

Abstract
The purpose of this study was to develop and test a non-viral gene delivery system that can be employed to deliver genes of interest into a pre-osteoblastic cell line. Human embryonic palatal mesenchymal (HEPM 1486) cells were transfected with vector-plasmid DNA (pDNA) complexes. We explored calcium phosphate and polyethylenimine (PEI) as non-viral vectors and compared their respective in vitro transfection efficacies. Plasmid DNA encoding luciferase protein (LUC) was complexed with PEI (with differing N/P ratios) and calcium phosphate (with differing Ca/P ratios) using established protocols. The complexes prepared were then characterized for size and surface charge using a Malvern Zetasizer Nano-ZS. The transfection efficiency and cytotoxicity of the prepared complexes were evaluated in HEPM cells. The PEI-pDNA complexes over the whole range of N/P ratios were found to be < 160 nm in size while the calcium phosphate-pDNA complexes were relatively bigger. The PEI-pDNA complexes prepared at a N/P ratio of 10 were found to have maximum transfection efficiency at 4 h of treatment with minimal cytotoxicity. The highest transfection efficiency obtained with calcium phosphate-pDNA complexes (Ca/P 200) was nearly 12-fold lower than that obtained with PEI-pDNA complexes (N/P 10). Following this, transgene expression in the HEPM cells treated with complexes prepared at a N/P ratio of 10 was further examined using pDNA coding for enhanced green fluorescent protein (EGFP-N1) or therapeutically relevant platelet-derived growth factor B (PDGF-B). In conclusion, PEI was a more effective vector for delivering genes of interest to pre-osteoblasts than calcium phosphate.
Keywords: Branched polyethylenimine, calcium phosphate, non-viral vectors, gene delivery, plasmid DNA, HEPM cells, transfection efficiency, cytotoxicity
1. Introduction
Gene therapy involves treatment of diseases by the delivery of foreign genetic material into specific cells of the host (Mulligan, 1993). The genetic material in the form of plasmid DNA encoding a functional gene for a therapeutic protein can be used to supplement or alter the expression of existing genes or replace a mutated gene within the affected host cells. Depending on the nature of the disease, either short term or long term gene expression may be needed. The main prerequisites of a successful gene therapy is that it should be safe, highly efficient, controllable and selective to the target cells (Verma et al., 2000).
Tissue engineering approaches for bone regeneration have included protein therapy to deliver osteogenic cytokines and growth factors such as bone morphogenetic proteins (BMPs) (Zegzula et al., 1997), delivery of genes encoding protein factors that promote bone growth (Fang et al., 1996), and implantation of osteogenic cells at sites of bone defects (Lieberman et al., 1999). These approaches may be combined with biomimetic scaffolds to enhance the regeneration process (Shea et al., 2000). The major drawback of protein therapy is that it requires supra-physiological doses of growth factors which is expensive and runs the risk of inducing toxicity (Endo et al., 2006, Fang et al., 1996). In addition, exogenously supplied proteins are susceptible to instability, rapid clearance due to short half-lives, and loss of bioactivity. The alternative use of gene therapy is less expensive because the in vitro production of plasmid DNA is relatively simple and economical as compared to commercial protein production (Horn et al., 1995). In addition, local cell-mediated production of growth factors in situ would promote efficient cell surface receptor targeting and require less protein to achieve similar levels of therapeutic effect when compared to protein therapy. Using implanted gene-activated matrices, prolonged plasmid gene expression and continuous protein production is achieved that stimulates osteogenesis and bone repair in vivo (Bonadio et al., 1999). Localized gene therapy also averts systemic toxicity that can occur as a result of dose dumping during protein therapy (Langer, 1998, Terrell et al., 1993).
Human embryonic palatal mesenchymal (HEPM) cells are osteogenic progenitors and therefore clinically relevant in bone tissue regeneration. HEPM cells have been widely used as in vitro model cells to study osteogenesis. The HEPM cells are also a good cell type to study palatal growth and closure (Yoneda and Pratt, 1982). In this study, as a proof of concept, HEPM cells were evaluated for their ability to internalize cationic complexes of plasmid DNA, undergo transfection and produce proteins of interest. The long term goal of this study is to develop a safe and efficient non-viral gene delivery system that can deliver multiple genes in vivo for periodontal, bone and other orthopedic applications. In this report, we show for the first time that HEPM cells can be genetically manipulated using cationic complexes of plasmid DNA to produce functional proteins.
2. Materials and methods
2.1. Reagents and plasmids
Branched polyethylenimine (PEI, mol. wt. 25 kDa) was purchased from Sigma-Aldrich® (St. Louis, MO). Analytical grade calcium chloride dehydrate and dextrose monohydrate was from Sigma-Aldrich®, sodium chloride and HEPES free acid from RPI Corp. (Mt. Prospect, IL), potassium chloride and sodium phosphate tribasic dodecahydrate from Fischer Scientific (Fair Lawn, NJ). Plasmid DNA (6.4 Kb) encoding the firefly luciferase reporter protein (pLUC) driven by cytomegalovirus (CMV) promoter/enhancer (VR1255 plasmid DNA), plasmid DNA (4.7 Kb) encoding the enhanced green fluorescent protein (pEGFP-N1) driven by CMV promoter/enhancer, and plasmid DNA (4.9 Kb) encoding the platelet derived growth factor B (pPDGF-B) were used in this study. The GenElute™ HP endotoxin-free plasmid maxiprep kit was obtained from Sigma-Aldrich® (St. Louis, MO). Luciferase assay system was purchased from Promega (Madison, WI). The microBCA™ protein assay kit was purchased from Pierce (Rockford, IL). The PDGF-BB ELISA kit was purchased from Quantikine® (R & D Systems®, Minneapolis, MN). All the reagents used for transmission election microscopy (TEM) were from Electron Microscopy Services (Ft. Washington, PA). Agarose was obtained from Bio-Rad Laboratories (Hercules, CA). All other chemicals and solvents used were of reagent grade. Human palatal mesenchyme stem cells (HEPM) were purchased from American Type Culture Collection (ATCC®, Manassas, VA). Eagle’s Minimum Essential Medium (EMEM) was obtained from ATCC® (Manassas, VA). Trypsin-EDTA (0.25%, 1X solution) and Dulbecco’s phosphate buffered saline (PBS) was purchased from Gibco® (Invitrogen™, Grand Island, NY). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals® (Lawrenceville, GA). Gentamycin sulfate (50 mg/ml) was purchased from Mediatech Inc. (Manassas, VA). MTS cell growth assay reagent (Cell Titer 96® AQueous One Solution cell proliferation assay) was purchased from Promega Corporation (Madison, WI). Alexa Fluor® 568 phalloidin was purchased from Invitrogen, NY. Triton X-100 was obtained from Sigma-Aldrich®. Vectashield®, Hardset™ mounting medium with 4’,6-diamidino-2-phenylindole (DAPI) was obtained from Vector Labs Inc., CA.
2.2. Preparation of plasmid DNA (pDNA) encoding luciferase protein (LUC), enhanced green fluorescent protein (EGFP-N1) or platelet-derived growth factor B (PDGF-B)
The chemically competent DH5α™ bacterial strain (Escherichia coli species) was transformed with the necessary pDNA to amplify the plasmid. The pDNA in the transformed cultures was then expanded by amplification of the E. coli cells in Lennox L Broth (LB Broth) overnight at 37°C in an incubator shaker at 300 rpm. The pDNA was extracted using GenElute™ HP endotoxin-free plasmid maxiprep kit. The extracted pDNA was analyzed using NanoDrop 2000 UV-Vis Spectrophotometer (Thernoscientific, Wilmington, DE) for purity by measuring the ratio of absorbance (A260 nm/A280 nm). The concentration of pDNA solution was determined by UV absorbance at 260 nm. The size and quality of the extracted pDNA was determined by agarose gel electrophoresis.
2.3. Fabrication of calcium phosphate-pDNA complexes
Calcium phosphate-pDNA (LUC) complexes were prepared by a standard method as described previously (Elangovan et al., 2013, Olton et al., 2007). Briefly, 500 µl of a calcium precursor solution in water comprised of 62 µl 2 M CaCl2.2H2O and 50 µg pDNA (LUC) was added drop wise, while slowly vortexing, to an equal volume of a phosphate precursor solution in water, pH 7.5, containing varying amounts of Na3PO4.12H2O ranging from 0.83 mM to 2.48 mM of phosphate to formulate complexes with different Ca/P ratios (Table 1). The calcium concentration in the calcium precursor solution was fixed at 248 mM. The mixture was incubated at room temperature for 20 min. The final volume of the complexes used in the transfection experiments was 20 µl containing 1 µg of pDNA.
