cruz), and rat anti-F4/80 (AbD Serotec) overnight at 4°C followed by 1 hour incubation with the appropriate horseradish peroxidase-conjugated secondary antibody (1:100, Santa Cruz) at room temperature. Slides were developed using Sigma Fast DAB chromogenic tablets (Sigma) and then counterstained with toluidine blue in order to increase the contrast with DAB staining.
In vitro ROS Detection
ROS production was measured with dihydro-rhodamine as previously described 29. Briefly, 1.5 × 105 mesangial cells were plated in six-well plates in Dulbecco's modified Eagle's medium (DMEM) containing 1% fetal calf serum (FCS). After 2 days, the cells were washed with PBS and then incubated in serum-free medium with 2 μM dihydro-rhodamine (Sigma) for additional 2 hours. The cells were then trypsinized and the generation of fluorescent rhodamine 123 was analyzed by a FACScan (BD Bioscience). Three independent experiments were performed in triplicate.
GTP-Rac Assay
PAK1-p21Rac binding domain (PBD)-conjugated glutathione Sepharose beads were prepared and used as previously described.28 24 hour serum starved cells were scraped in lysis buffer containing 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 200 mM NaCl, 1% NP-40, 5% glycerol, 0.05% Tween 20, 1 mM NaF, 1 mM Na3VO4, and proteinase inhibitor (Roche). Equal amount of cell lysates were incubated with 30 μl of PAK1-PBD-conjugated glutathione Sepharose beads at 4 °C for 1 hour. Beads were then centrifuged, washed with 25 mM Tris-HCl (pH 7.6)-30 mM MgCl2-40 mM NaCl-1 mM dithiothreitol-1% NP-40, suspended in 20 μl of Laemmli's sample buffer and analyzed by Western blotting as indicated below.
Western Blot Analysis
For analysis of collagen IV, collagen I, p47phox, NOX4, and N-Tyrosine levels, total kidney lysates were prepared in 62.5 mM Tris, pH 6.8, 2% SDS, and 10% glycerol by homogenization. For p47phox membrane localization, kidneys were homogenized in 20 mM Hepes pH 7.2 using a Polytron homogenizer. After addition of NaCl (100 mM final concentration) the homogenates were centrifuged at 3,000 rpm for 5 minutes to remove debris. The supernatant was collected and centrifuged at 55,000 rpm for 50 minutes in order to separate membrane (pellet) and cytosol (supernatant) rich fractions. For analysis of phosphorylated EGF receptor, glomeruli were isolated 1 week after adriamycin injection as described 69 and lysed in the buffer described above. For analysis of phosphorylated and total ERK, as well as collagen IV levels in mesangial cells, 24 hour serum starved cells were lysed in RIPA buffer (50mM Tris, pH 7.4, 150mM NaCl, 1% NP40, 0.25% Na-deoxycholate, 1× Roche complete mini protease inhibitor cocktail, and 4 mM Na Orthovanadate).
Equal amounts of total proteins (20-40 μg/lane) were resolved in 8% SDS-PAGE, transferred onto nitrocellulose, and probed with anti-collagen IV (MD Bioproducts), anti-collagen I (MD Bioproducts), anti-p47phox (Santa Cruz), anti-NOX4 (Abcam) anti-β-actin (Santa Cruz), anti-phosphorylated-EGF receptor and anti-EGF receptor (Santa Cruz), anti-Rac1 (Cell Signaling Technology), anti-ERK and anti-phosphorylated ERK (Cell Signaling Technology), and anti-N-Tyrosine (Saint Cruz) followed by the appropriate horseradish peroxidase-conjugated secondary antibodies.
In some experiments, collagen IV, collagen I, p47phox, NOX4, N-Tyrosine, phosphorylated and total EGF receptor bands, and β-actin bands were quantified by densitometry analysis and the levels of collagen IV, collagen I, N-Tyrosine, p47phox, NOX4, and phosphorylated EGF receptor expressed as collagen IV/β-actin, N-Tyrosine/β-actin, p47phox/β-actin, NOX4/β-actin or phosphorylated EGF receptor/EGF receptor.
In some experiments 1 μl spot urine was analyzed for presence of albuminuria on 10% SDS-PAGE and stained with SimplyBlue (Invitrogen)
Statistical Analysis
We used ANOVA and Tukey's Post Hoc Test to analyze the statistical differences between multiple groups. p≤0.05 was considered statistically significant. Unpaired two tailed t-test was used for comparison of two groups.
