

Abstract
The pharmacokinetics of adenosine 5′-triphosphate (ATP) was investigated in a clinical trial that included 15 patients with advanced malignancies (solid tumors). ATP was administered by continuous intravenous infusions of 8 h once weekly for 8 weeks. Three values of blood ATP levels were determined. These were total blood (erythrocyte) and blood plasma (extracellular) ATP pools along with the initial rate of release of ATP into the blood plasma. We found that values related to erythrocyte ATP pools showed great variability (diversity) among individuals (standard deviation of about 30–40 % of mean at baseline). It was discovered that erythrocyte baseline ATP pool sizes are unique to each individual and that they fall within a narrow range in each individual. At the end of an 8 h continuous intravenous infusion of ATP, intracellular erythrocyte ATP pools were increased in the range of 40–60 % and extracellular ATP declined from elevated levels achieved at the beginning and middle of the infusion, to baseline levels. The ability of erythrocytes to sequester exogenously administered ATP to this degree, after its initial conversion to adenosine in the blood plasma is unexpected, considering that some of the adenosine is likely to have been degraded by in vivo catabolic activities or taken up by organs. The data suggest that administration of ATP by short-term intravenous infusions, of up to 4 h, may be a favorable way for elevating extracellular ATP pools. A large fraction of the total exogenously administered ATP is sequestered into the intracellular compartments of the erythrocytes after an 8 h intravenous infusion. Erythrocytes loaded with ATP are known to release their ATP pools by the application of previously established agents or conditions applied locally or globally to circulating erythrocytes. Rapid degradation of intravenously administered ATP to adenosine and subsequent accumulation of ATP inside erythrocytes indicate the existence of very effective mechanisms for uptake of adenosine from blood plasma. These in vivo studies offer an understanding as to how both adenosine and ATP can act as purinergic transmission signals. ATP levels in blood are always accompanied by adenosine formed by catabolism of ATP. The continuous uptake of adenosine enables both to act in transmission of sometimes opposite functions.
Keywords: ATP, Extracellular ATP, Cancer, Pharmacokinetics, Human trial
Introduction
The discovery that extracellular adenosine 5′-triphosphate (ATP) acts as a neurotransmitter was made by Burnstock and reported in 1970 [1]. ATP possesses high metabolic lability, especially at the beta-gamma and alpha-beta phosphodiester bonds, which allows its intracellular pools (steady state concentrations) to fluctuate in response to extracellular conditions that affect growth or cellular metabolism. Extracellular activities, which catalyze the degradation of ATP to adenosine, are performed mostly by ecto-ATPDases (diphosphohydrolase, cd39, or apyrase) [2] followed by activities of 5′-nucleotidase (cd73) [3]. ATP evolved into a role of the major extracellular signal. The extracellular activities of ATP are in addition to its vast intracellular role in cellular energetics, as an intermediate in biosynthetic pathways and as an allosteric effector of protein activity. Polypeptides or steroid hormones on the other hand, because of their high metabolic stability, evolved into extracellular signals or endocrine hormones affecting responder tissues primarily by interacting with specific membrane receptors. ATP can also be regarded as a hormone, being sequestered in erythrocytes. Erythrocytes traverse the vascular bed and upon signals (chemical, mechanical, or physiological) release ATP, thus the stimulus-secretion action that characterizes endocrine action. This system can be used for release of ATP at sites when and where ATP is needed. As a therapeutic modality, ATP has the potential of being delivered for the physiological execution of vascular and extravascular functions (e.g., established activities in inhibition of platelet aggregation and thrombus formation) [4].
It is known that extracellular ATP, upon binding to its P2Y and P2X receptors [5] and its in vivo degradation product, adenosine, acting on its adenosine (P1) receptors [6], are major autocrine and paracrine signals controlling a myriad of physiological functions. It was demonstrated that very low levels of extracellular ATP kill some tumor cells with high specificity in vitro and in mice tumor models [7]. Extracellular ATP cytotoxicity was shown to involve permeabilization or pore formation of tumor cell membrane leading to tumor cell death [8]. It was assumed for a long time that the main source of ATP acting on purine receptors were damaged or dying cells. It is now clear, however, that ATP is released without prior damage, from many cell types, including the mature erythrocyte [7, 9, 10]. Of particular significance is the activity of extracellular ATP in mice tumor models, achieving “cures” in experiments where tumor burden is small [11, 12]. Thus, ATP may also possess a role in a biological tumor surveillance system. Recently published articles suggest that extracellular ATP is critical (obligatory) in inducing immunogenic cell death [13]. Erythrocyte ATP release to above baseline level is affected mainly by two factors: hypoxia (low oxygen tension) and cellular deformation. Erythrocytes are exposed to signaling factors when traversing the microcirculation [14]. These conditions, which exist in vivo are also known to stimulate release of erythrocytes’ ATP when applied in isolated controlled systems [14, 15]. ATP acts on vascular endothelial receptors, which, in turn, induce the synthesis of vasodilators. Although ATP is released in a paracrine manner, data presented here suggest that the erythrocyte ATP pool can act as an endocrine system, traversing the vascular bed and having the potential of being released for purposes such as bio-surveillance of developing tumors.
A major aspect of the present trial was establishing whether expansions of ATP pools in erythrocytes could provide sufficient ATP levels to be used therapeutically in advanced, terminal cancer, an immediately life-threatening disease for which no other therapeutic option exists. The other questions asked in the pharmacokinetics part of the trial are whether erythrocyte ATP pools can be increased meaningfully after 8 h of continuous intravenous infusions of ATP and are the rate of release of ATP from erythrocytes and the all important blood plasma pools of ATP increased concomitantly by infusions of exogenous ATP. What is the stability of extracellular ATP, and finally, is there any value that affects the variability (diversity) of ATP pool sizes in erythrocytes among individual patients and do erythrocyte ATP pools affect subsequent accumulation (sequestration) of exogenous ATP in erythrocytes.