Table 1.
Ca/P ratios with corresponding phosphate concentrations (in mm phosphate) used in preparing calcium phosphate-pDNA (LUC) complexes
| Ca/P ratio | Phosphate precursor mM Phosphate |
|---|---|
| 100 | 2.48 |
| 150 | 1.65 |
| 200 | 1.24 |
| 300 | 0.83 |
2.4. Fabrication of PEI-pDNA complexes
Complexes were prepared by adding 500 µl PEI solution in water drop wise to 500 µl pDNA (LUC/EGFP-N1/PDGF-B) solution in water containing 50 µg pDNA and mixed by vortexing for 20 s. The mixture was incubated at room temperature for 30 min to allow complex formation between the positively charged PEI (amine groups) and the negatively charged pDNA (phosphate groups). Complexes were fabricated using different N (nitrogen) to P (phosphate) ratios (molar ratio of amine groups of PEI to phosphate groups in pDNA backbone) by varying the PEI amounts while keeping the amount of pDNA constant (Table 2). The final volume of the complexes utilized in the transfection and biocompatibility experiments was 20 µl containing 1 µg of pDNA.
Table 2.
N/P ratios with corresponding PEI amounts used in preparing PEI-pDNA (LUC/EGFP-N1/PDGF-B) complexes
| N/P ratio | PEI amount (µg) |
|---|---|
| 1 | 0.13 |
| 5 | 0.65 |
| 10 | 1.30 |
| 15 | 1.95 |
| 20 | 2.6 |
2.5 In vitro characterization of pDNA (LUC) complexes
2.5.1. Size and polydispersity
Size measurements of the complexes in water were carried out using Zetasizer Nano-ZS (Malvern Instruments, Westborough, MA) and the mean hydrodynamic diameter of the samples was determined by cumulative analysis. The particle size and particle size distribution by intensity were measured by photon correlation spectroscopy (PCS) using dynamic laser light scattering (4 mW He-Ne laser with a fixed wavelength of 633 nm, 173 ° backscatter at 25 °C) in 10 mm diameter cells. All measurements were done in triplicate. Complexes with a N/P ratio of 10 were placed on carbon-coated grids for 1 min and negatively stained with 1 % uranyl acetate for 20 s. After drying, the samples were imaged with a JEOL JEM-1230 TEM.
2.5.2. Surface charge
Zeta potential (surface charge) determinations of the complexes in water were based on the electrophoretic mobility of the complexes using folded capillary cells in automatic mode of measurement duration using Zetasizer Nano-ZS. Surface charge measurements were performed by the laser scattering method using Smoluchowski model (Laser Doppler Micro-electrophoresis, He-Ne laser 633 nm at 25 °C). The mean value was recorded as the average of three measurements.
2.6. Cell culture
The HEPM cells were maintained in EMEM supplemented with 10 % FBS and 50 µg/ml gentamycin in a humidified incubator (Sanyo scientific Autoflow, IR Direct Heat CO2 Incubator) at 37°C containing 95% air and 5 % CO2. The cells were plated and grown as a monolayer in 75 cm2 polystyrene cell culture flasks (Corning Incorporated, Corning, NY) and subcultured (subcultivation ratio of 1:6) after 80–90 % confluence was achieved. Cell lines were started from frozen stocks and the medium was changed every 2–3 days. The passage number at which the cells were used in experiments ranged between 4 and 8.
2.7 In vitro evaluation of the transfection efficiency of pDNA (LUC) complexes
PEI-pDNA (LUC) complexes were prepared using N/P ratios of 1, 5, 10, 15 and 20. Calcium phosphate-pDNA (LUC) complexes were prepared using Ca/P ratios of 100, 150 200 and 300. Cells were seeded at a density of 80,000 cells/well in 24-well plates. The following day, at ∼ 70 % cell confluency, the cell culture media was changed to serum-free media and the treatments were gently vortexed and added drop wise into the wells. Each well was treated with 20 µl complexes containing 1 µg pDNA. The complexes were incubated with the cells for 4 h or 24 h. At the end of each treatment period, the cells were washed with 1X phosphate buffered saline (PBS) and fresh complete media was added to the cells until analysis. After a total incubation time of 48 h, the cells were washed with 1X PBS, and treated with 1X lysis buffer and subjected to 2 freeze-thaw cycles. The cells were scraped and centrifuged at 14,000 rpm for 5 min. Luciferase expression was detected by a standard luciferase assay system using a luminometer. The relative light units (RLU) values per mg of the total cell protein, indicative of the transfection efficiency, were normalized against the protein concentration in cell extracts using a microBCA protein assay kit. The values are expressed as mean ± SD for each treatment (n = 3).
2.8 In vitro evaluation of cytotoxicity of PEI- pDNA (LUC) complexes
Cell survival assays were conducted to demonstrate the effect of N/P ratio of the PEI-pDNA (LUC) complexes on the biocompatibility of complexes in HEPM cells over a period of time. Cells were seeded in clear polystyrene, flat bottom, 96-well plates (Costar®, Corning Inc, NY) at a density of 10,000 cells/well and allowed to attach overnight. The following day, the complete medium was replaced with serum-free media and the cells were treated with complexes containing 1 µg pDNA. Untreated cells were used as controls. Cells treated with PEI alone or uncomplexed pDNA alone served as additional controls. The complexes were incubated with the cells for 4 h or 24 h to mimic the conditions used in the transfection experiments. At the end of the treatment period, the cells were washed with 1X PBS and fresh complete media was added to the cells followed by addition of 20 µL of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H–tetrazolium) cell growth assay reagent. The plates were incubated at 37°C in a humidified 5 % CO2 atmosphere for 4 h. The amount of soluble formazan produced by reduction of MTS reagent by viable cells was measured spectrophotometrically using SpectraMax® Plus384 (Molecular Devices, Sunnyvale, CA) at 490 nm. The cell viability was expressed by the following equation: Cell viability (%) = (Absorbance intensity of treated cells / Absorbance intensity of untreated cells (control)) x 100. Values are expressed as mean ± SD for each treatment performed in triplicate.
2.9In vitro visualization of transfection with PEI-pDNA (EGFP-N1) complexes
To determine the qualitative fluorescence expressed by EGFP-N1, HEPM cells were plated at a density of 50,000 cells/well in clear, flat-bottom, 8-chambered glass slides with cover (Lab-Tek, Nunc™, NY) that were previously coated with 0.1 % poly-L-lysine. The cells were allowed to attach overnight and the following day, the cell culture medium was removed. The cells were treated with complexes containing 1 µg pDNA (N/P 10) in serum-free media. Untreated cells, cells treated with uncomplexed pDNA and cells treated with PEI alone were used as controls. The complexes were incubated with the cells for 4 h or 24 h. At the end of each treatment period, the cells were washed with 1X PBS and fresh complete media was added to the cells until analysis. After a total incubation time of 48 h, the cells were washed with 1X PBS. The cells were then fixed with 4 % paraformaldehyde (Hatfield, PA), followed by permeabilization of cells with 0.2 % Triton® X-100. The cells were later treated with phalloidin for fluorescently tagging the cellular actin with Alexa Fluor® 568 phalloidin. The specimen was finally mounted with Vectashield® Hardset™ mounting medium containing DAPI. The cells were washed with PBS during every step in the process. The cellular fluorescence was observed using confocal laser scanning microscopy (60X, Carl Zeiss 710, Germany) equipped with Zen 2009 imaging software. The images were processed using ImageJ® software (National Institutes of Health, MD).
2.10 In vitro evaluation of transfection with PEI-pDNA (PDGF-B) complexes
The transfection in HEPM cells was further evaluated by using pDNA encoding for the PDGF-B protein. PEI-pDNA complexes were prepared using N/P ratio of 10. The cells were plated at a seeding density of 80,000 cells/well in 24-well plates. The following day, at ∼ 70 % cell confluency, the cell culture media was changed to serum-free media and the treatments were gently vortexed and added drop wise into the wells. Each well was treated with complexes containing 1 µg pDNA. The complexes were incubated with the cells for 4 h. At the end of the treatment period, cells were washed with 1X PBS and fresh complete media was added to the cells until analysis. After a total incubation time of 48 h, the cell culture supernatants were then collected by centrifugation and analyzed for PDGF-BB using a PDGF-BB ELISA kit. Untreated cells and cells treated with naked, uncomplexed plasmids were employed as controls. The data is represented as mean ± SD for each treatment carried out in triplicate.
