

Abstract
Legumes are a highly diverse angiosperm family that include many agriculturally important species. To date, 21 complete chloroplast genomes have been sequenced from legume crops confined to the Papilionoideae subfamily. Here we report the first chloroplast genome from the Mimosoideae, Acacia ligulata, and compare it to the previously sequenced legume genomes. The A. ligulata chloroplast genome is 158,724 bp in size, comprising inverted repeats of 25,925 bp and single-copy regions of 88,576 bp and 18,298 bp. Acacia ligulata lacks the inversion present in many of the Papilionoideae, but is not otherwise significantly different in terms of gene and repeat content. The key feature is its highly divergent clpP1 gene, normally considered essential in chloroplast genomes. In A. ligulata, although transcribed and spliced, it probably encodes a catalytically inactive protein. This study provides a significant resource for further genetic research into Acacia and the Mimosoideae. The divergent clpP1 gene suggests that Acacia will provide an interesting source of information on the evolution and functional diversity of the chloroplast Clp protease complex.
Introduction
The Leguminosae (Fabaceae) are a large and economically important family of flowering plants. The family is separated into a number of subfamilies, with Papilionoideae and Mimosoideae being the most species-rich. The Papilionoideae has been the best studied of these subfamilies due to the fact that it includes a large number of agriculturally important species, such as soybean (Glycine max L.), chickpea (Cicer arietinum L.), the common bean (Phaseolus vulgaris L.) and mungbean (Vigna radiata L.).
The Mimosoideae includes genera such as Mimosa, Inga and Acacia. The genus Acacia (sensu stricto) is found across tropical, subtropical, warm temperate and arid climates. It occurs predominantly in Australia, although several species also occur in Southeast Asia and Madagascar [1]. With over 1,000 species, Acacia is the largest angiosperm genus in Australia [2]. Acacia species play an important ecological role both as a dominant component of many vegetation classes in Australia [3], particularly in the arid/semi-arid interior, and also internationally as invasive species [4–6]. Many Austalian acacias are also important sources of wood and wood products and are widely grown in the tropics and sub-tropics [7]. Previous genetic research on Acacia has focused largely on informing conservation and agro-forestry management, for example by identifying provenances for seed sourcing [8], examining mating systems and the level and distribution of genetic variation within species [9–11], establishing phylogeographic patterns [12], enhancing species identification through DNA barcoding [6, 13], and clarifying species relationships in phylogenetic studies [14–19].
In recent years, the benefits of whole genome sequencing to conservation and restoration genetics have become increasingly clear. These benefits include large-scale development of both neutral and adaptive markers and larger datasets for increased phylogenetic resolution [20–23]. Prior to the development of next-generation sequencing technologies, the time and cost associated with sequencing an entire genome was impractical for non-model species. However, in the last decade, the development of high throughput technologies has made whole genome sequencing increasingly practical and cost-effective, notably via high-throughput shallow sequencing of total DNA [24, 25].
To date, approximately 530 complete chloroplast genomes have been sequenced (see http://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=2759&opt=plastid), with 21 of these belonging to the Leguminosae (all 21 are Papilionoideae). The typical chloroplast genome comprises two inverted repeats (IRs) separated by a small single copy (SSC) and a large single copy (LSC) region [26]. In general, chloroplast genomes range in size from 120-160-kb and include 120–130 genes, many of which are essential for photosynthesis. The chloroplast’s role in photosynthesis has resulted in these features being highly conserved [27–29].
Compared to this typical chloroplast genome, many Papilionoideae chloroplast genomes display significant rearrangements, including the inversion of a 50-kb region of the LSC [30, 31], and the loss of one inverted repeat copy [32]. These features, as well as transfer of the rpl22 gene to the nucleus [33, 34], and intron loss in the clpP and rps12 genes [35, 36], have been well studied and their presence/absence mapped onto the current Leguminosae phylogeny [36].
Unlike Papilionoideae, Mimosoideae appears to display neither loss of the inverted repeat [37], nor the 50-kb inversion [30], however, no complete Mimosoideae chloroplast genomes have yet been sequenced. Here we report the complete chloroplast genome sequence of Acacia ligulata, a widespread species found throughout arid and semi-arid Australia. We discuss the chloroplast genome structure of A. ligulata including its gene content, inverted repeat organisation and repeat structure, and compare these with other legume chloroplast genomes. We also investigated the functionality of the A. ligulata clpP1 gene by exploring the transcript’s ability to be spliced, and the conservation of the catalytic triad.
Results and Discussion
Sequencing and assembly
Dried herbarium material of a specimen of Acacia ligulata Benth. was used for DNA extraction. Illumina sequencing of a library prepared from total DNA produced 2,216,882 paired-end reads with a read length of 100 nt. 5.26% of reads were assembled into 23 contigs showing homology to legume plastid DNA. Gaps between contigs were then filled by PCR amplification and Sanger sequencing. The complete assembled chloroplast genome of A. ligulata is typical in its general structure with a pair of IRs of 25,925 bp, an LSC of 88,576 bp and an SSC of 18,298 bp (Fig 1). Thus, unlike the chloroplast genomes of many of the Papilionoideae, the A. ligulata genome has inverted repeats and no inversions within the LSC. The total size of the A. ligulata chloroplast genome is 158,724 bp, 49.4% of which is non-coding DNA. The GC content for the whole genome is 36.2%, while that of the protein-coding, rRNA and tRNA genes is 37.4%, 55.3% and 53.2%, respectively. These values are similar to those in other Leguminosae genomes (see Table 1 for those used in our comparisons).
Fig 1. Genome Map of the Acacia ligulata Chloroplast.

Genes shown on the inside of the circle are transcribed in the clockwise direction and those shown on the outside of the circle are transcribed in the anticlockwise direction. Genes marked with an asterisk contain introns, with the introns indicated by clear boxes. The legend indicates the functional group to which each gene belongs. The figure was generated with OrganelleGenomeDRAW [79].