Haskell et al. reported a variability/range of 0.56–1.49 mM (mean ± 2 standard deviation) in total blood ATP pools at baseline among hospitalized patients without cancer and eight patients with advanced cancer [16]. Since ATP itself does not permeate cellular plasma membranes in normal non-transformed cells, the first step in the uptake of ATP is its enzymatic degradation to adenosine, which is taken up by erythrocytes and acts in expanding total erythrocyte ATP pools. The expansion of erythrocyte ATP pools by blood plasma adenosine is explained by the fact that the expanded ATP pools are not regulated by feedback but by substrate supply [7, 12, 17, 18]. Free adenosine is taken up by erythrocytes using two mechanisms depending on the type of nucleoside transporters (NTs). One operates by facilitated diffusion (equilibrative transport) and is dependent on nucleoside gradient and becoming saturated at about 10 μM and the other type of transporters (concentrative) whereby adenosine is transported by utilization of transmembrane sodium ion concentration gradients. Adenosine entering the erythrocyte is rapidly phosphorylated by cellular adenosine kinase, an enzyme also regulated by substrate supply. The metabolism of adenosine in blood was established in the 1960–1970s [19–22] and consists of rapid elimination of adenosine from the blood plasma by means of catabolism and cellular uptake. The two enzymes that act in catabolizing adenosine are cellular adenosine deaminase with a Ka for adenosine in the vicinity of 10 μM and cellular adenosine kinase with a Ka for adenosine of about 1 μM. These two enzymatic activities, along with the two erythrocytes nucleoside uptake systems, [23] metabolize high and low levels of adenosine in blood. We show in this publication that erythrocytes, which sequester adenosine in the form of expanded ATP pools, have a major role in enabling ATP and adenosine to act as purinergic signals. Since the presence of ATP in blood plasma is always accompanied by the presence of adenosine, its biological degradation product, only an effective removal of adenosine would enable adenosine and ATP to regulate opposing activities, such as inflammation by ATP as agonist and immunosuppression by adenosine as agonist. A further consideration is that adenosine has poor solubility in blood plasma in comparison to ATP, which has higher solubility by orders of magnitude. Erythrocytes were shown not to possess the major ecto-ATPDase (cd39) and lack the major 5′-nucleotidase (cd73) as well [24]. Erythrocytes possess a slow adenosine uptake system and are void of catabolic enzymatic activities required to convert the exogenously administered ATP to adenosine [24, 25].
Recently, it was shown that human red blood cells are capable of storing nitric oxide (NO) and that the red blood cell was not simply a sink for NO. NO and its related metabolic products in red blood cells produced an “erythrocrine” function or an “endocrine” function in these cells [26]. The classical definition of an endocrine organ involves a stimulus-secretion mechanism. The release of erythrocyte ATP in response to hypoxia/deformation complies with this definition.
Materials and methods
Clinical trial
A clinical phase I trial was performed between December 20, 2000 and July 18, 2002 at Dartmouth-Hitchcock Medical Center. The study was conducted in accordance with standards of current Good Clinical Practice, as defined by the International Conference on Harmonization, the Food and Drug Administration, and all applicable federal and local regulations. All patients participating in the trial signed an informed consent form, copy of which is submitted with this manuscript. The studies were approved by the institutional research ethics committee and were performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments.
Treatments
ATP was administered to each subject once weekly for eight consecutive weeks as an 8 h continuous intravenous infusion at rates of 50, 75, or 100 μg/kg min. ATP was provided as a sterile solution in single use vials. Each vial contained 2 g of ATP as a sodium salt in 20 mL of water for injection. The ATP solutions were adjusted to pH 6.7–7.2 with sodium hydroxide before sterilization. The final concentration of ATP in the vials was 100 mg/mL. Storage of the clinical solutions was at a controlled refrigerated temperature (2–8 °C). The administering solution required that the volume of one vial of ATP be aseptically removed using a syringe and added to a 250 mL bag of 0.5 % normal saline (0.45 % NaCl). The concentration of the final sterile solution was 8 mg/mL ATP. The solution was administered by continuous intravenous infusions using an Ivac or similar infusion device, through venous access in a peripheral vein. If venous access was a problem, either a Hickman catheter or its equivalent, or an Infusaport or its equivalent was inserted to provide vascular access.
Each patient received doses of ATP of 50–100 μg/kg min. The 50 μg/kg min dose could be reduced to 25 μg/kg/min if adverse effects occurred. The rate of infusion of ATP during each of the weekly 8-h infusions was determined according to the following set of guidelines:
In all patients, the initial 8 h of infusion (infusion 1 on day 0, week 1) was at 50 μg/kg min, a rate of infusion that has been generally well tolerated in other clinical trials of ATP in cancer patients [16]. Venous blood samples (5 mL) for pharmacokinetic assessments were obtained via an indwelling catheter in the limb contralateral to that used for the infusion of ATP.
Samples were collected in ethylenediaminetetraacetic acid (EDTA) tubes (2 × 2.5 mL purple tops) and immediately placed on ice. Samples were obtained at time 0 (pre-infusion) and at 4 h into the infusion, just before the end of the infusion (8 h) and at the following times: 8.5, 9, 10, 11, 12, 14, 16, 20, and 24 h.
Sample preparation
Samples of human blood were collected in 2.5 mL evacuated blood collection tubes (Vacutainer, Becton Dickinson Company) containing 0.025 mL of a 15 % EDTA (K3) solution. Samples were collected from the arm contralateral to that used to infuse the ATP. These samples were placed on ice and kept at 3–5 °C. Whole blood was diluted either 1/100 (for blood plasma ATP pools or rate of release of ATP into blood plasma determinations) or 1/1000 into phosphate-buffered saline (PBS), for erythrocyte cellular ATP pool determinations (lysing dilution). Measurement of diluted whole blood avoided artifacts produced when erythrocytes are separated by centrifugation. Concomitant erythrocytes counts were obtained using a Coulter counter. Both extracellular ATP and the rate of release of ATP into the blood plasma are referred to as initial values. Both blood values of ATP are determined initially after collection of the sample. In case of the rate of release of ATP, the presence of EDTA blocks the activities of all catabolic enzymes, and the accumulation of ATP on top of the initial levels of extracellular ATP (during a 5-min period) is used to calculate the rate of release of ATP from erythrocytes (in nM/min).
Analysis of ATP levels is very accurate for erythrocyte ATP pools (total blood) and less accurate for the other two values, which are lower by about four orders of magnitude.
The method of analyzing ATP employed the firefly luciferase assay. The assays were performed with a Packard Instrument Co. 12 detector luminometer. The diluted blood was analyzed in a 96-well plate (Packard Co.). The luciferin/luciferase reaction assay (Sigma) had a threshold for ATP detection in the subnanomolar concentration range using this system. Quadruplicate standards were measured over ATP concentration range from blank (fresh PBS), 10−14 to 10−9 mol per well with a complete set of standards comprising 24 of the total 96 wells. Additional sets of standards were to have been employed to correct for the presence of any added compounds such as EDTA or lysing reagents. In this assay, samples and standards were repeatedly analyzed at a rate of approximately two times per minute. The activities of ATPDase/cd39 (apyrase) and related extracellular enzymes were completely inhibited. As a result, the release rate of ATP from the erythrocytes could be monitored. Collection of samples was precisely timed to the internal clock of the luminometer. The time from sample collection to introduction of the samples to the luminometer was 5 min. Routinely, the diluted blood samples were measured again at 1 h after collection. The diluted samples kept on ice had an exponential decrease in total ATP. If the samples were not ready at the 5 min time point, it was possible to determine these levels by back extrapolation if three or more determinations of ATP levels were made. In general, ATP values were determined based on the average of 12 individual wells (12 replicates) per sample. With dual standards on a single plate, it was possible to simultaneously measure total blood ATP and erythrocyte ATP release rates.