2.11. Agarose gel retardation assay for PEI-pDNA (LUC) complexes
The condensation of pDNA by polycationic PEI polymer and the subsequent retention and stability of pDNA within the complexes (N/P ratio of 10) was assessed using gel electrophoresis. Samples were loaded into the wells of a 1 % agarose gel (in TAE buffer (1X) containing 5 µg/ml ethidium bromide) with BlueJuice gel loading buffer (2X final concentration). Electrophoresis was carried out at 80 mA in 1X TAE running buffer. The pDNA bands were visualized using a UV transilluminator.
2.12. Data presentation and statistical analysis
Numerical data was reported as mean ± standard deviation (S.D.) from at least three replicate samples. The graphs were generated using Prism 5.0 (GraphPad Software Inc., San Diego, CA). The data was compared by ANOVA followed by a Tukey post-test analysis. The differences between the groups were considered to be statistically significant when the P value was < 0.05. Statistical analyses were performed using Prism 5.0 software.
3. Results and discussion
Plasmid DNA is non-toxic and relatively high doses can be administered to achieve sustained gene expression and therapeutic quantities of protein production (Parker et al., 1995). The main concern with the use of pDNA is the safety and efficacy of the gene delivery vehicles. Both, viral and non-viral vectors systems can be used for gene transfer. Viral vectors such as adenoviruses and retroviruses are usually capable of high transfection efficiencies (Anderson, 1998). However, viral vectors have the potential to stimulate host immune responses, inflammatory and toxic reactions, random insertion of the viral genome and mutagenesis that hinders the clinical translatability of this approach (Endo et al., 2006). On the other hand, non-viral vectors lack such drawbacks and are easy to synthesize. Although non-viral gene delivery systems generally display lower transfection efficiencies when compared to viruses, they nevertheless have the potential to be applied to a wide array of applications in dental and craniofacial fields. In this study, we explored the non-viral delivery of different genes to HEPM cells using PEI-pDNA complexes. Polyethylenimine has proven to be an effective vehicle for pDNA in several applications (Boussif et al., 1995, Intra and Salem, 2008, Kircheis et al., 2001). Branched PEI has yielded significantly higher transfection efficiencies than linear PEI, and is therefore commonly used for polymer-mediated pDNA delivery. Attributes of branched PEI such as effective protonability and buffering capacity, high cationic charge density potential and tight condensation of pDNA contribute to its high transfection efficiency (Boussif et al., 1995, Godbey et al., 1999b). The transfection efficiencies of three different molecular weights of PEI (25, 50, and 800 kDa) were examined in vivo and it was found that 25 kDa PEI resulted in the highest transfection efficiency of all. PEI-pDNA complexes made using low molecular weight PEIs (10 kDa and lower) would readily dissociate and result in lower transfection efficiencies (Abdallah et al., 1996, Godbey et al., 1999c). We used a systematic approach to identify the optimal amine to phosphate (N/P) ratio at which PEI-pDNA complexes could generate maximum transfection in HEPM cells whilst maintaining low cytotoxicity. The transfection efficiency of PEI vector was also compared to an alternative non-viral calcium phosphate vector that is widely used for pDNA delivery due to its biocompatibility and biodegradability (Liu et al., 2011, Olton et al., 2007, O’Mahoney and Adams, 1994). The calcium/phosphate (Ca/P) ratio of the calcium phosphate-pDNA complexes is a measure of the amount of calcium to phosphate used in synthesizing a particular precipitate. In order to optimize the stoichiometry (N/P and Ca/P ratios) of these complexes, formulations with varying N/P and Ca/P ratios were fabricated and the influence of ratio stoichiometry on complex size, charge and stability, transfection efficiency and toxicity in HEPM cells was evaluated. The zeta potential and size of the pDNA complexes are very critical parameters governing the interaction of the complexes with the cells and their uptake into the cells via endocytosis. This ultimately affects the maximum transfection efficiency attained and cell viability (Bettinger et al., 1999, Naoto et al., 1986). The optimized complex ratio can improve the transfection efficiencies by overcoming the formulation and various other barriers in the intracellular and intra-nuclear transport of pDNA. Transfection efficiencies of these complexes varied significantly in different cell types compared and hence it was necessary that the transfection method be optimized for each cell line used.
3.1. Generation of pDNA encoding LUC, EGFP-N1 or PDGF-B proteins
The recovery and purity of the isolated pDNA was determined by spectrophotometric analysis. The ratio of absorbance at A260 nm/A280 nm (optical density of pDNA solution) was within the range of 1.8 to 2.0, as recommended by the manufacturer’s protocol.
3.2. Size and surface charge of calcium phosphate-pDNA (LUC) complexes
In preparing these complexes, the amount of calcium precursor (248 mM) and pDNA (1 µg) was maintained at a constant level, while the amount of phosphate precursor was varied so as to generate different Ca/P ratios: 100, 150, 200 and 300 (Table 1). This was done to assess the effect of Ca/P stoichiometry on the particle size of the resulting complexes. The size of the complexes was found to directly correlate with the Ca/P ratio (Figure 1a). The size decreased from 2317 ± 163 nm to 538 ± 6 nm with an increase in the Ca/P ratios from 100 (2.48 mM phosphate) to 300 (0.83 mM phosphate). With a decline in the phosphate concentration in the phosphate precursor solution from 2.48 mM to 0.83 mM (increasing Ca/P ratios), the pDNA bound and condensed more efficiently forming smaller, more stable complexes. The surface charge of the complexes prepared at Ca/P ratio of 200 was found to be - 11 ± 0.9 mV.
Figure 1.
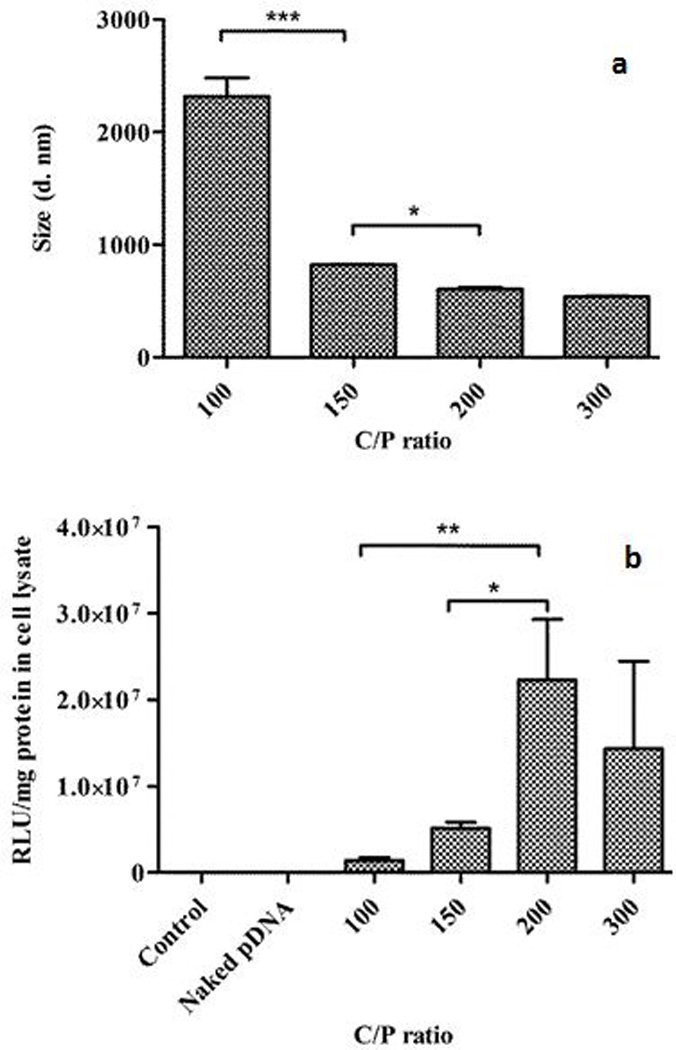
Effect of different Ca/P ratios on size of calcium phosphate-pDNA (LUC) complexes; Inset: Table displaying average size of the complexes (with standard deviation) corresponding to different Ca/P ratios (a) and transfection efficiency of calcium phosphate-pDNA (LUC) complexes in HEPM cells at 4 h of incubation time (b) (*p < 0.05; **p < 0.01; ***p < 0.001)
3.3In vitro gene expression by calcium phosphate-pDNA (LUC) complexes
Plasmid DNA was complexed with various Ca to P ratios (Table 1) to determine the influence of the Ca/P stoichiometry in achieving the optimum transfection efficiency in HEPM cells. Cells were treated with complexes containing 1 µg of pDNA for 4 h or 24 h and processed as described in the methods section. Untreated cells and cells treated with uncomplexed pDNA served as controls. The results demonstrated that Ca/P ratio of the complexes significantly affected the expression of luc gene in HEPM cells (p < 0.05). The amount of transfection obtained was higher for complexes with Ca/P ratio of 200 (1.24 mM phosphate) than with Ca/P ratios of 100 (2.48 mM phosphate), 150 (1.65 mM phosphate) or 300 (0.83 mM phosphate) (Figure 1b). The different transfection efficiencies obtained with different Ca/P ratios may partly be attributed to the sizes of these complexes. It has been reported that particle size of calcium phosphate-pDNA complexes significantly affects the extent of transfection obtained. Calcium phosphate complexes tend to aggregate with time, resulting in reduction in the level of transfection attained (Sokolova et al., 2006, Welzel et al., 2004). As the Ca/P ratios increased from 100 to 200, the sizes of complexes obtained decreased almost 4-fold from an average size of 2317 nm to an average size of 605 nm respectively, with the latter consequently having the highest transfection efficiency among the groups. Ca/P ratio of 100 exhibited the lowest transfection efficiency among the various ratios tested. Although Ca/P 300 had a small average particle size of 538 nm, the drop in the transfection achieved could be due to low pDNA binding capacity or low pDNA condensation capacity of Ca/P 300 (Olton et al., 2007). The lower transfection capabilities of Ca/P 100 and in particular, Ca/P 150 (average size of 823 nm) could also be due to the reduced binding and condensation of pDNA within the complexes. The gene expression attained at the second treatment time point of 24 h was much lower than that attained at 4 h of treatment (data not shown).