Supplementary Material
Supplemental Figure 1. Loss of p47phox ameliorates kidney injury induced by partial renal ablation. (A) Urine albumin excretion, expressed as fold of albumin/creatinine ratio over uninjured WT mice, was analyzed in WT, α1KO, p47phoxKO, and DKO mice 12 weeks after partial renal ablation. Values represent the mean ± SD of the mice indicated. Differences between injured WT and α1KO (*) or injured WT and p47phoxKO (**) or injured α1KO and DKO (δ) mice were significant (p≤0.05). (B) Periodic-acid-Schiff (PAS) and collagen IV staining of kidneys from WT, α1KO, p47phoxKO, and DKO mice 12 weeks after partial renal ablation. Loss of p47phox ameliorates glomerular damage and excessive collagen IV deposition observed in injured integrin α1KO mice. (C) Equal amount of kidney lysates (20 μg/lane) from WT, α1KO, p47phoxKO and DKO mice (n=3) 12 weeks after partial renal ablation were analyzed by Western blot for levels of collagen IV. (D) The collagen IV and β-actin bands were analyzed by densitometry analysis and the levels of collagen IV are expressed as collagen IV/β-actin ratio. The values represent the mean ± SD of 3 kidneys/genotype.
Acknowledgments
This work was supported by the VA Merit Review 1I01BX002025 (AP) and 1I01BX002196 (RZ); the National Institutes of Health Grants R01-CA162433 (AP), R01-DK095761 (AP), R01-DK083187 (RZ), R01-DK075594 (RZ), R01-DK383069221 (RZ); R01-DK095785 (RCH/MZ); NHLBI HL92870 (TSB), NHLBI HL085317 (TSB). RZ is an American Heart Association Established Investigator.
Footnotes
Disclosure: The authors declare no financial interest.
References
- 1.Schell C, Huber TB. New players in the pathogenesis of focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2012;27:3406–3412. doi: 10.1093/ndt/gfs273. [DOI] [PubMed] [Google Scholar]
- 2.Borza CM, Pozzi A. The role of cell-extracellular matrix interactions in glomerular injury. Exp Cell Res. 2012;318:1001–1010. doi: 10.1016/j.yexcr.2012.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Modlinger PS, Wilcox CS, Aslam S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin Nephrol. 2004;24:354–365. doi: 10.1016/j.semnephrol.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Pozzi A, Zent R. Integrins in Kidney Disease. J Am Soc Nephrol. 2013 doi: 10.1681/ASN.2013010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coaccioli S, Crapa G, Fantera M, et al. Oxidant/antioxidant status in patients with chronic HIV infection. Clin Ter. 2010;161:55–58. [PubMed] [Google Scholar]
- 6.Satoh M, Fujimoto S, Haruna Y, et al. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am J Physiol Renal Physiol. 2005;288:F1144–1152. doi: 10.1152/ajprenal.00221.2004. [DOI] [PubMed] [Google Scholar]
- 7.Zuo L, Otenbaker NP, Rose BA, et al. Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol Immunol. 2013;56:57–63. doi: 10.1016/j.molimm.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Sardina JL, Lopez-Ruano G, Sanchez-Sanchez B, et al. Reactive oxygen species: are they important for haematopoiesis? Crit Rev Oncol Hematol. 2012;81:257–274. doi: 10.1016/j.critrevonc.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 9.Touyz RM, Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension. Hypertens Res. 2011;34:5–14. doi: 10.1038/hr.2010.201. [DOI] [PubMed] [Google Scholar]
- 10.Massaad CA. Neuronal and vascular oxidative stress in Alzheimer's disease. Curr Neuropharmacol. 2011;9:662–673. doi: 10.2174/157015911798376244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 12.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31(Suppl 2):S170–180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 13.You YH, Okada S, Ly S, et al. Role of Nox2 in diabetic kidney disease. Am J Physiol Renal Physiol. 2013;304:F840–848. doi: 10.1152/ajprenal.00511.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bokoch GM, Zhao T. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid Redox Signal. 2006;8:1533–1548. doi: 10.1089/ars.2006.8.1533. [DOI] [PubMed] [Google Scholar]