Results
In assessing pharmacokinetics during a clinical trial, we noticed that the mean sizes of erythrocyte ATP pools or any other value closely associated with erythrocyte ATP pools had standard deviations of at least 30–40 % of the mean value. Three parameters of blood ATP levels were determined: total blood (erythrocytes) ATP pools, initial ATP release rate into blood plasma, and initial extracellular (blood plasma) ATP pools. Since the method of determining ATP levels at the patient bedside was consistent and reproducible with an estimated standard deviation of less than 10 %, we decided to investigate the possibility that the introduction of the large standard deviation was due to the variability (diversity) of individual patients’ pool size of ATP. The range of erythrocyte ATP pool sizes remains stable over a period of 8 weeks indicating an effective homeostatic system maintaining the size of the ATP pools. It was discovered that baseline erythrocyte ATP pool sizes are unique to each individual and are maintained for weeks within a narrow range. The range of individual patients’ erythrocyte ATP pool sizes was about 0.25 mM. Since ATP is not taken up directly by cells, the capacity of erythrocytes for uptake of adenosine is essential for the separation of ATP and adenosine functions. The variations in intra-erythrocyte ATP pool sizes (differences in ATP pools within one patient at different weeks) were smaller than variations in inter-erythrocyte ATP pool sizes (variations from one patient to another or the diversity of erythrocyte ATP pool sizes among patients). Stability of intra-erythrocyte pool sizes of ATP within a narrow range up to 8 weeks was observed even though the individual patients were nutritionally deficient due to their advanced disease. In order to minimize the biological degradation of ATP ex vivo, blood samples were drawn at the patients’ bedside into EDTA tubes, which were maintained at ice temperature. The biological degradation of ATP is in a form of dephosphorylation, which is catalyzed by vascular ATPDase/cd39 diphosphohydrolase (apyrase) along with non-specific phosphatases followed by the action of 5′-nucleotidase/cd73. The initial (first determination after blood withdrawal) rate of release of ATP into blood plasma and the initial size of extracellular (blood plasma) ATP pools, before, during, and after 8 h of continuous intravenous infusions of ATP in advanced cancer patients, were determined. Tables 1, 2, and 3 outline data for the three values of ATP in whole blood, which were measured during the trial. The column headed by N specifies the number of patients receiving intravenous infusions of ATP on week 1, week 3, or week 8, respectively. Time 0 or pre-infusion sizes of ATP pools are equal to baseline values. The mean values for baseline (pre-infusion) intracellular ATP pools in total blood were determined as 0.615 ± 0.229, 0.610 ± 0.248, and 0.573 ± 0.164 mM of ATP. All values reported here represent constitutive release of ATP, namely no support by in vivo stimulants of ATP release (hypoxia, shear stress) or by exogenous ATP releasing agents [14, 27]. The expansions of erythrocyte ATP pools before, during, and after infusions of ATP are illustrated in Fig. 1.
Table 1.
Erythrocyte intracellular ATP pools before, during, and after intravenous infusions of ATP level (mM)
| Visit | Time point | N | Mean | Std. Dev. | Median | Minimum | Maximum |
|---|---|---|---|---|---|---|---|
| Week 1 (infusion 1) | Time “0” (pre-infusion) | 15 | 0.61 | 0.22 | 0.55 | 0.31 | 1.10 |
| 4 h into infusion | 14 | 0.80 | 0.34 | 0.75 | 0.40 | 1.50 | |
| 8 h (before the end) | 15 | 0.89 | 0.32 | 0.70 | 0.58 | 1.60 | |
| 8.5 h | 15 | 0.89 | 0.30 | 0.81 | 0.56 | 1.52 | |
| 9 h | 14 | 0.91 | 0.44 | 0.76 | 0.52 | 1.90 | |
| 10 h | 14 | 0.82 | 0.32 | 0.73 | 0.46 | 1.50 | |
| 11 h | 15 | 0.82 | 0.42 | 0.68 | 0.46 | 2.00 | |
| 12 h | 15 | 0.84 | 0.39 | 0.69 | 0.48 | 1.80 | |
| 14 h | 11 | 0.85 | 0.42 | 0.73 | 0.46 | 1.70 | |
| 16 h | 10 | 0.90 | 0.44 | 0.75 | 0.48 | 1.80 | |
| 20 h | 11 | 0.77 | 0.24 | 0.67 | 0.49 | 1.20 | |
| 24 h | 15 | 0.71 | 0.26 | 0.62 | 0.44 | 1.40 | |
| Week 3 (infusion 3) | Time “0” (pre-infusion) | 12 | 0.61 | 0.24 | 0.56 | 0.24 | 1.00 |
| 4 h into infusion | 11 | 0.84 | 0.49 | 0.54 | 0.35 | 1.66 | |
| 8 h (before the end) | 11 | 0.95 | 0.45 | 0.77 | 0.49 | 1.60 | |
| 8.5 h | 11 | 0.98 | 0.44 | 0.96 | 0.40 | 1.66 | |
| 9 h | 11 | 0.90 | 0.39 | 0.95 | 0.40 | 1.60 | |
| 10 h | 10 | 0.90 | 0.39 | 1.00 | 0.45 | 1.60 | |
| 11 h | 11 | 0.88 | 0.51 | 0.67 | 0.41 | 1.84 | |
| 12 h | 11 | 0.90 | 0.53 | 0.82 | 0.36 | 2.10 | |
| 14 h | 9 | 0.85 | 0.48 | 0.60 | 0.40 | 1.70 | |
| 16 h | 9 | 0.85 | 0.41 | 0.74 | 0.36 | 1.70 | |
| 20 h | 8 | 0.85 | 0.60 | 0.61 | 0.52 | 2.30 | |
| 24 h | 11 | 0.68 | 0.33 | 0.58 | 0.33 | 1.50 | |
| Week 8 (infusion 8) | Time “0” (pre-infusion) | 6 | 0.57 | 0.16 | 0.56 | 0.36 | 0.81 |
| 4 h into infusion | 5 | 0.70 | 0.14 | 0.77 | 0.53 | 0.86 | |
| 8 h (before the end) | 5 | 0.82 | 0.24 | 0.89 | 0.53 | 1.06 | |
| 8.5 h | 3 | 1.00 | 0.35 | 0.98 | 0.66 | 1.36 | |
| 9 h | 4 | 1.03 | 0.34 | 1.15 | 0.53 | 1.28 | |
| 10 h | 4 | 0.96 | 0.30 | 1.08 | 0.52 | 1.17 | |
| 11 h | 4 | 0.97 | 0.32 | 1.01 | 0.55 | 1.32 | |
| 12 h | 4 | 0.92 | 0.29 | 1.05 | 0.48 | 1.12 | |
| 14 h | 5 | 0.98 | 0.37 | 0.95 | 0.48 | 1.52 | |
| 16 h | 5 | 0.89 | 0.46 | 0.70 | 0.49 | 1.69 | |
| 20 h | 5 | 0.96 | 0.39 | 0.90 | 0.50 | 1.58 | |
| 24 h | 5 | 0.70 | 0.10 | 0.75 | 0.55 | 0.81 |
Table 2.