Table 1. GenBank Accession Numbers and References for All Taxa Used in the Phylogenetic and Genomic Comparison of Acacia ligulata.
| Species | Family | GenBank accession | Genome size (bp) | Reference |
|---|---|---|---|---|
| Cicer arietinum | Leguminosae | NC_011163 | 125,319 | [36] |
| Glycine canescens | Leguminosae | KC893635 | 152,518 | Unpub. |
| Glycine cyrtoloba | Leguminosae | KC893632 | 152,381 | Unpub. |
| Glycine dolichocarpa | Leguminosae | KC893636 | 152,804 | Unpub. |
| Glycine falcata | Leguminosae | KC563637 | 153,023 | Unpub. |
| Glycine max | Leguminosae | NC_007942 | 152,218 | [38] |
| Glycine soja | Leguminosae | KF611800 | 152,217 | Unpub. |
| Glycine stenophita | Leguminosae | KC893634 | 152,618 | Unpub. |
| Glycine syndetika | Leguminosae | KC893638 | 152,783 | Unpub. |
| Glycine tomentella | Leguminosae | KC893633 | 152,728 | Unpub. |
| Lathyrus sativus | Leguminosae | NC_014063 | 121,020 | [34] |
| Lotus japonicus | Leguminosae | AP002983 | 150,519 | [39] |
| Lupinus luteus | Leguminosae | NC_014063 | 151,894 | [40] |
| Medicago truncatula | Leguminosae | NC_003119 | 124,033 | Unpub. |
| Millettia pinnata | Leguminosae | NC_016708 | 152,968 | [41] |
| Phaseolus vulgaris | Leguminosae | NC_009259 | 150,285 | [42] |
| Pisum sativum | Leguminosae | NC_014057 | 122,169 | [34] |
| Trifolium subterraneum | Leguminosae | EU849487 | 144,763 | [43] |
| Vigna angularis | Leguminosae | AP012598 | 151,683 | Unpub. |
| Vigna radiata | Leguminosae | NC_013843 | 151,271 | [44] |
| Vigna unguiculata | Leguminosae | JQ755301 | 152,415 | Unpub. |
| Pyrus pyrifolia | Maleae | NC_015996 | 159,922 | [45] |
| Morus indica | Moraceae | DQ226511 | 158,484 | [46] |
| Castanea mollissima | Fagaceae | NC_014674 | 160,799 | [47] |
| Cucumis sativus | Cucurbitaceae | DQ119058 | 155,527 | [48] |
| Eucalyptus globulus | Myrtaceae | KC180787 | 160,267 | [49] |
Genome content and order
The A. ligulata chloroplast genome contains 109 unique genes, including 76 unique protein-coding genes, 4 unique rRNA genes and 29 unique tRNA genes. As is seen throughout the Leguminosae, the rpl22 gene is absent from the A. ligulata plastid genome following an ancient transfer to the nuclear genome [33]. The inverted repeat of the A. ligulata chloroplast genome results in the complete duplication of the rpl2, rpl23, ycf2, ndhB and rps7 genes, as well as exons 1 and 2 of rps12, all four rRNA genes and seven tRNA genes. As is also seen in those Leguminosae species that retain their inverted repeat, the IR of the A. ligulata chloroplast runs roughly 450 bp into the ycf1 gene. This feature has been shown to distinguish legume chloroplasts from many other angiosperms, which typically have 1,000 bp or more of the ycf1 gene included in their IR [38]. Of those legumes that do retain the inverted repeat, that of A. ligulata is larger than in Lupinus, Glycine, Lotus and Millettia, but smaller than in Phaseolus and Vigna (Fig 2). The rps19 gene of A. ligulata is found partially within the IR, with 101 bp being repeated. This is consistent with Glycine and Lotus that also display partial duplication of the rps19 gene. However, this feature varies throughout the Leguminosae, with the duplication of the entire gene in Phaseolus and Vigna, while rps19 is not within the IR for Millettia and Lupinus.
Fig 2. Structure of the LSC/IR junction regions in legume genera.

Protein coding regions are indicated by grey boxes with genes below the line being transcribed right to left and those below the line transcribed left to right. The number of base pairs between the end of the gene and the IR is indicated for genes on either side of the junction, unless the junction coincides with the end of a gene.
Eleven protein-coding genes and seven tRNA genes contained at least one intron, with clpP1, rps12 and ycf3 each containing two introns. This is in contrast to Cicer arietinum, Medicago truncatula, Trifolium subterraneum, Pisum sativum and Lathyrus sativus, all of which have lost an intron in both clpP1 and rps12 [36]. The largest intron was found in trnK-UUU (2,544 bp), spanning the entire matK gene, whilst trnL-UAA contains the smallest intron (543 bp). Two sets of open reading frames overlap: atpA and atpE overlap by four nucleotides whilst psbC and psbD overlap by 17 nucleotides, taking the start codon of psbC to be the GTG codon at position 36,432, based on the results on psbC translation in tobacco [50].
Repeat content
The 59 sets of direct and indirect repeats of 30 bp or longer in the A. ligulata chloroplast genome are listed in S1 Table (not including the large IRs). These include 29 forward repeats, seven reverse repeats, four complementary repeats and 19 palindromic repeats. Repeats were found in the rpl16, ndhA, ycf3 and clpP1 introns, and in the accD, psaA and psaB genes. Compared to other legumes, A. ligulata has a typical repeat content. The Trifolium subterraneum plastid genome contains by far the greatest number of repeats with 500 repeats in total, while Millettia pinnata and Lupinus luteus have the fewest, with 33 and 34 repeats, respectively (Fig 3).
Fig 3. Acacia ligulata Chloroplast Genome Repeat Content Compared to that of Other Legume Genomes.

Repeats are separated into groups according to their size, and the total number of repeats is shown above the bars.
One of these repeats, in the psbJ-petA spacer, is a tandem duplication of 60 bp. This is shorter than the longest tandem repeats found in other legumes: for example, some tandem repeats in Cicer arietinum, Medicago truncatula and Trifolium subterraneum are well over 100 bp in length. The A. ligulata chloroplast genome contains another 31 tandem repeats of 10 bp or more in length (S2 Table). Ten were found within genes, including sets in the ndhA, atpF and clpP1 introns. The remaining repeats were found within intergenic spacer regions. Two sets of tandem repeats observed in A. ligulata are also found in other legumes: repeat 13 is also in the rps12-trnV spacer regions of Lotus japonicus, Millettia pinnata and Lupinus luteus, whereas repeat 21 is also in the ycf2 genes of Millettia pinnata and Lupinus luteus.
Phylogenetic analysis
Phylogenetic reconstruction of the 74 concatenated A. ligulata chloroplast genes, with introns removed, supported previous phylogenetic hypotheses based on both genome rearrangement [36] and the matK gene [32, 51], that place Acacia sister to a clade containing all the Papilionoideae legume taxa. Lupinus is sister to a clade containing two subclades, one containing Cicer, Medicago, Trifolium, Pisum and Lathyrus, and a second containing Millettia, Phaseolus, Vigna and Glycine (Fig 4). All nodes are strongly supported and the phylogeny generated from concatenated genes of the chloroplast genomes is congruent with all but 7 of the 74 trees built from individual chloroplast genes (data not shown).
Fig 4. Phylogenetic Tree Constructed from 74 Concatenated Chloroplast Genes Showing the Position of Acacia ligulata.