- 15.Katsuyama M, Matsuno K, Yabe-Nishimura C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J Clin Biochem Nutr. 2012;50:9–22. doi: 10.3164/jcbn.11-06SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Eid AA, Ford BM, Block K, et al. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem. 2010;285:37503–37512. doi: 10.1074/jbc.M110.136796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sedeek M, Callera G, Montezano A, et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2010;299:F1348–1358. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 19.Eid AA, Ford BM, Bhandary B, et al. mTOR Regulates Nox4-Mediated Podocyte Depletion in Diabetic Renal Injury. Diabetes. 2013 doi: 10.2337/db12-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Babelova A, Avaniadi D, Jung O, et al. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med. 2012;53:842–853. doi: 10.1016/j.freeradbiomed.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 21.Nlandu Khodo S, Dizin E, Sossauer G, et al. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol. 2012;23:1967–1976. doi: 10.1681/ASN.2012040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitada M, Koya D, Sugimoto T, et al. Translocation of glomerular p47phox and p67phox by protein kinase C-beta activation is required for oxidative stress in diabetic nephropathy. Diabetes. 2003;52:2603–2614. doi: 10.2337/diabetes.52.10.2603. [DOI] [PubMed] [Google Scholar]
- 23.Salguero G, Akin E, Templin C, et al. Renovascular hypertension by two-kidney one-clip enhances endothelial progenitor cell mobilization in a p47phox-dependent manner. J Hypertens. 2008;26:257–268. doi: 10.1097/HJH.0b013e3282f09f79. [DOI] [PubMed] [Google Scholar]
- 24.Liu GC, Fang F, Zhou J, et al. Deletion of p47phox attenuates the progression of diabetic nephropathy and reduces the severity of diabetes in the Akita mouse. Diabetologia. 2012;55:2522–2532. doi: 10.1007/s00125-012-2586-1. [DOI] [PubMed] [Google Scholar]
- 25.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 26.Mathew S, Chen X, Pozzi A, et al. Integrins in renal development. Pediatr Nephrol. 2012;27:891–900. doi: 10.1007/s00467-011-1890-1. [DOI] [PubMed] [Google Scholar]
- 27.Pozzi A, Zent R. Integrins: sensors of extracellular matrix and modulators of cell function. Nephron Exp Nephrol. 2003;94:e77–84. doi: 10.1159/000072025. [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Abair TD, Ibanez MR, et al. Integrin alpha1beta1 controls reactive oxygen species synthesis by negatively regulating epidermal growth factor receptor-mediated Rac activation. Mol Cell Biol. 2007;27:3313–3326. doi: 10.1128/MCB.01476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Moeckel G, Morrow JD, et al. Lack of integrin alpha1beta1 leads to severe glomerulosclerosis after glomerular injury. Am J Pathol. 2004;165:617–630. doi: 10.1016/s0002-9440(10)63326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zent R, Yan X, Su Y, et al. Glomerular injury is exacerbated in diabetic integrin alpha1-null mice. Kidney Int. 2006;70:460–470. doi: 10.1038/sj.ki.5000359. [DOI] [PubMed] [Google Scholar]
- 31.Borza CM, Su Y, Chen X, et al. Inhibition of integrin alpha2beta1 ameliorates glomerular injury. J Am Soc Nephrol. 2012;23:1027–1038. doi: 10.1681/ASN.2011040367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Honore S, Kovacic H, Pichard V, et al. Alpha2beta1-integrin signaling by itself controls G1/S transition in a human adenocarcinoma cell line (Caco-2): implication of NADPH oxidase-dependent production of ROS. Exp Cell Res. 2003;285:59–71. doi: 10.1016/s0014-4827(02)00038-1. [DOI] [PubMed] [Google Scholar]
- 33.Gao XP, Standiford TJ, Rahman A, et al. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox-/- and gp91phox-/- mice. J Immunol. 2002;168:3974–3982. doi: 10.4049/jimmunol.168.8.3974. [DOI] [PubMed] [Google Scholar]
- 34.Han W, Li H, Cai J, et al. NADPH oxidase limits lipopolysaccharide-induced lung inflammation and injury in mice through reduction-oxidation regulation of NF-kappaB activity. J Immunol. 2013;190:4786–4794. doi: 10.4049/jimmunol.1201809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eid AA, Gorin Y, Fagg BM, et al. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes. 2009;58:1201–1211. doi: 10.2337/db08-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jha JC, Gray SP, Barit D, et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol. 2014;25:1237–1254. doi: 10.1681/ASN.2013070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen A, Sheu LF, Ho YS, et al. Experimental focal segmental glomerulosclerosis in mice. Nephron. 1998;78:440–452. doi: 10.1159/000044974. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Wang YP, Tay YC, et al. Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 2000;58:1797–1804. doi: 10.1046/j.1523-1755.2000.00342.x. [DOI] [PubMed] [Google Scholar]