Initial ATP release rates from erythrocytes into the blood plasma (extracellular). Initial ATP release rates (nM/min)
| Visit | Time point | N | Mean | Std. Dev. | Median | Minimum | Maximum |
|---|---|---|---|---|---|---|---|
| Week 1 (infusion 1) | Time “0” (pre-infusion) | 13 | 15.45 | 13.40 | 10.10 | 2.90 | 46.90 |
| 4 h into infusion | 13 | 33.40 | 33.72 | 23.40 | 1.60 | 134.30 | |
| 8 h (before the end) | 13 | 47.22 | 34.51 | 31.75 | 9.25 | 124.90 | |
| 8.5 h | 12 | 41.35 | 30.37 | 32.42 | 7.90 | 90.10 | |
| 9 h | 13 | 25.40 | 17.10 | 26.40 | 3.40 | 57.60 | |
| 10 h | 13 | 13.95 | 9.41 | 10.40 | 4.00 | 36.69 | |
| 11 h | 15 | 14.74 | 11.81 | 11.70 | 2.33 | 45.50 | |
| 12 h | 15 | 12.56 | 9.74 | 10.20 | 3.20 | 36.11 | |
| 14 h | 9 | 12.06 | 14.64 | 7.14 | 1.50 | 47.46 | |
| 16 h | 10 | 15.33 | 16.69 | 10.20 | 4.50 | 61.80 | |
| 20 h | 9 | 20.06 | 18.33 | 12.62 | 5.40 | 57.90 | |
| 24 h | 13 | 17.45 | 10.38 | 14.80 | 4.50 | 37.80 | |
| Week 3 (infusion 3) | Time “0” (pre-infusion) | 10 | 18.22 | 11.65 | 13.35 | 5.74 | 44.34 |
| 4 h into infusion | 9 | 55.41 | 79.91 | 35.10 | 5.80 | 265.20 | |
| 8 h (before the end) | 9 | 48.28 | 24.62 | 49.00 | 13.60 | 96.00 | |
| 8.5 h | 10 | 38.78 | 21.00 | 39.50 | 9.30 | 72.00 | |
| 9 h | 9 | 34.99 | 23.01 | 37.33 | 6.90 | 69.50 | |
| 10 h | 9 | 27.34 | 20.45 | 21.60 | 2.60 | 61.00 | |
| 11 h | 11 | 18.88 | 16.19 | 13.36 | 1.90 | 53.30 | |
| 12 h | 10 | 14.66 | 10.69 | 13.15 | 2.90 | 34.90 | |
| 14 h | 6 | 10.39 | 12.17 | 5.80 | 2.70 | 34.54 | |
| 16 h | 6 | 10.22 | 10.34 | 7.90 | 3.70 | 30.50 | |
| 20 h | 6 | 11.70 | 9.38 | 9.20 | 3.50 | 28.30 | |
| 24 h | 7 | 15.88 | 9.71 | 11.90 | 6.01 | 34.75 | |
| Week 8 (infusion 8) | Time “0” (pre-infusion) | 3 | 10.96 | 5.51 | 11.50 | 5.20 | 16.20 |
| 4 h into infusion | 3 | 28.70 | 12.41 | 29.50 | 15.90 | 40.70 | |
| 8 h (before the end) | 4 | 27.93 | 14.79 | 32.14 | 7.34 | 40.10 | |
| 8.5 h | 3 | 11.26 | 2.90 | 11.20 | 8.40 | 14.20 | |
| 9 h | 3 | 20.16 | 24.22 | 10.10 | 2.60 | 47.80 | |
| 10 h | 4 | 5.80 | 5.55 | 3.65 | 1.90 | 14.00 | |
| 11 h | 4 | 15.70 | 11.72 | 14.35 | 3.90 | 30.20 | |
| 12 h | 4 | 14.10 | 11.67 | 10.60 | 4.40 | 31.00 | |
| 14 h | 5 | 19.60 | 21.49 | 9.90 | 2.80 | 52.90 | |
| 16 h | 5 | 11.68 | 8.29 | 9.00 | 2.60 | 24.60 | |
| 20 h | 5 | 14.52 | 10.73 | 15.50 | 2.20 | 30.90 | |
| 24 h | 5 | 16.20 | 11.57 | 9.90 | 6.00 | 32.40 |
Table 3.
Initial extracellular (blood plasma) ATP concentration (μM)
| Visit | Time point | N | Mean | Std. Dev. | Median | Minimum | Maximum |
|---|---|---|---|---|---|---|---|
| Week 1 (infusion 1) | Time “0” (pre-infusion) | 13 | 0.08 | 0.09 | 0.04 | 0.05 | 0.36 |
| 4 h into infusion | 13 | 0.15 | 0.18 | 0.19 | 0.02 | 0.54 | |
| 8 h (before the end) | 13 | 0.49 | 0.71 | 0.70 | 0.03 | 2.75 | |
| 8.5 h | 10 | 0.65 | 1.02 | 0.23 | 0.04 | 3.32 | |
| 9 h | 10 | 0.35 | 0.41 | 0.15 | 0.01 | 1.17 | |
| 10 h | 12 | 0.26 | 0.50 | 0.09 | 0.04 | 1.80 | |
| 11 h | 12 | 0.11 | 0.13 | 0.06 | 0.01 | 0.40 | |
| 12 h | 11 | 0.08 | 0.09 | 0.04 | 0.02 | 0.29 | |
| 14 h | 8 | 0.02 | 0.08 | 0.02 | 0.02 | 0.23 | |
| 16 h | 10 | 0.08 | 0.09 | 0.05 | 0.00 | 0.29 | |
| 20 h | 8 | 0.09 | 0.08 | 0.05 | 0.00 | 0.22 | |
| 24 h | 12 | 0.13 | 0.14 | 0.07 | 0.00 | 0.73 | |
| Week 3 (infusion 3) | Time “0” (pre-infusion) | 10 | 0.06 | 0.04 | 0.05 | 0.00 | 0.16 |
| 4 h into infusion | 9 | 0.78 | 1.51 | 0.20 | 0.00 | 4.68 | |
| 8 h (before the end) | 9 | 0.44 | 0.50 | 0.24 | 0.03 | 1.48 | |
| 8.5 h | 9 | 0.51 | 0.66 | 0.20 | 0.05 | 1.75 | |
| 9 h | 9 | 0.41 | 0.47 | 0.17 | 0.04 | 1.30 | |
| 10 h | 9 | 0.28 | 0.42 | 0.06 | 0.01 | 1.21 | |
| 11 h | 10 | 0.12 | 0.14 | 0.06 | 0.01 | 0.44 | |
| 12 h | 8 | 0.08 | 0.08 | 0.06 | 0.00 | 0.27 | |
| 14 h | 6 | 0.03 | 0.02 | 0.03 | 0.01 | 0.07 | |
| 16 h | 5 | 0.07 | 0.09 | 0.04 | 0.08 | 0.24 | |
| 20 h | 5 | 0.11 | 0.10 | 0.07 | 0.02 | 0.24 | |
| 24 h | 8 | 0.05 | 0.04 | 0.04 | 0.02 | 0.15 | |
| Week 8 (infusion 8) | Time “0” (pre-infusion) | 3 | 0.35 | 0.49 | 0.08 | 0.05 | 0.92 |
| 4 h into infusion | 3 | 0.37 | 0.31 | 0.39 | 0.06 | 0.68 | |
| 8 h (before the end) | 4 | 0.37 | 0.27 | 0.32 | 0.10 | 0.75 | |
| 8.5 h | 3 | 0.15 | 0.16 | 0.08 | 0.03 | 0.34 | |
| 9 h | 3 | 0.27 | 0.35 | 0.12 | 0.02 | 0.68 | |
| 10 h | 4 | 0.04 | 0.03 | 0.04 | 0.01 | 0.09 | |
| 11 h | 4 | 0.03 | 0.03 | 0.02 | 0.00 | 0.08 | |
| 12 h | 4 | 0.04 | 0.02 | 0.04 | 0.02 | 0.07 | |
| 14 h | 5 | 0.08 | 0.05 | 0.06 | 0.01 | 0.17 | |
| 16 h | 5 | 0.07 | 0.05 | 0.03 | 0.02 | 0.15 | |
| 20 h | 5 | 0.11 | 0.14 | 0.05 | 0.04 | 0.37 | |
| 24 h | 4 | 0.08 | 0.14 | 0.01 | 0.00 | 0.30 |
Fig. 1.