Phylogenetic reconstruction was performed using MrBayes with a General Time Reversible model with gamma and invariant sites. Posterior probabilities are indicated above the branches where they differ from 1.
Although not included in the concatenated phylogeny due to its loss in Pisum sativum, a phylogeny was also built for the ycf4 gene (S1 Fig). The ycf4 gene has previously been identified as a region of hypermutation in the Papilionoideae. Although this gene is typically 555 bp long, it has gained an additional several hundred bp in Glycine max, Lotus japonicus and Lathyrus [34, 39, 52]. Acacia ligulata does not display an elevated rate of divergence in this gene, as shown by the short branch length similar to those in the outgroup ycf4 genes.
Divergence in clpP1
In contrast to ycf4, the clpP1 gene is highly divergent in Acacia, as indicated by the unusually long branch length leading to A. ligulata in the tree based on clpP1 sequences (Fig 5). In order to determine the selective pressures influencing the divergence of the A. ligulata clpP1 gene, the non-synonymous versus synonymous nucleotide substitution ratio (dN/dS) was calculated using an alignment of clpP1 coding sequences (S2 Fig). A model using one dN/dS ratio across the clpP1 phylogeny (Fig 6) was compared to a model in which a separate ratio was calculated for the A. ligulata branch. The two-ratios model was found to be a significantly better fit to our nucleotide data than the one-ratio model (likelihood ratio test, P < 0.00001). In this model, the branches leading to the clpP1 genes of all species excluding A. ligulata were found to exhibit a low dN/dS ratio (0.30), indicative of purifying selection, as would be expected for such a highly conserved gene. In contrast, the branch leading to A. ligulata showed a dN/dS ratio (1.07) statistically indistinguishable from that in a model where the dN/dS ratio was fixed as 1 (likelihood ratio test, P > 0.99). This suggests that the clpP1 sequence in A. ligulata may not be under selection at all. An absence of detectable selection is generally considered a strong sign of a pseudogene [53]; however, none of the sequence changes lead to frameshifts or premature stop codons that would clearly indicate that clpP1 is a pseudogene.
Fig 5. Relative branch lengths leading to Acacia ligulata in different gene trees.
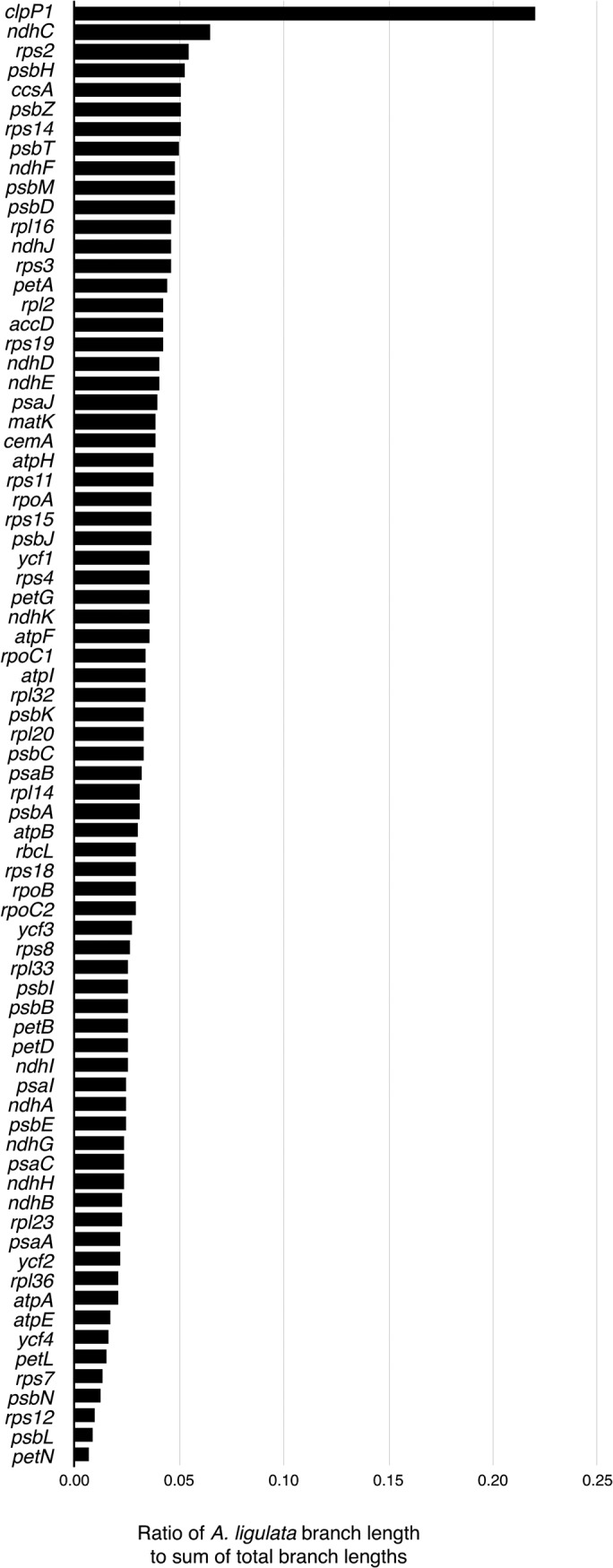
Phylogenetic reconstructions were performed separately for each individual gene alignment using MrBayes with a General Time Reversible model with gamma and invariant sites. The bar chart indicates the proportion of the total branch length in each tree contributed by the branch leading to Acacia ligulata.
Fig 6. Phylogenetic Tree of the clpP1 Gene Showing High Divergence in Acacia ligulata.

Phylogenetic reconstruction was performed using MrBayes with a General Time Reversible model with gamma and invariant sites. Posterior probabilities are indicated above the branches where they differ from 1.