3.4. Size and surface charge of PEI-pDNA (LUC) complexes
The PEI-pDNA (LUC) complexes were fabricated using a constant amount (1 µg) of pDNA and varying amounts of PEI in order to generate N/P ratios of 1, 5, 10, 15 and 20 (Table 2, Figure 2a). With increasing N/P ratios, the sizes of the complexes progressively decreased from 157 ± 1 nm for N/P 1 to 82 ± 2 nm for N/P 20 (Figure 2b) whilst the surface charge of these complexes increased from - 22 ± 2 mV for N/P 1 to + 45 ± 0.3 mV for N/P 20 (Figure 2c). These findings were the results of complexation, coating and condensation of the entire length of the pDNA by PEI (Dunlap et al., 1997, Godbey et al., 1999a, Hsu and Uludağ, 2008, Ogris et al., 1998). Complexes prepared at a N/P ratio of 10 were typically in the 100 nm size range with a net surface charge of ∼ + 35 mV. The width of the particle size distribution is given by the polydispersity index (PDI). Lower values of PDI indicate narrow size distribution and homogeneity of the sample. In our study, the PDI was < 0.3, confirming the relative uniformity of the particle size distribution. Transmission electron microscopy images of complexes prepared at a N/P ratio of 10 showed the formation of discrete, spherical particles of ∼ 50–100 nm in size (Figure 2d).
Figure 2.
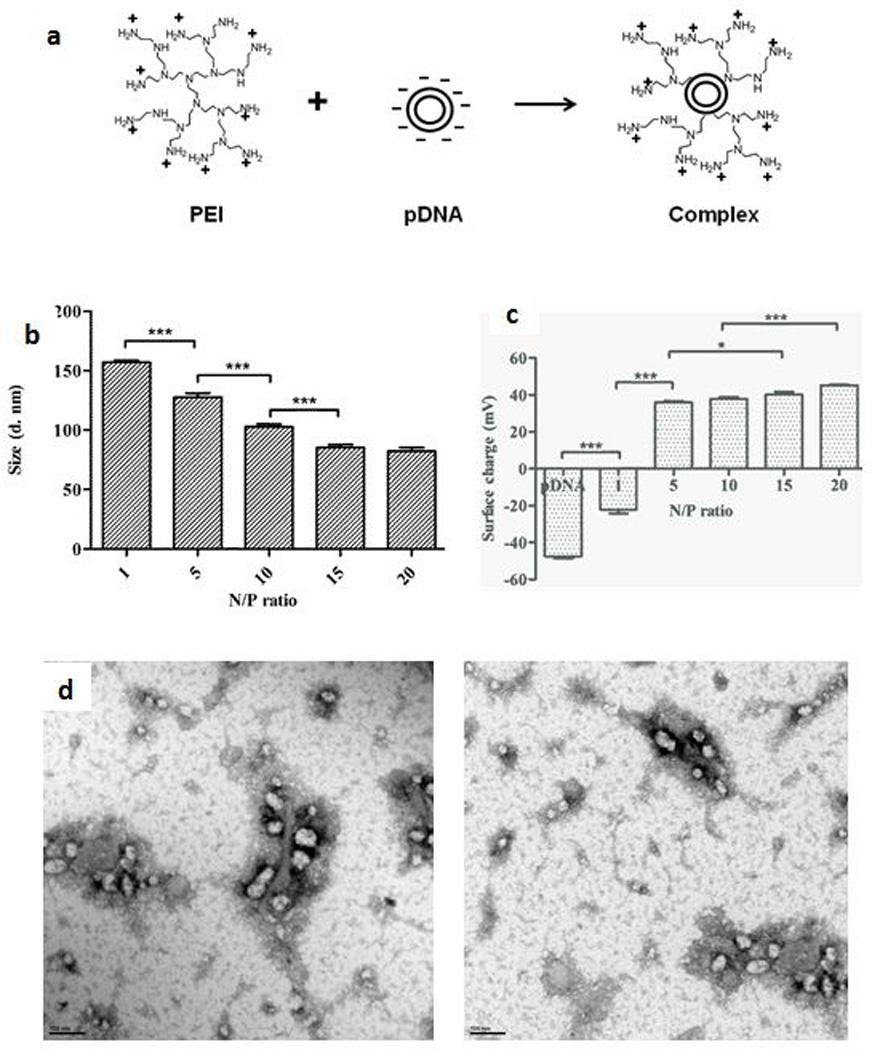
Characterization of PEI-based gene delivery system. Schematic showing the formation of PEI-pDNA complexes (a), effect of the various N/P ratios on size of PEI-pDNA (LUC) complexes; Inset: Table displaying average size of the complexes (with standard deviation) corresponding to different N/P ratios (b) and surface charge of PEI-pDNA (LUC) complexes; Inset: Table displaying average surface charge of the complexes (with standard deviation) corresponding to different N/P ratios (c), TEM images of PEI-pDNA (LUC) complexes prepared at a N/P ratio of 10 (d) (*p < 0.05; ***p < 0.001)
3.5 In vitro gene expression by PEI-pDNA (LUC) complexes
Plasmid DNA was complexed with various amounts of PEI (Table 2) to determine the N/P ratio that would yield maximum transfection efficiency and optimum levels of gene expression in HEPM cells. Cells were treated with the complexes containing 1 µg of pDNA for 4 h or 24 h and processed as described before. Untreated cells were the controls while cells treated with PEI alone were the negative controls. Cells treated with uncomplexed pDNA served as a control comparison with complex-treated cells. The luc gene expression levels were dependent on the transfection efficiencies of the N/P ratios used in this experiment (Figure 3a). The transfection efficiency increased as the N/P ratio increased from 1 (0.13 µg PEI) to 10 (1.30 µg PEI). Transfection efficiencies then dropped when the N/P ratios increased to ≥ 15. The different transfection efficiencies obtained with different N/P ratios could be attributed to the size and surface charge of the complexes, extent of pDNA binding and condensation, and toxicity of the complexes. Complexes prepared with a N/P ratio of 1 had a size of 157 nm and a surface charge of - 22 mV. The pDNA binding and condensation capacity of complexes with a N/P ratio of 1 is also lower than complexes prepared at higher N/P ratios, thus contributing to the instability of the complex (Dunlap et al., 1997, Hsu and Uludağ, 2008). These factors resulted in low transfection efficiencies of complexes. Although complexes prepared at a N/P ratio of 5 had a net positive surface charge, the size of the complexes remained larger than complexes prepared at a N/P ratio of 10. Incubation of the complexes prepared at a N/P ratio of 5 with the cells for 24 h showed an increase in the level of transgene expression obtained relative to that obtained at 4 h of incubation. This may be attributed to higher uptake and entry of the complexes into the cells over time. After 4 h of treatment, complexes prepared at a N/P ratio of 10 were able to transfect HEPM cells more efficiently than complexes prepared at other N/P ratios. This may be a result of their small size (100 nm) and high positive surface charge (+ 35 mV) compared to complexes with N/P ratios < 10, and the negative impact on cell viability due to PEI (see below) for N/P ratios > 10. However, the transgene expression decreased in cells treated with complexes prepared at a N/P ratio of 10 at 24 h due to induction of cytotoxicity by PEI (see below). For the same reason, even though complexes prepared at N/P ratios of 15 and 20 had small sizes and highly positive surface charges, the amount of transgene expression in the cells declined with these treatments. For complexes prepared at a N/P ratio of 15, the gene expression generated was found to be lower than gene expression generated by complexes prepared at a N/P ratio of 10 at both 4 h or 24 h of treatment. The transfection resulting at 4 h of treatment from complexes prepared at a N/P ratio of 20 was lower than the transfection generated from complexes prepared at a N/P ratio of 15, which decreased further at 24 h of incubation time. The optimal transfection efficiency obtained at 4 h of treatment with PEI-pDNA complexes at a N/P ratio of 10 was nearly 12-fold higher than that obtained with calcium phosphate-pDNA complexes (Ca/P ratio of 200). Possible reasons for this observation are the high positive surface charge and smaller sizes of the PEI-pDNA complexes, accompanied by strong pDNA binding and condensation capacity of PEI, which are factors that favor high transfection efficiency of gene delivery vectors.