Whole blood ATP pools pre- and post-infusion The mean pre-infusion and immediate post-infusion whole blood ATP concentrations, in patients for whom this parameter was measured at weeks 1, 3, and 8 (N = 15 in week 1, N = 11 in week 3, N = 6 in week 8). A one-way ANOVA analysis for these data showed a significant difference in pre- and post-infusion whole blood ATP concentrations (p = 0.0099)
The size of erythrocyte ATP pools is maintained by a homeostatic system, which overcomes the comorbidity of this patient population. It is the turnover of ATP, which is likely to be affected by the metabolic status of the individual. The homeostatic system maintaining the erythrocyte ATP pool size is most likely determined by genetic factors. Individual patient’s data (Table 4) outlines the erythrocyte ATP pool sizes for times 0 (pre-infusion), 9, and 24 h after the start of the infusion. Comparing patient’s time 0 ATP pool size on week 1 with time 0 ATP pools on week 3 and time 0 ATP pools on week 8 illustrates the stability of erythrocyte ATP pool sizes with time. The pool sizes of erythrocyte ATP at 24 h are larger than the pool sizes at time 0 but are invariably close to time 0 values. At time 24 h after the start of the infusion (16 h after its termination), most but not all of the sequestered erythrocyte ATP has been constitutively released into the blood plasma. These data support the steadiness and stability of erythrocyte ATP pool size during an 8-week period. Based on Table 4, the narrow range in which the pool size of erythrocyte ATP (intra-erythrocyte) fluctuates is close to 0.25 mM. The range of variability of erythrocyte ATP pool sizes (inter-erythrocyte) among the 15 patients participating in the trial is demonstrably larger than 0.25 mM.
Table 4.
Total blood (RBC) ATP levels (mM)
| Week 1 | Week 3 | Week 8 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient | Time 0 | 9 h | 24 h | Time 0 | 9 h | 24 h | Time 0 | 9 h | 24 h |
| 1 | 0.50 | 0.78 | 0.60 | ||||||
| 2 | 0.35 | 0.66 | 0.44 | 0.46 | 0.74 | 0.49 | 0.45 | 0.53 | 0.55 |
| 3 | 0.46 | 0.55 | 0.53 | 0.35 | 0.48 | 0.50 | 0.59 | 1.28 | 0.79 |
| 4 | 0.60 | 1.00 | 0.90 | 0.47 | 1.14 | 0.70 | 0.70 | 1.26 | 0.75 |
| 5 | 0.90 | 1.80 | 1.40 | ||||||
| 6 | 0.55 | 0.75 | 0.65 | 0.24 | 0.40 | 0.33 | |||
| 7 | 0.58 | 0.98 | 0.62 | 0.75 | 0.95 | 0.58 | 0.81 | 1.10 | 0.81 |
| 8 | 0.55 | 0.94 | 0.57 | ||||||
| 9 | 1.10 | 1.90 | 1.00 | 1.00 | 1.60 | 1.50 | |||
| 10 | 0.47 | 0.55 | 0.50 | 0.42 | 0.62 | 0.42 | 0.36 | 0.53 | N/A |
| 11 | 0.31 | 0.69 | 0.62 | 0.46 | 0.54 | 0.43 | |||
| 12 | 0.53 | 0.62 | 0.54 | ||||||
| 13 | 0.54 | 0.63 | 0.51 | 0.51 | 1.28 | 0.76 | |||
| 14 | 0.80 | 1.30 | 0.90 | 0.90 | 1.66 | 0.87 | |||
| 15 | 0.99 | 1.28 | 0.95 | 0.93 | 1.57 | 0.90 | |||
Erythrocyte ATP pool sizes can be expanded in a meaningful and potentially useful way by continuous intravenous infusions of 50 or 100 μg/kg min of ATP for several hours (Figs. 1 and 2a). During 8 h of infusion of ATP, the actual amount of ATP taken up by the erythrocytes can be calculated as follows: 0.4 mM, which is the sequestered or expanded size of erythrocyte pools of ATP after expansion of the pools X 510, molecular weight of ATP X 5, 5 L of blood in an adult, equals 1020 mg for the lower dose (Figs. 1 and 2a). The amounts of ATP administered during 8 h of continuous intravenous infusion are 50 μg/kg min × 70 kg, for an average adult weight × 60 min × 8 h equals 1680 mg. Therefore, the total amount of ATP sequestered in erythrocytes after intravenous infusion of the lower dose of ATP was 1020 mg compared to the total amount of ATP administered, which was calculated to be 1680 mg. For the higher dose of ATP, the amount of ATP sequestered was calculated to be 1180 mg of ATP versus 3360 mg, which were administered. The amount of ATP sequestered in erythrocytes as a percent of the amount administered was therefore calculated to be 40 % for the higher dose of infused ATP (100 μg/kg min) and 53 % of ATP delivered intravenously for the lower dose of ATP (50 μg/kg min). Therefore, the sequestered erythrocyte ATP pools amount to about 40–50 % or more of the total dose of the infused ATP. The loaded up, sequestered ATP in erythrocytes at the end of the infusion decay to baseline level or undergo induced release, which is expected to produce higher amounts of ATP per unit time in a specific microenvironment. Two factors determine the levels of infused ATP at the maximally tolerated dose (MTD). One is the rate of ATP infused, which is limited to a maximum tolerated dose of 100 μg/kg min, determined by adverse effects. The second factor is a maximum dose of ATP administered per cycle based on total time of continuous intravenous infusion of ATP. The total amount of ATP administered can reach about 36 g of ATP administered intravenously, continuously over a period of 96 h per cycle once every 4 weeks [16]. Both values, rate of infusion and total amount of ATP infused per cycle, were determined by the spectrum of adverse effects [16].
Fig. 2.