- 39.Gorin Y, Block K. Nox as a target for diabetic complications. Clin Sci (Lond) 2013;125:361–382. doi: 10.1042/CS20130065. [DOI] [PubMed] [Google Scholar]
- 40.Bollee G, Flamant M, Schordan S, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med. 2011;17:1242–1250. doi: 10.1038/nm.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang J, Liu N, Tolbert E, et al. Sustained Activation of EGFR Triggers Renal Fibrogenesis after Acute Kidney Injury. Am J Pathol. 2013 doi: 10.1016/j.ajpath.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf-Goldberg T, Barbul A, Ben-Dov N, et al. Low electric fields induce ligand-independent activation of EGF receptor and ERK via electrochemical elevation of H(+) and ROS concentrations. Biochim Biophys Acta. 2013;1833:1396–1408. doi: 10.1016/j.bbamcr.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 43.Cosgrove D, Meehan DT, Delimont D, et al. Integrin alpha1beta1 regulates matrix metalloproteinases via P38 mitogen-activated protein kinase in mesangial cells: implications for Alport syndrome. Am J Pathol. 2008;172:761–773. doi: 10.2353/ajpath.2008.070473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reich H, Tritchler D, Herzenberg AM, et al. Albumin activates ERK via EGF receptor in human renal epithelial cells. J Am Soc Nephrol. 2005;16:1266–1278. doi: 10.1681/ASN.2004030222. [DOI] [PubMed] [Google Scholar]
- 45.Rybak AP, Ingram AJ, Tang D. Propagation of human prostate cancer stem-like cells occurs through EGFR-mediated ERK activation. PLoS One. 2013;8:e61716. doi: 10.1371/journal.pone.0061716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dong QZ, Wang Y, Tang ZP, et al. Derlin-1 is overexpressed in non-small cell lung cancer and promotes cancer cell invasion via EGFR-ERK-mediated up-regulation of MMP-2 and MMP-9. Am J Pathol. 2013;182:954–964. doi: 10.1016/j.ajpath.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 47.Yang H, Guo H, Fan K, et al. Clearance of Propionibacterium acnes by kupffer cells is regulated by osteopontin through modulating the expression of p47phox. Mol Immunol. 2011;48:2019–2026. doi: 10.1016/j.molimm.2011.06.435. [DOI] [PubMed] [Google Scholar]
- 48.Makni-Maalej K, Chiandotto M, Hurtado-Nedelec M, et al. Zymosan induces NADPH oxidase activation in human neutrophils by inducing the phosphorylation of p47phox and the activation of Rac2: involvement of protein tyrosine kinases, PI3Kinase, PKC, ERK1/2 and p38MAPkinase. Biochem Pharmacol. 2013;85:92–100. doi: 10.1016/j.bcp.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 49.Ford BM, Eid AA, Gooz M, et al. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. Am J Physiol Renal Physiol. 2013 doi: 10.1152/ajprenal.00522.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pendyala S, Natarajan V. Redox regulation of Nox proteins. Respiratory physiology & neurobiology. 2010;174:265–271. doi: 10.1016/j.resp.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang M, Brewer AC, Schroder K, et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bataller R, Schwabe RF, Choi YH, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bodiga S, Zhong JC, Wang W, et al. Enhanced susceptibility to biomechanical stress in ACE2 null mice is prevented by loss of the p47(phox) NADPH oxidase subunit. Cardiovasc Res. 2011;91:151–161. doi: 10.1093/cvr/cvr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel VB, Wang Z, Fan D, et al. Loss of p47phox Subunit Enhances Susceptibility to Biomechanical Stress and Heart Failure Because of Dysregulation of Cortactin and Actin Filaments. Circ Res. 2013;112:1542–1556. doi: 10.1161/CIRCRESAHA.111.300299. [DOI] [PubMed] [Google Scholar]
- 55.Zoja C, Abbate M, Remuzzi G. Progression of renal injury toward interstitial inflammation and glomerular sclerosis is dependent on abnormal protein filtration. Nephrol Dial Transplant. 2014 doi: 10.1093/ndt/gfu261. [DOI] [PubMed] [Google Scholar]