Figure 3.
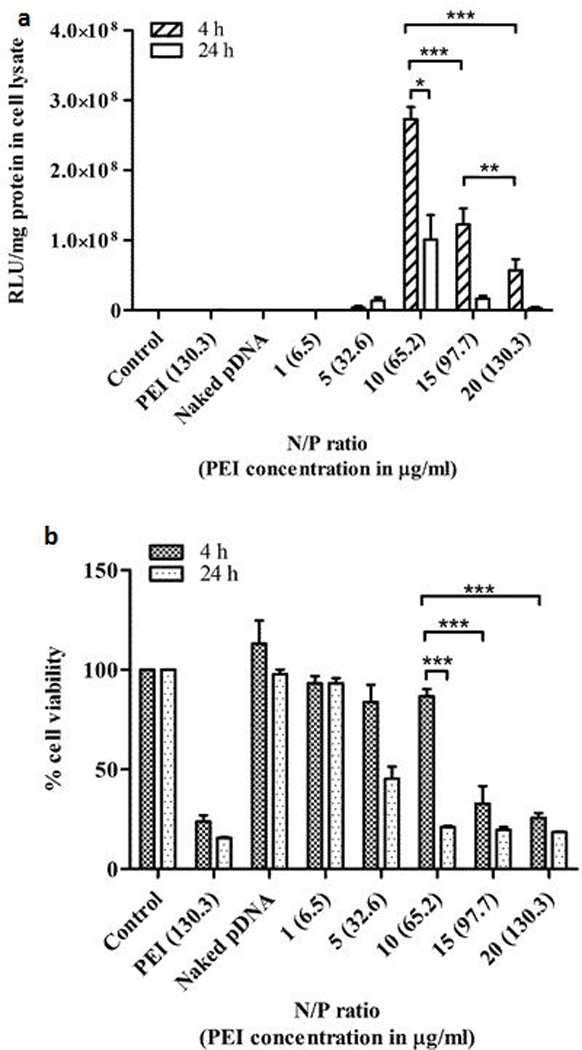
Transfection efficiency (a) and biocompatibility (PEI toxicity) (b) of PEI-pDNA (LUC) complexes fabricated at different N/P ratios in HEPM cells at 4 h or 24 h (*p < 0.05; **p < 0.01; ***p < 0.001)
Polyethylenimine is able to condense pDNA efficiently, delivers pDNA into the cells by ionic interactions with the cell membrane and releases pDNA into the cytoplasm from endocytic vesicles. Within the acidic endo-lysosomal compartments, protonation of PEI causes PEI to act as a ‘proton sponge’, thus triggering osmotic swelling and vesicle disruption (Boussif et al., 1995). In contrast, calcium phosphate-pDNA complexes are reported to enter the cells through the cell membrane calcium ion channel-mediated endocytosis (Bisht et al., 2005). When the complexes are prepared by calcium phosphate co-precipitation reaction technique, they aggregate rapidly with time and increase in particle size, and consequently the transfection reproducibility and efficiency is decreased (Sokolova et al., 2006). Transfection efficiencies of the calcium phosphate-pDNA complexes strongly depend on the formulation parameters including pH, temperature, and standing time between preparation and application, therefore resulting in inherently poor method reproducibility compared with PEI-based transfection methods (Jordan and Wurm, 2004, Seelos, 1997, Sokolova et al., 2006, Urabe et al., 2000). In addition, inefficient pDNA binding and condensing capacities, reduced cellular and nuclear uptake, limited endosomal escape, and very little protection of pDNA from nuclease degradation can all lead to lower levels of gene expression while using calcium phosphate as vector, when compared to PEI (Orrantia and Chang, 1990). Polyethylenimine-pDNA complexes were therefore more effective for delivering genes of interest to HEPM cells than the calcium phosphate-pDNA complexes.
3.6 In vitro cell viability assay for PEI-pDNA (LUC) complexes
Cell viability assays were performed to assess the effect of co-culture of PEI-pDNA (LUC) complexes with HEPM cells on cell survival at 4 h or 24 h incubation time. Plasmid DNA (1 µg) was complexed with different amounts of PEI (Table 2) to generate complexes with varying N/P ratios. The cells were treated with complexes for 4 h or 24 h and processed as described in the methods section. Untreated cells, cells treated with uncomplexed pDNA and PEI-treated cells served as controls. We quantified the percent cell viability at both the treatment time points of 4 h or 24 h and we observed a PEI dose- and time-dependent cytotoxicity (Figure 3b). The fact that complexes prepared at a N/P ratio of 10 were able to transfect HEPM cells more efficiently than complexes prepared at N/P ratios ≥ 15 at 4 h is consistent with the cell viability data (Figures 3a and 3b). The cell viability results at 4 h showed relatively low cellular toxicity (13 %) for complexes prepared at a N/P ratio of 10 when compared to the cytotoxicity induced by complexes prepared at N/P ratios ≥ 15. Approximately 87 % of the cells were viable at 4 h when incubated with complexes prepared at a N/P ratio of 10. However, the cell viability decreased to 20 % at 24 h for cells incubated with N/P 10 complexes. This effect on cell viability explains the corresponding decrease in transfection efficiency observed for N/P 10 complexes at 24 h. Similarly, the amount of PEI used in complexes prepared at N/P ratios of 15 and 20 caused a dramatic decrease in cell survival at both 4 h or 24 h and consequently had a significant negative impact on their transfection efficiencies (p < 0.05) (described above). This data clearly indicates that the amount of PEI used for transfecting the HEPM cells and the incubation time are critical factors determining the degree of transfection efficiency and toxicity. Complexes prepared at N/P ratios of 1 and 5 were relatively non-toxic to the cells, but they displayed low transfection efficiencies. Of all the complexes prepared, only the PEI-pDNA complexes prepared at a N/P ratio of 10 demonstrated high transfection efficiencies at 4 h of treatment time with minimum cytotoxicity. Hence, only the complexes prepared at a N/P ratio of 10 were used for cell transfections in the subsequent in vitro experiments.
3.7 In vitro gene expression by PEI-pDNA (EGFP-N1) complexes
The transfection in HEPM cells was further investigated by confocal microscopy. Plasmid DNA encoding for the EGFP-N1 protein (1 µg) was complexed with 1.3 µg PEI to form complexes at a N/P ratio of 10. Cells were exposed to the complexes for 4 h or 24 h and further processed as described above. Using fluorescent markers, we detected successful transfection occurring in cells at both the treatment time points of 4 h or 24 h (Figure 4). The confocal images (Z-series, 63 X) revealed the transfected cells appearing green as a whole as evidenced by expression of the EGFP-N1 fluorescent protein throughout the cytoplasm. In these fixed cells, phalloidin permeated the plasma membrane to stain the cytoplasmic F-actin in red. The cell nuclei were stained blue by DAPI. The control cells (untreated, or incubated with naked pDNA or PEI alone) did not show any green fluorescence, thus ruling out transfection with naked pDNA or autofluorescence from the cells, pDNA or PEI. Confocal microscopy imaging, along with the quantitative results obtained earlier, confirms the ability of PEI-pDNA complexes to transfect HEPM cells efficiently.