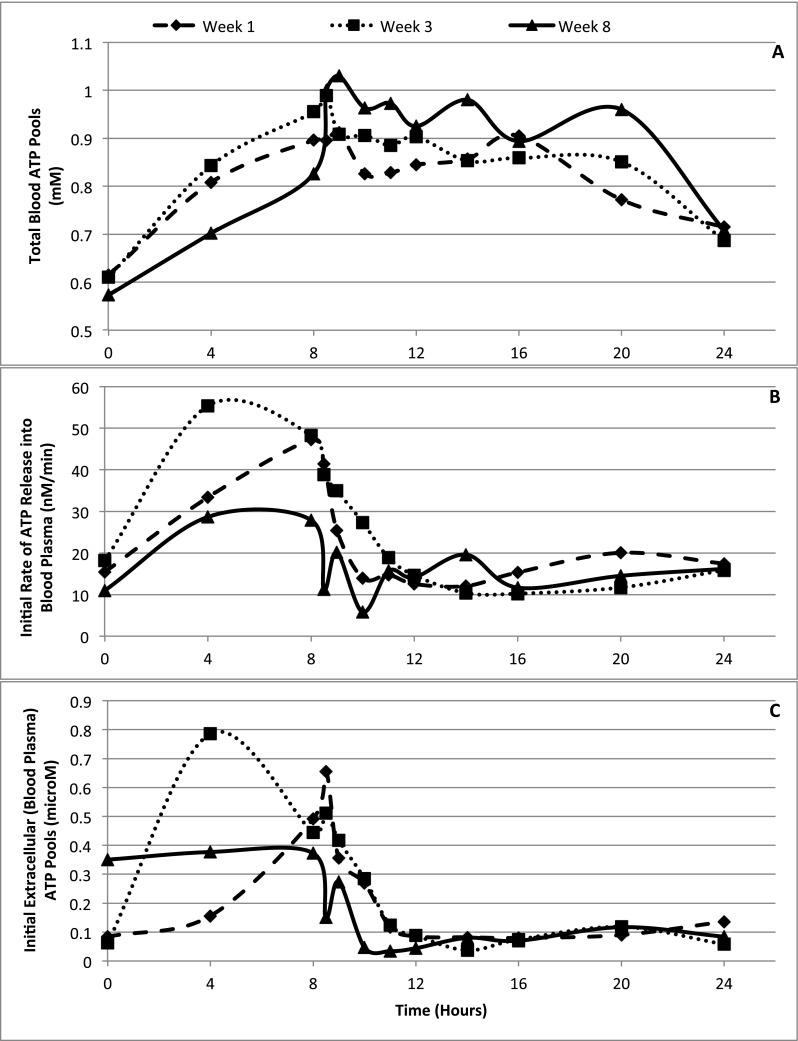
Summary of pharmacokinetics of ATP in advanced cancer patients
The ATP taken up by erythrocytes is constitutively released into the blood plasma. Enhanced release of the expanded erythrocyte ATP pools in vivo by locally stimulating signals [14] or by erythrocyte ATP release stimulants, added exogenously, could potentially open a field to new therapeutic approaches. The baseline pool size of adenosine in blood plasma is lower than that of ATP, with the latter reported here as 0.116 ± 0.042 μM of ATP. Another report found baseline steady state (pool size) levels of blood plasma ATP to be 0.072 μM of ATP [28].
The levels of extracellular ATP increase upon initiation of ATP infusion (Fig. 2c). A plot of extracellular ATP pools possesses similarities but is not parallel to the increase in release of ATP (Fig. 2b, Table 2). By the time the infusion was terminated at 8 h, which is the beginning of the large increases in intracellular erythrocyte ATP pools (Fig. 2a, Table 1), extracellular ATP pool was rapidly declining. By 10–12 h after initiation of the infusion whereby erythrocytes had a maximum load of intracellular ATP, extracellular ATP pools declined to about baseline levels. The mechanism that can account for the rapid decay of blood plasma extracellular ATP with its peak well before termination of the infusion relies on the ability of rising blood plasma levels of ATP to induce expression of catabolic enzymes acting on extracellular ATP. Intravenous infusions dispose ATP directly into the blood plasma compartment (extracellular). Initiating increases in blood plasma ATP pools at the beginning of the intravenous infusions of ATP is an effective mode of producing immediate increases in extracellular ATP pools. The peak blood plasma ATP pool is expected before effective ATP-stimulated expression of catabolic enzymatic activities mostly in the form of ecto-enzymes acting on ATP (17, 19). These results also suggest that administration of ATP will be most effective in achieving extracellular levels of ATP when carried out by short-term intravenous infusions or by intravenous push injections (Fig. 3).
Fig. 3.
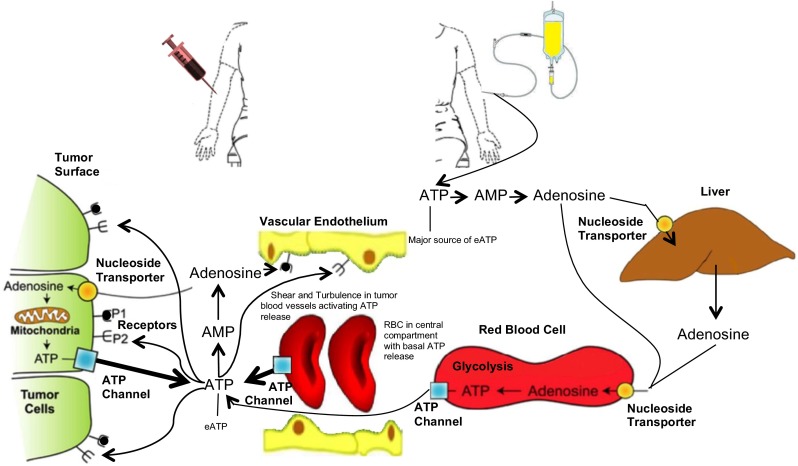
Proposed pathways of ATP and adenosine before, during, and after continuous intravenous infusions of ATP in humans. The major source of extracellular ATP (eATP) is the infused ATP, which is delivering ATP directly into the blood plasma compartment. After a short infusion time, the elevated eATP pools induce higher activities of catabolic ecto-enzymes. At the termination of an 8 h continuous intravenous infusion of ATP, the majority of the exogenously administered ATP is sequestered in the erythrocytes and all the eATP has been degraded. Blood samples were collected from the arm contralateral to the infusion arm. Intravenous infusions of ATP were at rates of 50–100 μg/kg min of ATP
It is important to note that the data, except for results reported in Fig. 1, did not reach statistical significance, potentially because the standard deviation was too big. Any value dependent on erythrocyte ATP pool sizes will reflect the variability of these pools in individual patients. The expansions of erythrocyte ATP pools by intravenous infusions of ATP did achieve statistical significance when comparing the pool sizes before and after intravenous infusion.