The clpP1 gene encodes a serine protease that is a subunit of the Clp protease [54]. Deletion of the clpP1 gene in both tobacco and Chlamydomonas reinhardtii shows that the gene product is absolutely essential [55–57] and indeed it is one of the few genes consistently conserved in non-photosynthetic parasitic or mycoheterotrophic plants that have greatly reduced chloroplast genomes [58–61]. The poor conservation of this gene in A. ligulata was therefore a surprise. Sequence alignments revealed that a hitherto invariant aspartate (part of the typical protease catalytic triad) has been mutated to a valine in A. ligulata (Fig 7). This mutation cannot be reversed by RNA editing and would imply that the gene product cannot be catalytically active. Mutation of the corresponding aspartate to alanine in bacterial ClpP1 orthologues eliminates proteolytic activity [62]. To verify that the clpP1 gene is actually expressed, we analysed A. ligulata clpP1 transcripts by RT-PCR (Fig 8). Transcripts were easily detected and both introns can be correctly spliced out (verified by sequencing of the products obtained using cDNA as a template), although many transcripts retain intron 1 (Fig 8B). Despite the divergent sequence, this suggests that the clpP1 protein might still be synthesised. In plastids, the Clp complex consists of a heterotetradecameric core composed of two rings of seven subunits [63]. The R-ring consists of three copies of catalytically active ClpP1 (the only subunit encoded by the plastid genome) and single copies of the catalytically inactive ClpR3, ClpR4, ClpR5 and ClpR6 subunits [64]. The P-ring consists of ClpP3, ClpP4, ClpP5, ClpP6 in the ratio 1:2:3:1 [64]. It is possible therefore, that the A. ligulata plastid clpP1 gene product assembles into a Clp complex whose proteolytic function is assured by nucleus-encoded ClpP subunits in the P-ring. However, loss of the ClpP1 active site would completely remove the catalytic activity from the R-ring. To our knowledge, the effects of a loss of activity of a specific ClpP subunit (as opposed to loss of the whole subunit) has not been tested in plants. Lack of expression of individual ClpP subunits leads to severe phenotypes (lethal in the case of ClpP1, ClpP4 and ClpP5; plants lacking ClpP3 can grow heterotrophically, but very slowly; reviewed in [63]).
Fig 7. Alignment of a Region of the ClpP1 Protein Sequence.

Alignment of a portion of the ClpP1 protein sequence from Arabidopsis thaliana, Acacia ligulata, other legumes and outgroups. The three residues of the catalytic triad at amino acid positions 102, 127 and 178 are indicated by black boxes. They are invariant apart from the mutation of aspartate 178 to valine in A. ligulata.
Fig 8. Splicing of the Acacia ligulata clpP1 Transcript.

(A) Schematic representation of the clpP1 transcript showing unspliced and spliced forms. Primer positions are indicated by arrows and the predicted size of PCR products are shown. (B) Ethidium bromide stained 1.0% agarose gel showing PCR amplified products of (1) Acacia ligulata DNA with Primer A + Primer C; (2) Acacia ligulata cDNA with Primer A + Primer C; (3) negative control for Primer A + Primer C; (4) Acacia ligulata DNA with Primer D + Primer F; (5) Acacia ligulata cDNA with Primer D + Primer F; and (6) negative control for Primer D + Primer F.
Search for a nuclear clpP1 gene
It seemed possible that the clpP1 gene has been transferred to the nucleus, and is functionally expressed from this new location in A. ligulata, as suggested in other rare cases where the chloroplast gene appears to be non-functional [65]. In order to identify any potentially nuclear clpP1 sequences, raw reads were compared to the chloroplast clpP1 gene of Lupinus luteus, the closest relative to Acacia with an available clpP1 sequence. Given that a functional transfer of a chloroplast sequence to the nuclear genome would most likely require loss of the two introns, the spliced Lupinus luteus sequence was used for the search. No reads aligning across the splice junctions were found (Table 2). So that the likelihood of identifying nuclear clpP1 reads given the low coverage expected could be ascertained, this analysis was repeated using nuclear CLP gene sequences from Medicago truncatula and Glycine max (the closest relatives of Acacia with sequenced nuclear genomes) as reference sequences. For some of the genes, reads potentially encoding Clp subunits were identified (Table 2). These reads confirm that the A. ligulata nuclear genome does encode subunits for a probable plastid Clp protease, but the low coverage precludes us from concluding whether or not these nuclear genes include a clpP1 paralogue.
Table 2. Results of searches for nucleus-encoded subunits of a plastid Clp complex in the Acacia ligulata sequences.
| Accession | Species | Gene | Length (in bp) | No. of hits | Min. identity | E-value range | % coverage |
|---|---|---|---|---|---|---|---|
| NC_023090 | L. luteus | clpP1 | 591 | 0 | |||
| XM_003624370 | M. truncatula | CLPP3 | 1165 | 1 | 80% | 0.086 | 8.67% |
| XM_003612554 | M. truncatula | clpP4 | 1172 | 3 | 81% | 1.3 – 9e-05 | 17.23% |
| XM_003591344 | M. truncatula | CLPP5 | 1185 | 0 | |||
| XM_003625930 | M. truncatula | CLPP6 | 1163 | 3 | 89% | 1e-06 – 4e-13 | 21.66% |
| XM_003592441 | M. truncatula | clpR1 | 1564 | 1 | 91% | 1e-07 | 6.65% |
| XM_003608743 | M. truncatula | CLPR2 | 1026 | 0 | |||
| XM_003626156 | M. truncatula | CLPR3 | 1416 | 0 | |||
| XM_006600793 | G. max | ClpR4 | 1286 | 1 | 90% | 1e-25 | 7.85% |
The choice of and nomenclature of these subunits follows the current understanding of the structure of the chloroplast Clp complex [63].
Conclusions
Investigations of the A. ligulata chloroplast genome revealed that it resembles a typical angiosperm chloroplast genome, with respect to structure and gene content. The large inversions and deletions observed in the Papilionoideae are not present in the A. ligulata chloroplast genome. Our well-resolved phylogenetic analysis supports existing proposed phylogenies for the Leguminosae. The most unusual feature of the genome is the highly divergent clpP1 gene. Our analysis of this gene suggests that the gene is expressed, but the protein product may not be catalytically active.
Methods
DNA sequencing
Dried phyllode material was obtained from a specimen of Acacia ligulata Benth. (Fabaceae) held at the Western Australian Herbarium (voucher number: PERTH07807864; collected at Lorna Glen, Western Australia, in 2006). Total genomic DNA was extracted using a CTAB protocol [11]. DNA quantity and quality were assessed using a NanoDrop spectrophotometer (ND-1000; Thermo Fisher Scientific, USA), and agarose gel electrophoresis, respectively. Genome library preparation was performed using a Nextera DNA Sample Preparation Kit (Illumina, San Diego, USA), following the manufacturer’s directions. The library was prepared for sequencing using the cBOT cluster generation system and PE V3 flow cell and cluster chemistry (Illumina). The library was sequenced on a single lane in paired-end mode using the HiSeq2000 platform and V3 SBS kit (Illumina). Library preparation and sequencing were both performed at the Ramaciotti Centre for Gene Function Analysis (Sydney, Australia; http://devspace.ddtoo.com/).