Figure 4.
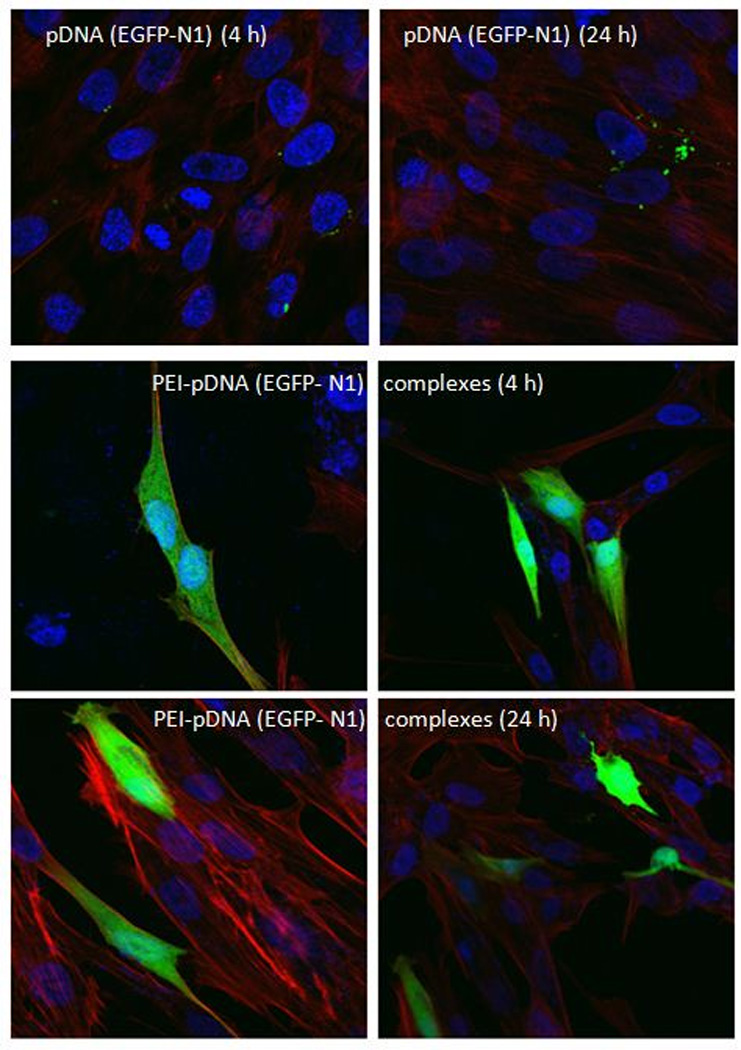
Confocal laser scanning microscopic images (Z-series, 63 X) demonstrating EGFP-N1 protein expression in HEPM cells at 4 h or 24 h after transfection with plasmids alone (upper row), PEI-pDNA (EGFP-N1) complexes prepared at a N/P ratio of 10 after 4 h (middle row) or 24 h (lower row) of transfection: nuclei (blue, DAPI-stained); cytoplasm and cell membrane (red, phalloidin-stained); and EGFP-N1 (green). Scale bar, 20µm.
3.8 In vitro gene expression by PEI-pDNA (PDGF-B) complexes
Since our future goals are ultimately targeted towards stimulation of new bone tissue formation, we investigated the transgene nuclear delivery and expression of growth factor protein, PDGF-B. Platelet-derived growth factor plays an important role in angiogenesis, is chemotactic and mitogenic for osteoblast and vascular endothelial cells, and stimulates osteoblast type-1 collagen synthesis and extracellular matrix (Battegay et al., 1994, Bolander, 1992). Based on the prior N/P ratio optimization experiments, we evaluated the efficiency of PEI-pDNA complexes prepared at a N/P ratio of 10. One microgram of pDNA was complexed with 1.3 µg PEI to form complexes at a N/P ratio of 10. Cells were exposed to the complexes for 4 h and processed as described for other transfection experiments. The PDGF-BB ELISA test demonstrated that the PDGF-BB levels (PDGF-BB conc. 56 pg/ml) were 3.5-fold higher in cells treated with complexes prepared at a N/P ratio of 10 than with uncomplexed, naked pDNA (PDGF-BB conc. 16 pg/ml) (Figure 5). These results confirmed the ability of PEI-pDNA complexes to transfect HEPM cells with genes that are therapeutically relevant to bone regeneration.
Figure 5.
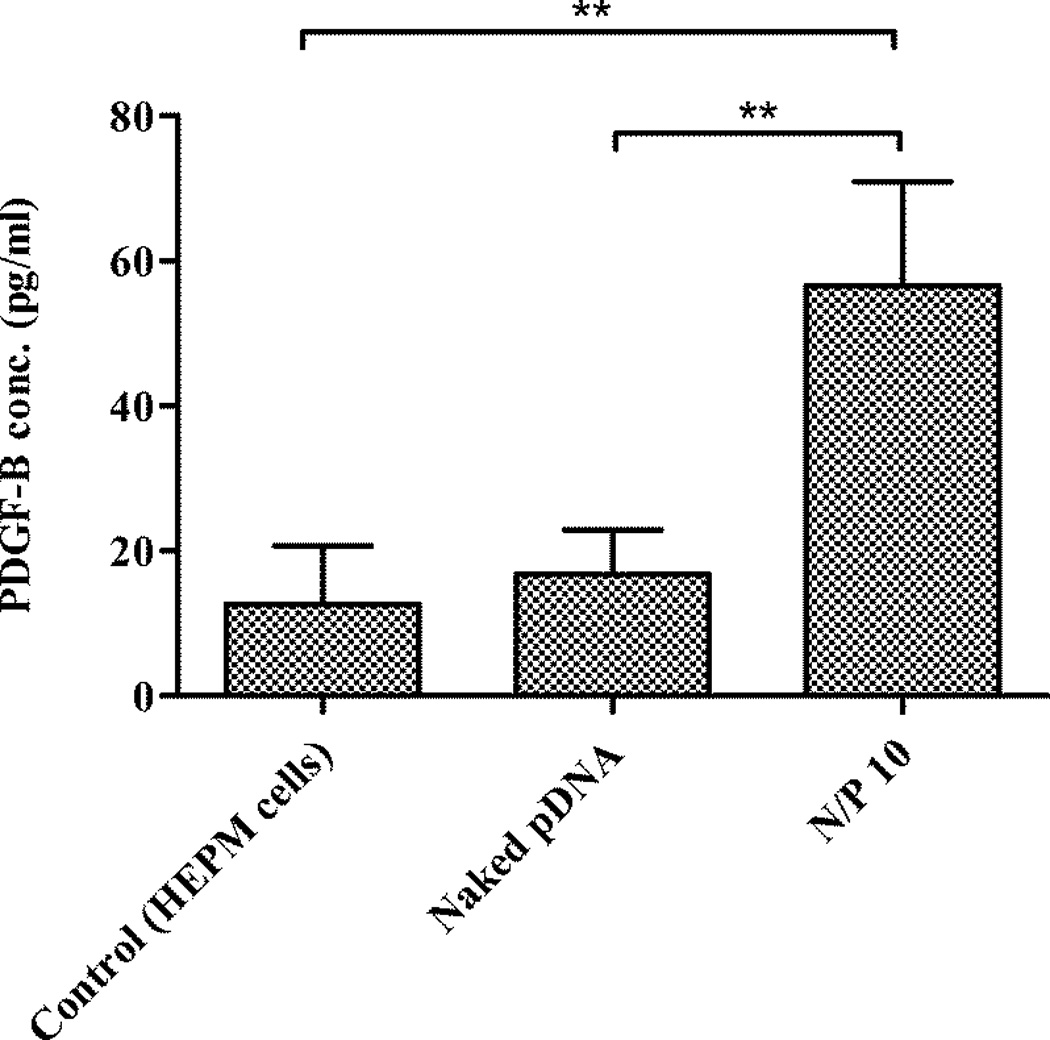
Expression of PDGF-BB protein in HEPM cells at 4 h after transfection of the cells with PEI-pDNA (PDGF-B) complexes prepared at a N/P ratio of 10 (**p < 0.01)
3.9. Gel retardation assay for PEI-pDNA (LUC) complexes
We evaluated the structural integrity of the complexes prepared at a N/P ratio of 10 by determining the electrophoretic mobility of the complexes on a 1 % agarose gel. To establish conditions similar to those used in the transfection experiments, complexes with N/P 10 were incubated with cell culture growth medium (containing 10 % fetal bovine serum) for 1 h at 37°C. This was performed to assess the effect of degradative enzymes such as DNases, and proteins present in the growth medium on complex dissociation and pDNA degradation. Uncomplexed pDNA was used as the control. Gel retardation assay demonstrated the stability of pDNA within the complexes (Figure 6). The uncomplexed pDNA moved freely in the gel (towards the positive terminal) as discrete bands, whereas the complexed pDNA was retarded in the loading well of the gel. This result suggests that PEI entraps pDNA and restricts its mobility in the gel. Upon treatment with the cell culture growth medium, the free plasmid appeared to undergo enzymatic digestion as visualized by fragmentation, smear and band dislocation. By contrast, the complex structure and the incorporated pDNA were completely unaffected. The complexes containing pDNA remained in the wells with no sign of decomplexation. The presence of serum DNases and proteins in the growth medium had no adverse effect on the complex conformation or the entrapped pDNA. Thus, PEI was able to effectively bind, package and condense the pDNA, consequently forming strong and stable complexes with pDNA. The condensation of DNA by PEI enables the formation of compact particles for endocytosis and also protects the entrapped pDNA from intracellular nuclease digestion (He et al., 2003).