Discussion
The findings disclosed in the present article are the following:
-
(A)
Erythrocytes maintain an intracellular pool size of ATP unique to each individual. The fluctuations in these pool sizes are within a range of standard deviations of 30–40 % of the mean of total blood (erythrocytes) ATP pool size. Patients suffering from advanced, terminal cancer having abnormal metabolism and poor nutritional status are nevertheless shown to maintain their erythrocyte ATP pool size (steady state level) for at least 8 weeks. Their comorbidities and compromised performance status are likely linked to reductions in the turnover of ATP whereas the pool size, steady state levels of ATP in erythrocytes remain constant.
-
(B)
Continuous intravenous infusions of ATP for 8 h once weekly enable the expansion of erythrocyte ATP pools. It is possible to achieve meaningful significant increases (40–60 %) in the size of these intracellular ATP pools. The half-life of the expanded ATP is calculated here to be about 8 h.
-
(C)
The amount of ATP, which accumulates in expanding erythrocyte ATP pools, is relatively substantial and equals about 40–60 % of the total ATP administered, depending on the level of the dose administered. Total blood ATP pools increase linearly during intravenous infusion of ATP and reach a maximum by the end of the infusion.
-
(D)
Extracellular, blood plasma pools of ATP increase during initial hours of infusion well before increases in the sequestration of the intracellular pools of ATP. The formation of extracellular ATP requires administration of ATP for a short period of time. Extracellular ATP is short lived and is degraded before the large increases of erythrocyte ATP pools materialize at or after termination of the infusion (8 h). It may be advantageous to deliver ATP, if ATP infusions are found to be of therapeutic benefit, by short-time intravenous infusions.
While summarizing the results of our human study of ATP in the treatment of advanced cancer, we came across the large standard deviation and diversity in the sizes of erythrocyte ATP pools. This variability was assigned to differences among individuals in the size of their erythrocyte ATP pools rather than to fluctuations in sizes of erythrocyte ATP pools in each individual. We then looked at the erythrocyte ATP pools of advanced cancer patients who participated in the trial. Fifteen patients took the week 1 ATP infusions, and 11 patients had week 3 infusion carried out. Five out of a total of 15 patients completed all eight ATP infusion cycles as ATP analysis was performed on weeks 1, 3, and 8. We found that during these 8 weeks, their pre-infusions, or “time 0”, erythrocyte ATP pool size was stable in a narrow range around a single value. The size of erythrocyte ATP pools was found unique to individual patients and was fairly reproducible within the standard deviation of blood sample withdrawal and analysis of blood ATP pools. A homeostatic system inside and outside the erythrocytes must be in place in order to maintain the stability of the erythrocyte ATP pool sizes at baseline levels (time 0 or pre).
The diversity in red blood cell distribution width (RDW) within the erythrocyte population of an individual has been known for a long time. RDW is recorded in every full blood count (FBC). RDW is indicative of size diversity among the erythrocyte population of an individual. RDW is a strong independent predictor of mortality in several disease states and in healthy older individuals where high RDW is a negative predictor of survival [29]. There are no data to our knowledge regarding the relationship of ATP pools to cellular size of erythrocytes. Recently however, quantitative measurement of the absolute ATP concentrations in individual cells has been achieved in bacterial cell populations [30].
The use of erythrocyte ATP pools for therapeutic purposes is supported by the findings that major human morbidity is related to failure of ATP release from erythrocytes [14]. Early studies showing anti-neoplastic activity of exogenously administered ATP in mice yielded information regarding the metabolism and pathways of adenine nucleotides in vivo [7, 11, 12]. Exogenous ATP is able to elevate blood plasma, total blood (erythrocytes) and liver ATP pools with the anti-tumor activities seemingly related to the size of blood plasma ATP pools, which, in turn, are not directly related in size to the erythrocyte ATP pools. Elevation of these pools also acted in the significant inhibition in the weight loss aspect of cancer cachexia [7, 11, 12]. The release of baseline levels of erythrocyte ATP pools into the blood plasma is enhanced in vivo by low oxygen tension and exposure of erythrocytes to shear stress along vessel walls and mechanical deformation in small capillary vessels. Other agents were proposed as well for stimulation of release of erythrocyte ATP pools [14]. These include compounds that induce increases in erythrocyte cyclic AMP levels along with vasoactive agents such as PGI2 (prostacyclin) [27].
Conceivably, the use of externally administered compounds to stimulate fast release of erythrocyte ATP pools into the blood plasma, after or before termination of push intravenous infusions, could result in a burst of ATP into the extracellular compartment, opening the way to a new therapeutic modality. We have shown here that erythrocyte ATP pools can be increased by 40 and 53 % during infusion of ATP, depending on the dose of infused ATP. These sequestered pools of ATP are released constitutively over a period of 24 h (three half-lives). Their proposed release during a much shorter period of time would increase the available concentration of extracellular ATP in blood plasma and affect binding to P2 receptors, which require blood plasma ATP concentrations higher than those present at baseline (Fig. 3).
Mouse tumor models suggested a non-immune (chemical) as well as immune components for the anti-tumor activities of blood plasma ATP. The non-immune path has been shown to involve permeabilization (pore formation) of tumor cell membrane by low levels of extracellular ATP leading to tumor cell death [8, 11, 12, 31, 32]. Mechanisms of ATP release through specific membrane ATP channels are unique to specific cell types. ATP release from erythrocytes [11, 12] contrary to release from tumor cells may involve different or overlapping ATP channels [33, 34]. In addition to the mechanism of pore formation, recent experimental data suggest that extracellular ATP is critical in inducing immunogenic cell death. The activity of some chemotherapeutic agents active in cancer is now being linked to the chemotherapy-induced release of ATP from tumors. Acting in a paracrine mode, the released ATP attracts dendritic cells and cytolytic T cells by chemotaxis. At the site of the tumor, extracellular ATP activates dendritic cell conversion into mature immune cells with the ability to present tumor-associated antigens, while recruiting and protecting other myeloid differentiated cells [13].
Extracellular ATP is particularly effective in killing low burden tumors in animals [11, 12]. This property, along with recent claims that blood flow is greatly impaired when passing through small growing tumors, forms the foundation for considering another in vivo activity of ATP. Small developing tumors are notorious for their compressed tumor vessels producing deformed erythrocytes due to solid stress and a hypoxic microenvironment [35]. The stress in the tumor microenvironment is also a result of interstitial fluid accumulation. Hypoxia and erythrocytes’ deformation are conditions known to stimulate release of erythrocyte ATP pools [14]. The sudden decrease in oxygen tension in the vicinity of the developing tumor along with the severe impairment of perfusion pressure in the tumor are expected to serve as a stimulus for the increases in release of erythrocyte ATP pools into the blood plasma in a stimulus-secretion manner.
Results reported here were obtained by in vivo continuous intravenous infusions of ATP and in the presence of rising initial levels of extracellular ATP. These initial elevated extracellular ATP pools were the outcome of the intravenous administration, placing the infused ATP directly into the extracellular (blood plasma) compartment. Although it was known that erythrocytes have a poor ability to metabolize ATP to adenosine and a slow uptake system for the uptake of nucleosides in whole blood [36], there are some proposed mechanisms arising from the present study as to how the majority of the administered exogenous ATP ends up in the intracellular compartment of erythrocytes. It was shown previously that ATP has remarkable stability in whole blood but is degraded within one cycle after bolus injection in vivo [36]. Experimental results reported here would be helpful in identifying mechanisms explaining the ability of infused ATP escaping degradation inside the vascular bed and becoming sequestered in the erythrocytes.