Genome assembly
Overlapping paired-end reads were merged using the software FLASH version 1.2.7 [33] and merged reads were assembled using Velvet version 1.2.08 [34], with k-mer values ranging from 51 to 71 and a coverage cut-off of 10. MUMmer version 3.0 [35] was used to compare the assembled chloroplast contigs with the closest related complete chloroplast genome sequence available, Inga leiocalycina Benth. (Koenen et al. unpublished data). Based on the alignments, contigs were ordered and then merged to produce a single draft genome. Finally, reads were mapped to the assembly using Bowtie 2 [66], and visually inspected for discrepancies using Tablet version 1.13.07.31 [67]. Gaps between contigs were filled by PCR amplification with primers that were designed based on the contig sequences (S3 Table). Reactions were performed in 25 μL reactions using 1X PCR Polymerisation Buffer (Fisher-Biotec, Wembley, Australia), 1.5 mM MgCl2, 1.5 μM each forward and reverse primer (GeneWorks; Thebarton, Australia), 0.5 U Taq DNA polymerase (Fisher-Biotec) and 40 ng/μL template DNA. The cycling profile used was: 5 mins at 95°C; followed by 30 secs at 95°C, 45 secs at the annealing temperature (available in S3 Table), and 2 mins at 72°C for 35 cycles; then 4 mins at 72°C.
PCR products were purified prior to sequencing (QIAquick PCR Purification Kit; QIAGEN; Chadstone, Australia), according to the manufacturer’s instructions. Sequencing reactions were performed with forward and reverse primers in separate 10 μL reactions (ABI BigDYE V3.1 Ready-Reaction Kit; Applied Biosystems, USA), following the manufacturer’s directions, and analysed on a 3730XL DNA Analyser (Applied Biosystems). PCR purification and sequencing reactions were performed at the Australian Genome Research Facility (Perth, Australia). Forward and reverse sequences were aligned and manually assessed for incorrect base calls using the CodonCode Aligner software (version 3.7.1; CodonCode Corporation, http://www.codoncode.com/aligner/).
Genome content
The genome was annotated by comparison with other annotated genomes, particularly from other legumes, using NCBI Blast [68]. All tRNA sequences were also checked against the PlantRNA database [69]. The complete A. ligulata genome has been deposited into EMBL (accession number: LN555649). GC content for all species was calculated in Geneious (version 6.0.5; created by BioMatters; available from http://www.geneious.com/). The number and location of all tandem repeats greater than 10 bp were detected for all Leguminosae species using the Phobos Tandem Repeat Finder plugin in Geneious. Additionally, the number of forward, reverse, complementary and palindromic repeats were also detected using REPuter [70]. In order to allow comparison between our analysis and previous repeat analyses in legumes [38, 40, 43, 44], we removed one copy of the IR prior to analysis. Repeats greater than 30 bp were then detected using a Hamming distance of 3, corresponding to a sequence identity of over 90%.
Phylogenetic analyses
Seventy-four protein coding genes were extracted from 21 taxa within the Fabaceae as well as several outgroups, including Eucalyptus globulus, Pyrus pyrifolia, Cucumis sativus, Morus indica and Castanea mollissima. The accD and ycf4 genes were not used in this analysis due to their absence in Trifolium subterraneum and Pisum sativum, respectively. All genome sequences were obtained from GenBank (accession numbers in Table 1). Nucleotide sequences were aligned using MAFFT [71] in Geneious. The model of molecular evolution for each gene was determined using the jModelTest [72] function in MetaPiga version 3.1 [73] (models selected can be seen in S4 Table). The alignments from the 74 genes were concatenated and Bayesian inference was performed using MrBayes [74]. Data were analysed with a Gamma model of rate heterogeneity, the proportion of invariable sites was estimated, and for concatenated multilocus datasets, the alignment was partitioned and branch lengths optimised on a per locus basis.
Bayesian analyses were conducted using MrBayes version 3.2 [75] and were run in parallel on the Fornax supercomputer (located at iVEC@UWA) utilising the BEAGLE library [76]. The Fornax computer consists of 96 computer nodes, each with two six-core Intel Xeon X5650CPUs, a NVIDIA Tesla C2075 GPU and 74 GB of memory. Analyses were run for 10 million generations with sampling every 1,000 generation, partitioned datasets and parameter estimation for each partition unlinked. Each analysis consisted of two independent runs, each utilising twelve chains, eleven cold and one hot. Convergence between runs was monitored by finding a plateau in the likelihood score (standard deviation of split frequencies < 0.0015) and the potential scale reduction factor (PSRF) approaching one. Convergence of other parameters within the runs was also checked using Tracer version 1.5.4 [77], with ESS values above 200 for each run. The first 25% of each run was discarded as burn-in for the estimation of consensus topology and the posterior probability for each node. Bayesian run files are available from the authors upon request.
Assessing clpP1 divergence
Analysis of selection was performed across the clpP1 gene using the codeml package in PAML [78]. dN/dS, the ratio of non-synonymous to synonymous nucleotide substitition, was calculated using an alignment of the clpP1 coding sequences in conjunction with the previously identified phylogeny of clpP1 (Fig 4). We compared the one-ratio model to a branch-specific model, in which the value of dN/dS was separately estimated for A. ligulata. A likelihood ratio test was used to evaluate the model of best fit. A model assuming neutral selection (dN/dS fixed to 1) across all branches was also calculated to determine the significance of the A. ligulata dN/dS value.
Analysis of clpP1 RNA
Acacia ligulata phyllodes were frozen in liquid nitrogen and ground using a ball mill (Retsch; Haan, Germany). Total RNA was extracted using the QIAGEN RNeasy Plant Mini Kit according to the manufacturer’s instructions (buffer RLC was added to the tissue powder). Contaminating genomic DNA was removed using the TURBO DNA-free kit (Ambion) and the treated RNA was assessed for any remaining genomic DNA contamination by standard PCR (primers A, C, D and F). RNA quantity and quality were assessed using a NanoDrop spectrophotometer (ND-1000), and the Agilent 2100 Bioanalyzer (Agilent, USA), respectively. cDNA was generated from 0.5 μg of total RNA using the Superscript III reverse transcriptase (Invitrogen, Australia) and random primers, according to the manufacturer’s instructions.
PCR primers were designed based on the A. ligulata DNA sequence in order to test for intron splicing (S5 Table). Reactions were performed in 20 μL volumes using 1X PCR buffer (Invitrogen), 2.5 mM Mg2+, 0.2 mM dNTPs (Invitrogen), 0.2 μM forward and reverse primers and Platinum Taq DNA polymerase (Invitrogen). The PCR cycling profile was: 5 mins at 94°C, followed by 30 secs at 94°C, 30 secs at 55°C and 1 min at 72°C for 35 cycles, then 10 mins at 72°C. Multiple products were obtained in one case (lane 4 of Fig 8B). Attempts to improve the stringency of the reaction by designing new primers and adjusting the annealing temp or Mg++ concentration were not successful. We obtained the same set of multiple products when pure plasmid containing the 998 bp amplicon was used as a template, so the multiple products are not due to additional copies of the gene elsewhere in the genome. To verify the identification of PCR products generated, products were purified from the gel using the QIAquick Gel Extraction kit (QIAGEN). Purified amplicons were cloned using a pGEM-T Easy vector (Promega, Australia). Plasmid DNA was extracted using the QIAprep Spin Miniprep Kit (QIAGEN), and then sequenced as described above (Macrogen Inc.).