Figure 6.
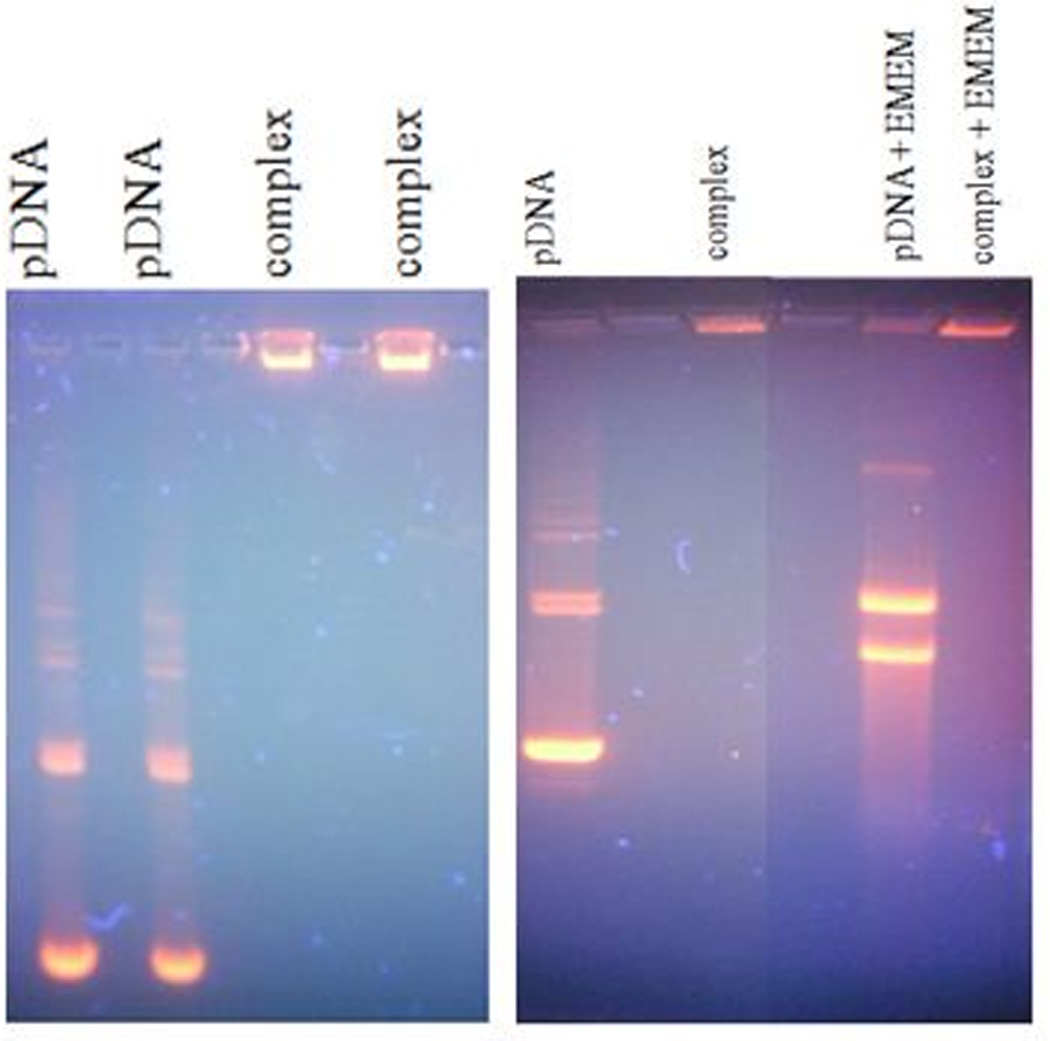
Agarose gel electrophoresis images of pDNA and PEI-pDNA (LUC) complexes prepared at a N/P ratio of 10
4. Conclusions
Cationic complexes incorporating pDNAs encoding the LUC, EGFP-N1 or PDGF-B proteins were successfully prepared. Two different non-viral vectors, PEI and calcium phosphate, were empirically assessed as gene delivery vectors. It was found that PEI is a more effective vector for delivering genes of interest to pre-osteoblastic HEPM cells than calcium phosphate. The cell-complex incubation time, amount of PEI in complexes, size and surface charge of the complexes were critical parameters for achieving efficient transfections. In vitro experiments demonstrated that PEI-pDNA complexes prepared at a N/P ratio of 10 were able to transfect LUC, EGFP-N1 and PDGF-B genes efficiently into HEPM cells with relatively lower cytotoxicity than complexes prepared at other N/P ratios. Future studies will explore the in vivo transfection efficacy of PEI-pDNA complexes prepared at a N/P ratio of 10 for the purpose of bone regeneration.
Acknowledgements
This study is supported by a University of Iowa Start-up Grant, ITI Foundation for the Promotion of Implantology, Switzerland (ITI Research Grant No. 855 2012), the American Cancer Society (RSG-09-015-01-CDD), and the National Cancer Institute at the National Institutes of Health (1R21CA13345-01/R21CA128414-01A2/UI Mayo Clinic Lymphoma SPORE).