- 56.van den Berg JG, van den Bergh Weerman MA, Assmann KJ, et al. Podocyte foot process effacement is not correlated with the level of proteinuria in human glomerulopathies. Kidney Int. 2004;66:1901–1906. doi: 10.1111/j.1523-1755.2004.00964.x. [DOI] [PubMed] [Google Scholar]
- 57.Orikasa M, Matsui K, Oite T, et al. Massive proteinuria induced in rats by a single intravenous injection of a monoclonal antibody. J Immunol. 1988;141:807–814. [PubMed] [Google Scholar]
- 58.Liu G, Kaw B, Kurfis J, et al. Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J Clin Invest. 2003;112:209–221. doi: 10.1172/JCI18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sedeek M, Gutsol A, Montezano AC, et al. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin Sci (Lond) 2013;124:191–202. doi: 10.1042/CS20120330. [DOI] [PubMed] [Google Scholar]
- 60.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 61.Tung WH, Hsieh HL, Lee IT, et al. Enterovirus 71 induces integrin beta1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: role of HO-1/CO in viral replication. J Cell Physiol. 2011;226:3316–3329. doi: 10.1002/jcp.22677. [DOI] [PubMed] [Google Scholar]
- 62.Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol. 2002;158:357–368. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taddei ML, Parri M, Mello T, et al. Integrin-mediated cell adhesion and spreading engage different sources of reactive oxygen species. Antioxid Redox Signal. 2007;9:469–481. doi: 10.1089/ars.2006.1392. [DOI] [PubMed] [Google Scholar]
- 64.Lin LJ, Grimme JM, Sun J, et al. The antagonistic roles of PDGF and integrin alphavbeta3 in regulating ROS production at focal adhesions. Biomaterials. 2013;34:3807–3815. doi: 10.1016/j.biomaterials.2013.01.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Volpe JP, Cleaver JE. Xeroderma pigmentosum variant cells are resistant to immortalization. Mutat Res. 1995;337:111–117. doi: 10.1016/0921-8777(95)00015-c. [DOI] [PubMed] [Google Scholar]
- 66.Zhang MZ, Wang S, Yang S, et al. Role of blood pressure and the renin-angiotensin system in development of diabetic nephropathy (DN) in eNOS-/- db/db mice. Am J Physiol Renal Physiol. 2012;302:F433–438. doi: 10.1152/ajprenal.00292.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yao B, Harris RC, Zhang MZ. Intrarenal dopamine attenuates deoxycorticosterone acetate/high salt-induced blood pressure elevation in part through activation of a medullary cyclooxygenase 2 pathway. Hypertension. 2009;54:1077–1083. doi: 10.1161/HYPERTENSIONAHA.109.137174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takagi N, Tanizawa T, Kon V, et al. Mineralocorticoid Receptor Blocker Protects against Podocyte-Dependent Glomerulosclerosis. Nephron extra. 2012;2:17–26. doi: 10.1159/000334961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen X, Whiting C, Borza C, et al. Integrin alpha1beta1 regulates epidermal growth factor receptor activation by controlling peroxisome proliferator-activated receptor gamma-dependent caveolin-1 expression. Mol Cell Biol. 2010;30:3048–3058. doi: 10.1128/MCB.00892-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Loss of p47phox ameliorates kidney injury induced by partial renal ablation. (A) Urine albumin excretion, expressed as fold of albumin/creatinine ratio over uninjured WT mice, was analyzed in WT, α1KO, p47phoxKO, and DKO mice 12 weeks after partial renal ablation. Values represent the mean ± SD of the mice indicated. Differences between injured WT and α1KO (*) or injured WT and p47phoxKO (**) or injured α1KO and DKO (δ) mice were significant (p≤0.05). (B) Periodic-acid-Schiff (PAS) and collagen IV staining of kidneys from WT, α1KO, p47phoxKO, and DKO mice 12 weeks after partial renal ablation. Loss of p47phox ameliorates glomerular damage and excessive collagen IV deposition observed in injured integrin α1KO mice. (C) Equal amount of kidney lysates (20 μg/lane) from WT, α1KO, p47phoxKO and DKO mice (n=3) 12 weeks after partial renal ablation were analyzed by Western blot for levels of collagen IV. (D) The collagen IV and β-actin bands were analyzed by densitometry analysis and the levels of collagen IV are expressed as collagen IV/β-actin ratio. The values represent the mean ± SD of 3 kidneys/genotype.