Search for nuclear clp genes
A search database was created from all A. ligulata reads using the BLAST package version 2.2.10 [68]. The L. luteus reference was then compared to the A. ligulata database using blastn. In order to identify nuclear sequences rather than chloroplast sequences, the results were assessed for reads which aligned to the reference across the splice junctions. This analysis was repeated using nucleus-encoded Clp subunits of M. truncatula and G. max as reference sequences (Table 2). Potential matches were confirmed by comparing the reads to a database of plant sequences using tblastx and verifying that the best matches were nuclear CLP genes.
Supporting Information
Phylogenetic reconstruction was performed using MrBayes with a General Time Reversible model with gamma and invariant sites. Posterior probabilities are indicated above the branches.
(EPS)
Nucleotide sequences were aligned using MAFFT in Geneious.
(PDF)
The table lists repeated sequences of 30 or more nucleotides in length. The type of repeat (C, complementary; P, palindromic; F, forward; R, reverse) is indicated.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
Our thanks go to Hayden Walker for his aid with genome assembly, to Joe Miller for his advice on Acacia biology and phylogeny, and to Erik Koenen for providing the unpublished Inga leiocalycina genome sequence.
Data Availability
The complete A. ligulata genome has been deposited into the EMBL database (accession number: LN555649).
Funding Statement
This work was supported by the Australian Research Council (http://www.arc.gov.au) through an Australian Postgraduate Award to AVW, a Discovery Early Career Researcher Award (DE120101117) to KAH and Centre of Excellence grant CE140100008 to IS. Funding was also provided by Karara Mining Limited (http://www.kararamining.com.au). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bui EN, González-Orozco CE, Miller JT: Acacia, climate, and geochemistry in Australia. Plant Soil 2014, 381:161–175. [Google Scholar]
- 2.Australian plant census website. Available: http://www.anbg.gov.au/chah/apc/index.html. Accessed 2015 Apr 8.
- 3. Hnatiuk RJ, Maslin BR: Phytogeography of Acacia in Australia in relation to climate and species-richness. Aust J Bot 1988, 36:361–383. [Google Scholar]
- 4. Gallagher RV, Leishman MR, Miller JT, Hui C, Richardson DM, Suda J et al. : Invasiveness in introduced Australian acacias: the role of species traits and genome size. Divers Distrib 2011, 17:884–897. [Google Scholar]
- 5. Miller JT, Murphy DJ, Brown GK, Richardson DM, González-Orozco CE: The evolution and phylogenetic placement of invasive Australian Acacia species. Divers Distrib 2011, 17:848–860. [Google Scholar]
- 6. Newmaster SG, Ragupathy S: Testing plant barcoding in a sister species complex of pantropical Acacia (Mimosoideae, Fabaceae). Mol Ecol Resour 2009, 9:172–180. [DOI] [PubMed] [Google Scholar]
- 7. Griffin AR, Midgley SJ, Bush D, Cunningham PJ, Rinaudo AT: Global uses of Australian acacias—recent trends and future prospects. Divers Distrib 2011, 17:837–847. [Google Scholar]
- 8. Broadhurst LM, Young AG, Thrall PH, Murray BG: Sourcing seed for Acacia acinacea, a key revegetation species in south eastern Australia. Conserv Genet 2006, 7:49–63. [Google Scholar]
- 9. Butcher P, Harwood C, Quang TH: Studies of mating systems in seed stands suggest possible causes of variable outcrossing rates in natural populations of Acacia mangium . For Genet 2004, 11:303–309. [Google Scholar]
- 10. Butcher P, Williams E, Whitaker D, Ling S, Speed T, Moran G: Improving linkage analysis in outcrossed forest trees–an example from Acacia mangium . Theor Appl Genet 2002, 104:1185–1191. [DOI] [PubMed] [Google Scholar]
- 11. Butcher PA, Moran GF, Perkins HD: RFLP diversity in the nuclear genome of Acacia mangium . Heredity 1998, 81:205–213. [Google Scholar]
- 12. Byrne M, Macdonald B, Coates D: Phylogeographical patterns in chloroplast DNA variation within the Acacia acuminata (Leguminosae: Mimosoideae) complex in Western Australia. J Evol Biol 2002, 15:576–587. [Google Scholar]
- 13. Nevill PG, Wallace MJ, Miller JT, Krauss SL: DNA barcoding for conservation, seed banking and ecological restoration of Acacia in the Midwest of Western Australia. Mol Ecol Resour 2013, 13:1033–1042. 10.1111/1755-0998.12060 [DOI] [PubMed] [Google Scholar]
- 14. Miller JT, Seigler D: Evolutionary and taxonomic relationships of Acacia sl (Leguminosae: Mimosoideae). Aust Syst Bot 2012, 25:217–224. [Google Scholar]
- 15.Mishler BD, Knerr N, Gonzalez Orozco CE, Thornhill AH, Laffan SW, Miller JT: Phylogenetic measures of biodiversity and neo- and paleo-endemism in Australian Acacia Nat Commun 2014, 10.1038/ncomms5473 [DOI] [PubMed]
- 16. Brown G, Ariati S, Murphy D, Ladiges P: Bipinnate acacias (Acacia subg. Phyllodineae sect. Botrycephalae) of eastern Australia are polyphyletic based on DNA sequence data. Aust Syst Bot 2006, 19:315–326. [Google Scholar]
- 17. Brown G, Murphy D, Miller J, Ladiges P: Acacia s.s. and its relationship among tropical legumes, tribe Ingeae (Leguminosae: Mimosoideae). Syst Bot 2008, 33:739–751. [Google Scholar]
- 18. Murphy DJ, Miller JT, Bayer RJ, Ladiges PY: Molecular phylogeny of Acacia subgenus Phyllodineae (Mimosoideae: Leguminosae) based on DNA sequences of the internal transcribed spacer region. Aust Syst Bot 2003, 16:19–26. [Google Scholar]
- 19. Miller JT, Bayer RJ: Molecular phylogenetics of Acacia (Fabaceae: Mimosoideae) based on the chloroplast matK coding sequence and flanking trnK intron spacer regions. Am J Bot 2001, 88:697–705. [PubMed] [Google Scholar]
- 20. Allendorf FW, Hohenlohe PA, Luikart G: Genomics and the future of conservation genetics. Nat Rev Genet 2010, 11:697–709. 10.1038/nrg2844 [DOI] [PubMed] [Google Scholar]
- 21. Angeloni F, Wagemaker N, Vergeer P, Ouborg J: Genomic toolboxes for conservation biologists. Evol Appl 2012, 5:130–143. 10.1111/j.1752-4571.2011.00217.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Avise JC: Perspective: conservation genetics enters the genomics era. Conserv Genet 2010, 11:665–669. [Google Scholar]
- 23. Williams AV, Nevill PG, Krauss SL: Next generation restoration genetics: applications and opportunities. Trends Plant Sci 2014, 19:529–537. 10.1016/j.tplants.2014.03.011 [DOI] [PubMed] [Google Scholar]