References
- Abdallah B, Hassan A, Benoist C, Goula D, Behr JP, Demeneix BA. A powerful nonviral vector for in vivo gene transfer into the adult mammalian brain: polyethylenimine. Hum Gene Ther. 1996;7:1947–1954. doi: 10.1089/hum.1996.7.16-1947. [DOI] [PubMed] [Google Scholar]
- Anderson WF. Human gene therapy. Nature. 1998;392:25–30. doi: 10.1038/32058. [DOI] [PubMed] [Google Scholar]
- Battegay EJ, Rupp J, Iruela-Arispe L, Sage EH, Pech M. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J Cell Biol. 1994;125:917–928. doi: 10.1083/jcb.125.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettinger T, Remy J-S, Erbacher P. Size reduction of galactosylated PEI/DNA complexes improves lectin-mediated gene transfer into hepatocytes. Bioconjugate chemistry. 1999;10:558–561. doi: 10.1021/bc990006h. [DOI] [PubMed] [Google Scholar]
- Bisht S, Bhakta G, Mitra S, Maitra A. pDNA loaded calcium phosphate nanoparticles: highly efficient non-viral vector for gene delivery. International Journal of Pharmaceutics. 2005;288:157–168. doi: 10.1016/j.ijpharm.2004.07.035. [DOI] [PubMed] [Google Scholar]
- Bolander ME. Regulation of Fracture Repair by Growth Factors. Experimental Biology and Medicine. 1992;200:165–170. doi: 10.3181/00379727-200-43410a. [DOI] [PubMed] [Google Scholar]
- Bonadio J, Smiley E, Patil P, Goldstein S. Localized, direct plasmid gene delivery in vivo: prolonged therapy results in reproducible tissue regeneration. Nat Med. 1999;5:753–759. doi: 10.1038/10473. [DOI] [PubMed] [Google Scholar]
- Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap DD, Maggi A, Soria MR, Monaco L. Nanoscopic structure of DNA condensed for gene delivery. Nucleic Acids Research. 1997;25:3095–3101. doi: 10.1093/nar/25.15.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elangovan S, Jain S, Tsai PC, Margolis HC, Amiji M. Nano-sized calcium phosphate particles for periodontal gene therapy. J Periodontol. 2013;84:117–125. doi: 10.1902/jop.2012.120012. [DOI] [PubMed] [Google Scholar]
- Endo M, Kuroda S, Kondo H, Maruoka Y, Ohya K, Kasugai S. Bone regeneration by modified gene-activated matrix: effectiveness in segmental tibial defects in rats. Tissue Eng. 2006;12:489–497. doi: 10.1089/ten.2006.12.489. [DOI] [PubMed] [Google Scholar]
- Fang J, Zhu YY, Smiley E, Bonadio J, Rouleau JP, Goldstein SA, McCauley LK, Davidson BL, Roessler BJ. Stimulation of new bone formation by direct transfer of osteogenic plasmid genes. Proc Natl Acad Sci U S A. 1996;93:5753–5758. doi: 10.1073/pnas.93.12.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbey W, Wu K, Hirasaki G, Mikos A. Improved packing of poly (ethylenimine)/DNA complexes increases transfection efficiency. Gene therapy. 1999a;6:1380–1388. doi: 10.1038/sj.gt.3300976. [DOI] [PubMed] [Google Scholar]
- Godbey WT, Wu KK, Mikos AG. Poly(ethylenimine) and its role in gene delivery. Journal of Controlled Release. 1999b;60:149–160. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- Godbey WT, Wu KK, Mikos AG. Size matters: molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J Biomed Mater Res. 1999c;45:268–275. doi: 10.1002/(sici)1097-4636(19990605)45:3<268::aid-jbm15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- He XX, Wang K, Tan W, Liu B, Lin X, He C, Li D, Huang S, Li J. Bioconjugated nanoparticles for DNA protection from cleavage. J Am Chem Soc. 2003;125:7168–7169. doi: 10.1021/ja034450d. [DOI] [PubMed] [Google Scholar]
- Horn NA, Meek JA, Budahazi G, Marquet M. Cancer gene therapy using plasmid DNA: purification of DNA for human clinical trials. Hum Gene Ther. 1995;6:565–573. doi: 10.1089/hum.1995.6.5-565. [DOI] [PubMed] [Google Scholar]
- Hsu C, Uludağ H. Effects of size and topology of DNA molecules on intracellular delivery with non-viral gene carriers. BMC biotechnology. 2008;8:23. doi: 10.1186/1472-6750-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intra J, Salem AK. Characterization of the transgene expression generated by branched and linear polyethylenimine-plasmid DNA nanoparticles in vitro and after intraperitoneal injection in vivo. Journal of Controlled Release. 2008;130:129–138. doi: 10.1016/j.jconrel.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan M, Wurm F. Transfection of adherent and suspended cells by calcium phosphate. Methods. 2004;33:136–143. doi: 10.1016/j.ymeth.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Kircheis R, Wightman L, Schreiber A, Robitza B, Rössler V, Kursa M, Wagner E. Polyethylenimine/DNA complexes shielded by transferrin target gene expression to tumors after systemic application. Gene therapy. 2001;8:28–40. doi: 10.1038/sj.gt.3301351. [DOI] [PubMed] [Google Scholar]
- Langer R. Drug delivery and targeting. Nature. 1998;392:5–10. [PubMed] [Google Scholar]
- Lieberman JR, Daluiski A, Stevenson S, Wu L, McAllister P, Lee YP, Kabo JM, Finerman GA, Berk AJ, Witte ON. The effect of regional gene therapy with bone morphogenetic protein-2-producing bone-marrow cells on the repair of segmental femoral defects in rats. J Bone Joint Surg Am. 1999;81:905–917. doi: 10.2106/00004623-199907000-00002. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang T, He F, Liu Q, Zhang D, Xiang S, Su S, Zhang J. An efficient calcium phosphate nanoparticle-based nonviral vector for gene delivery. International journal of nanomedicine. 2011;6:721. doi: 10.2147/IJN.S17096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan R. The basic science of gene therapy. Science. 1993;260:926–932. doi: 10.1126/science.8493530. [DOI] [PubMed] [Google Scholar]
- Naoto O, Yamaguchi N, Yamaguchi N, Shibamoto S, Fumiaki I, Nango M. The fusogenic effect of synthetic polycations on negatively charged lipid bilayers. Journal of biochemistry. 1986;100:935–944. doi: 10.1093/oxfordjournals.jbchem.a121806. [DOI] [PubMed] [Google Scholar]
- O’Mahoney JV, Adams TE. Optimization of experimental variables influencing reporter gene expression in hepatoma cells following calcium phosphate transfection. DNA and cell biology. 1994;13:1227–1232. doi: 10.1089/dna.1994.13.1227. [DOI] [PubMed] [Google Scholar]
- Ogris M, Steinlein P, Kursa M, Mechtler K, Kircheis R, Wagner E. The size of DNA/transferrin-PEI complexes is an important factor for gene expression in cultured cells. Gene therapy. 1998;5:1425–1433. doi: 10.1038/sj.gt.3300745. [DOI] [PubMed] [Google Scholar]
- Olton D, Li J, Wilson ME, Rogers T, Close J, Huang L, Kumta PN, Sfeir C. Nanostructured calcium phosphates (NanoCaPs) for non-viral gene delivery: Influence of the synthesis parameters on transfection efficiency. Biomaterials. 2007;28:1267–1279. doi: 10.1016/j.biomaterials.2006.10.026. [DOI] [PubMed] [Google Scholar]
- Orrantia E, Chang PL. Intracellular distribution of DNA internalized through calcium phosphate precipitation. Experimental cell research. 1990;190:170–174. doi: 10.1016/0014-4827(90)90181-9. [DOI] [PubMed] [Google Scholar]
- Parker SE, Vahlsing HL, Serfilippi LM, Franklin CL, Doh SG, Gromkowski SH, Lew D, Manthorpe M, Norman J. Cancer gene therapy using plasmid DNA: safety evaluation in rodents and non-human primates. Hum Gene Ther. 1995;6:575–590. doi: 10.1089/hum.1995.6.5-575. [DOI] [PubMed] [Google Scholar]
- Seelos C. A critical parameter determining the aging of DNA-calcium-phosphate precipitates. Analytical biochemistry. 1997;245:109–111. doi: 10.1006/abio.1996.9948. [DOI] [PubMed] [Google Scholar]
- Shea LD, Wang D, Franceschi RT, Mooney DJ. Engineered bone development from a pre-osteoblast cell line on three-dimensional scaffolds. Tissue Eng. 2000;6:605–617. doi: 10.1089/10763270050199550. [DOI] [PubMed] [Google Scholar]
- Sokolova VV, Radtke I, Heumann R, Epple M. Effective transfection of cells with multi-shell calcium phosphate-DNA nanoparticles. Biomaterials. 2006;27:3147–3153. doi: 10.1016/j.biomaterials.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Terrell TG, Working PK, Chow CP, Green JD. Pathology of recombinant human transforming growth factor-beta 1 in rats and rabbits. Int Rev Exp Pathol. 1993;34 Pt B:43–67. doi: 10.1016/b978-0-12-364935-5.50009-2. [DOI] [PubMed] [Google Scholar]
- Urabe M, Kume A, Tobita K, Ozawa K. DNA/calcium phosphate precipitates mixed with medium are stable and maintain high transfection efficiency. Analytical biochemistry. 2000;278:91–92. doi: 10.1006/abio.1999.4429. [DOI] [PubMed] [Google Scholar]
- Verma IM, Naldini L, Kafri T, Miyoshi H, Takahashi M, Blömer U, Somia N, Wang L, Gage FH. Gene Therapy: Promises, Problems and Prospects. In: Boulyjenkov V, Berg K, Christen Y, editors. Genes and Resistance to Disease. Springer Berlin Heidelberg; 2000. [Google Scholar]
- Welzel T, Radtke I, Meyer-Zaika W, Heumann R, Epple M. Transfection of cells with custom-made calcium phosphate nanoparticles coated with DNA. Journal of Materials Chemistry. 2004;14:2213–2217. [Google Scholar]
- Yoneda T, Pratt RM. Glucocorticoid receptors in palatal mesenchymal cells from the human embryo: relevance to human cleft palate formation. J Craniofac Genet Dev Biol. 1982;1:411–423. [PubMed] [Google Scholar]
- Zegzula HD, Buck DC, Brekke J, Wozney JM, Hollinger JO. Bone Formation with Use of rhBMP-2 (Recombinant Human Bone Morphogenetic Protein-2)*. The Journal of Bone & Joint Surgery. 1997;79:1778–1790. doi: 10.2106/00004623-199712000-00003. [DOI] [PubMed] [Google Scholar]