- 24. Kane N, Sveinsson S, Dempewolf H, Yang J, Zhang D, Engels J et al. : Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am J Bot 2012, 99:320–329. 10.3732/ajb.1100570 [DOI] [PubMed] [Google Scholar]
- 25. Nock C, Waters D, Edwards M, Bowen S, Rice N, Cordeiro G et al. : Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol J 2011, 9:328–333. 10.1111/j.1467-7652.2010.00558.x [DOI] [PubMed] [Google Scholar]
- 26. Jansen RK, Ruhlman TA: Plastid Genomes of Seed Plants In: Genomics of Chloroplasts and Mitochondria: Advances in Photosynthesis and Respiration. Edited by Bock R, Knoop V, vol. 35: Springer Science and Business Media; 2012: 103–126. [Google Scholar]
- 27. Goremykin VV, Holland B, Hirsch-Ernst KI, Hellwig FH: Analysis of Acorus calamus chloroplast genome and its phylogenetic implications. Mol Biol Evol 2005, 22:1813–1822. [DOI] [PubMed] [Google Scholar]
- 28. Jansen RK, Cai Z, Raubeson LA, Daniell H, dePamphilis CW, Leebens-Mack J et al. : Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci U S A 2007, 104:19369–19374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raubeson L, Peery R, Chumley T, Dziubek C, Fourcade HM, Boore J et al. : Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus . BMC Genomics 2007, 8:174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doyle JJ, Doyle JL, Ballenger JA, Palmer JD: The distribution and phylogenetic significance of a 50-kb chloroplast DNA inversion in the flowering plant family Leguminosae. Mol Phylogenet Evol 1996, 5:429–538. [DOI] [PubMed] [Google Scholar]
- 31. Bruneau A, Doyle JJ, Palmer JD: A chloroplast DNA inversion as a subtribal character in the Phaseoleae (Leguminosae). Syst Bot 1990:378–386. [Google Scholar]
- 32. Wojciechowski MF, Lavin M, Sanderson MJ: A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am J Bot 2004, 91:1846–1862. [DOI] [PubMed] [Google Scholar]
- 33. Gantt JS, Baldauf SL, Calie PJ, Weeden NF, Palmer JD: Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J 1991, 10:3073–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Magee AM, Aspinall S, Rice DW, Cusack BP, Semon M, Perry AS et al. : Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res 2010, 20:1700–1710. 10.1101/gr.111955.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doyle JJ, Doyle JL, Palmer JD: Multiple independent losses of two genes and one intron from legume chloroplast genomes. Syst Bot 1995:272–294.
- 36. Jansen RK, Wojciechowski MF, Sanniyasi E, Lee S-B, Daniell H: Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol Phylogenet Evol 2008, 48:1204–1217. 10.1016/j.ympev.2008.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lavin M, Doyle JJ, Palmer JD: Evolutionary significance of the loss of the chloroplast-DNA inverted repeat in the Leguminosae subfamily Papilionoideae. Evolution 1990, 44:390–402. [DOI] [PubMed] [Google Scholar]
- 38. Saski C, Lee SB, Daniell H, Wood TC, Tomkins J, Kim HG et al. : Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol Biol 2005, 59:309–322. [DOI] [PubMed] [Google Scholar]
- 39. Kato T, Kaneko T, Sato S, Nakamura Y, Tabata S: Complete structure of the chloroplast genome of a legume, Lotus japonicus . DNA Res 2000, 7:323–330. [DOI] [PubMed] [Google Scholar]
- 40. Martin GE, Rousseau-Gueutin M, Cordonnier S, Lima O, Michon-Coudouel S, Naquin D et al. : The first complete chloroplast genome of the Genistoid legume Lupinus luteus: evidence for a novel major lineage-specific rearrangement and new insights regarding plastome evolution in the legume family. Ann Bot 2014, 113:1197–1210. 10.1093/aob/mcu050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kazakoff SH, Imelfort M, Edwards D, Koehorst J, Biswas B, Batley J et al. : Capturing the biofuel wellhead and powerhouse: the chloroplast and mitochondrial genomes of the leguminous feedstock tree Pongamia pinnata . PLoS ONE 2012, 7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo X, Castillo-Ramirez S, Gonzalez V, Bustos P, Fernandez-Vazquez JL, Santamaria RI et al. : Rapid evolutionary change of common bean (Phaseolus vulgaris L) plastome, and the genetic diversification of legume chloroplasts. BMC Genomics 2007, 8:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cai Z, Guisinger M, Kim H-G, Ruck E, Blazier JC, McMurtry V et al. : Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J Mol Evol 2008, 67:696–704. [DOI] [PubMed] [Google Scholar]
- 44. Tangphatsornruang S, Sangsrakru D, Chanprasert J, Uthaipaisanwong P, Yoocha T, Jomchai N et al. : The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: structural organization and phylogenetic relationships. DNA Res 2010, 17:11–22. 10.1093/dnares/dsp025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Terakami S, Matsumura Y, Kurita K, Kanamori H, Katayose Y, Yamamoto T et al. : Complete sequence of the chloroplast genome from pear (Pyrus pyrifolia): genome structure and comparative analysis. Tree Genet Genomes 2012, 8:841–854. [Google Scholar]
- 46. Ravi V, Khurana JP, Tyagi AK, Khurana P: The chloroplast genome of mulberry: complete nucleotide sequence, gene organization and comparative analysis. Tree Genet Genomes 2006, 3:49–59. [Google Scholar]
- 47. Jansen RK, Saski C, Lee S-B, Hansen AK, Daniell H: Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol Biol Evol 2011, 28:835–847. 10.1093/molbev/msq261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim J-S, Jung JD, Lee J-A, Park H-W, Oh K-H, Jeong W-J et al. : Complete sequence and organization of the cucumber (Cucumis sativus L. cv. Baekmibaekdadagi) chloroplast genome. Plant Cell Rep 2006, 25:334–340. [DOI] [PubMed] [Google Scholar]
- 49. Bayly MJ, Rigault P, Spokevicius A, Ladiges PY, Ades PK, Anderson C et al. : Chloroplast genome analysis of Australian eucalypts—Eucalyptus, Corymbia, Angophora, Allosyncarpia and Stockwellia (Myrtaceae). Mol Phylogenet Evol 2013, 69:704–716. 10.1016/j.ympev.2013.07.006 [DOI] [PubMed] [Google Scholar]
- 50. Kuroda H, Suzuki H, Kusumegi T, Hirose T, Yukawa Y, Sugiura M: Translation of psbC mRNAs starts from the downstream GUG, not the upstream AUG, and requires the extended Shine-Dalgarno sequence in tobacco chloroplasts. Plant Cell Physiol 2007, 48:1374–1378. [DOI] [PubMed] [Google Scholar]
- 51. The Legume Phylogeny Working Group: Legume phylogeny and classification in the 21st century: Progress, prospects and lessons for other species-rich clades. Taxon 2013, 62:217–248. [Google Scholar]
- 52. Reverdatto S, Beilinson V, Nielsen N: The rps16, accD, psaI, ORF 203, ORF 151, ORF 103, ORF 229 and petA gene cluster in the chloroplast genome of soybean (PGR95-051). Plant Physiol 1995, 109:338. [Google Scholar]
- 53. Svensson O, Arvestad L, Lagergren J: Genome-wide survey for biologically functional pseudogenes. PLoS Comput Biol 2006, 2:e46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maurizi MR, Clark WP, Kim SH, Gottesman S: Clp P represents a unique family of serine proteases. J Biol Chem 1990, 265:12546–12552. [PubMed] [Google Scholar]
- 55. Shikanai T, Shimizu K, Ueda K, Nishimura Y, Kuroiwa T, Hashimoto T: The chloroplast clpP gene, encoding a proteolytic subunit of ATP-dependent protease, is indispensable for chloroplast development in tobacco. Plant Cell Physiol 2001, 42:264–273. [DOI] [PubMed] [Google Scholar]
- 56. Kuroda H, Maliga P: The plastid clpP1 protease gene is essential for plant development. Nature 2003, 425:86–89. [DOI] [PubMed] [Google Scholar]
- 57. Huang C, Wang S, Chen L, Lemieux C, Otis C, Turmel M et al. : The Chlamydomonas chloroplast clpP gene contains translated large insertion sequences and is essential for cell growth. Mol Gen Genet 1994, 244:151–159. [DOI] [PubMed] [Google Scholar]
- 58. Delannoy E, Fujii S, Colas des Francs-Small C, Brundrett M, Small I: Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol Biol Evol 2011, 28:2077–2086. 10.1093/molbev/msr028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Funk HT, Berg S, Krupinska K, Maier UG, Krause K: Complete DNA sequences of the plastid genomes of two parasitic flowering plant species, Cuscuta reflexa and Cuscuta gronovii . BMC Plant Biol 2007, 7:45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wolfe KH, Morden CW, Palmer JD: Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant. Proc Natl Acad Sci U S A 1992, 89:10648–10652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McNeal JR: Parallel loss of plastid introns and their maturase in the genus Cuscuta . PLoS ONE 2009, 4:e5982 10.1371/journal.pone.0005982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zeiler E, List A, Alte F, Gersch M, Wachtel R, Poreba M et al. : Structural and functional insights into caseinolytic proteases reveal an unprecedented regulation principle of their catalytic triad. Proc Natl Acad Sci U S A 2013, 110:11302–11307. 10.1073/pnas.1219125110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nishimura K, van Wijk KJ: Organization, function and substrates of the essential Clp protease system in plastids. Biochim Biophys Acta 2014, 10.1016/j.bbabio.2014.11.012 [DOI] [PubMed]
- 64. Olinares PD, Kim J, Davis JI, van Wijk KJ: Subunit stoichiometry, evolution, and functional implications of an asymmetric plant plastid ClpP/R protease complex in Arabidopsis. Plant Cell 2011, 23:2348–2361. 10.1105/tpc.111.086454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Martínez-Alberola F, del Campo EM, Lázaro-Gimeno D, Mezquita-Claramonte S, Molins A, Mateu-Andrés I et al. : Balanced gene losses, duplications and intensive rearrangements led to an unusual regularly sized genome in Arbutus unedo chloroplasts. PLoS ONE 2013, 8:e79685 10.1371/journal.pone.0079685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Langmead B, Salzberg SL: Fast gapped-read alignment with Bowtie 2. Nat Meth 2012, 9:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Milne I, G. S, Bayer M, Cock P, Pritchard L, Cardle L, Shaw P et al. : Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 2013, 14:193–202. 10.1093/bib/bbs012 [DOI] [PubMed] [Google Scholar]
- 68. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215:403–410. [DOI] [PubMed] [Google Scholar]
- 69. Cognat V, Pawlak G, Duchêne A-M, Daujat M, Gigant A, Salinas T et al. : PlantRNA, a database for tRNAs of photosynthetic eukaryotes. Nucleic Acids Res 2013, 41:D273–D279. 10.1093/nar/gks935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R: REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 2001, 29:4633–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Katoh K, Misawa K, Kuma Ki, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002, 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Posada D: jModelTest: Phylogenetic Model Averaging. Mol Biol Evol 2008, 25:1253–1256. 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- 73. Helaers R, Milinkovitch MC: MetaPIGA v2. 0: maximum likelihood large phylogeny estimation using the metapopulation genetic algorithm and other stochastic heuristics. BMC Bioinformatics 2010, 11:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17:754–755. [DOI] [PubMed] [Google Scholar]
- 75. Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S et al. : MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 2012, 61:539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ayres DL, Darling A, Zwickl DJ, Beerli P, Holder MT, Lewis PO et al. : BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst Biol 2012, 61:170–173. 10.1093/sysbio/syr100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rambaut A, Suchard M, Drummond A: Tracer website. Available: http://tree.bio.ed.ac.uk/software/tracer/. Accessed 2015 Apr 8.
- 78. Yang Z: PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007, 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- 79. Lohse M, Drechsel O, Kahlau S, Bock R: OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res 2013, 41:W575–581. 10.1093/nar/gkt289 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic reconstruction was performed using MrBayes with a General Time Reversible model with gamma and invariant sites. Posterior probabilities are indicated above the branches.
(EPS)
Nucleotide sequences were aligned using MAFFT in Geneious.
(PDF)
The table lists repeated sequences of 30 or more nucleotides in length. The type of repeat (C, complementary; P, palindromic; F, forward; R, reverse) is indicated.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
The complete A. ligulata genome has been deposited into the EMBL database (accession number: LN555649).