Acknowledgments
We thank Dr. Adaling Ogilvie for a critical review of the manuscript.
Conflict of interest
A clinical trial entitled “A Phase I Study of the Safety and Pharmacokinetics of Adenosine 5’-Triphosphate (ATP) When Administered by Intravenous Infusion on a Multiple Weekly Dose Schedule to Patients with Advanced Malignancies (Solid Tumors)” was funded by ATP Therapeutics LLC. E.R. and E.H.A. have financial interest in the development of purine-based drugs.
References
- 1.Burnstock G, Campbell G, Satchell D, Smythe A. Evidence that adenosine triphosphate or a related nucleotide is the transmitter substance released by non-adrenergic inhibitory nerves in the gut. Br J Pharmacol. 1970;40:668–688. doi: 10.1111/j.1476-5381.1970.tb10646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang TF Guidotti GJ. CD39 is an ecto-(Ca2+,Mg2+)-apyrase. J Biol Chem. 1996;271:9898–9901. doi: 10.1074/jbc.271.17.9898. [DOI] [PubMed] [Google Scholar]
- 3.van den Bosch R, Geuze H, du Maine AP, Strous GJ. Transport and metabolism of 5’-nucleotidase in a rat hepatoma cell line. Eur J Biochem. 1986;160(1):49–54. doi: 10.1111/j.1432-1033.1986.tb09938.x. [DOI] [PubMed] [Google Scholar]
- 4.Rapaport E. Mechanisms of anticancer activities of adenine nucleotides in tumor-bearing hosts. Ann NY Acad Sci. 1990;603:142–149. doi: 10.1111/j.1749-6632.1990.tb37668.x. [DOI] [PubMed] [Google Scholar]
- 5.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- 6.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rapaport E, Fontaine J. Generation of extracellular ATP in blood and its mediated inhibition of host weight loss in tumor bearing mice. Biochem Pharmacol. 1989;38:4261–4266. doi: 10.1016/0006-2952(89)90524-8. [DOI] [PubMed] [Google Scholar]
- 8.Rapaport E. Treatment of human tumor cells with ADP or ATP yields arrest of growth in the S phase of the cell cycle. J Cell Physiol. 1983;114:279–283. doi: 10.1002/jcp.1041140305. [DOI] [PubMed] [Google Scholar]
- 9.Sterling KM, Jr, Shah S, Kim RJ, Johnston NI, Salikhova AY, Abraham EH. Cystic fibrosis transmembrane conductance regulator in human and mouse red blood cell membranes and its interaction with ecto-apyrase. J Cell Biochem. 2004;91:1174–1182. doi: 10.1002/jcb.20017. [DOI] [PubMed] [Google Scholar]
- 10.Abraham EH, Prat AG, Gerweck L, Seneveratne T, Arceci RJ, Kramer R, Guidotti G, Cantiello HF. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proc Natl Acad Sci U S A. 1993;90:312–316. doi: 10.1073/pnas.90.1.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapaport E. Experimental cancer therapy in mice by adenine nucleotides. Eur J Cancer Clin Oncol. 1988;24:1491–1497. doi: 10.1016/0277-5379(88)90340-9. [DOI] [PubMed] [Google Scholar]
- 12.Rapaport E, Fontaine J. Anticancer activities of adenine nucleotides in mice are mediated through expansion of erythrocyte ATP pools. Proc Natl Acad Sci U S A. 1989;86:1662–1666. doi: 10.1073/pnas.86.5.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Adjemian S, Yang H, Catani JPP, Hannani D, Martins I, et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. OncoImmunol. 2013;2:e24568. doi: 10.4161/onci.24568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sprague RS, Ellsworth ML. Erythrocyte-derived ATP and perfusion distribution: role of intracellular and intercellular communication. Microcirculation. 2012;19:430–439. doi: 10.1111/j.1549-8719.2011.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergfeld GR, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc Res. 1992;26:40–47. doi: 10.1093/cvr/26.1.40. [DOI] [PubMed] [Google Scholar]
- 16.Haskell CM, Wong M, Williams A, Lee LY. Phase 1 trial of extracellular adenosine 5′-triphosphate in patients with advanced cancer. Med Pediatr Oncol. 1996;27:165–173. doi: 10.1002/(SICI)1096-911X(199609)27:3<165::AID-MPO6>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 17.Lund P, Cornell NW, Krebs HA. Effect of adenosine on the adenine nucleotides content and metabolism of hepatocytes. Biochem J. 1975;152:593–599. doi: 10.1042/bj1520593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rapaport E, Zamecnik PC. Incorporation of adenosine into ATP: formation of compartmentalized ATP. Proc Natl Acad Sci U S A. 1976;73:3122–3125. doi: 10.1073/pnas.73.9.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lerner MH, Lowy BA. Formation of adenosine in rabbit liver and its possible role as a direct precursor to the formation precursor of erythrocytes adenine nucleotides. J Biol Chem. 1974;249:959–966. [PubMed] [Google Scholar]
- 20.Henderson JF, LePage GA. Transport of adenine-8-C14 among mouse tissues by blood cells. J Biol Chem. 1959;234:3219–3223. [PubMed] [Google Scholar]
- 21.Prichard JF, Chavez-Peon F, Berlin RD. Purines: supply from liver to tissue. Am J Physiol. 1970;219:1263–1267. doi: 10.1152/ajplegacy.1970.219.5.1263. [DOI] [PubMed] [Google Scholar]
- 22.Sevigny J, Robson SC, Waelkens E, Csizmadial E, Smith RN, Lemmens R. Identification and characterization of a novel hepatic canalicular ATP diphosphohydrolase. J Biol Chem. 2000;275:5640–5647. doi: 10.1074/jbc.275.8.5640. [DOI] [PubMed] [Google Scholar]
- 23.Kichenin K, Seman M. Chronic oral administration of ATP modulates nucleoside transport and purine metabolism in rats. J Pharmacol Exp Ther. 2000;294:126–133. [PubMed] [Google Scholar]
- 24.Bonner F, Borg N, Burghoff S, Schrader J. Resident cardiac immune cells and expression of the ectonucleotidase enzymes cd39 and cd73 after ischemic injury. PLoS ONE. 2012;7(4):e34730. doi: 10.1371/journal.pone.0034730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hochachka PW. Intracellular convection, homeostasis and metabolic regulation. J Exp Biol. 2003;206:2001–2009. doi: 10.1242/jeb.00402. [DOI] [PubMed] [Google Scholar]
- 26.Cortese-Krott MM, Kelm M. Endothelial nitric oxide synthase in red blood cells: key to a new erythrocrine function? Redox Biol. 2014;2:251–258. doi: 10.1016/j.redox.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montalbetti N, Leal Denis MF, Pignataro OP, Kobatake E, Lazarowski ER, Schwarzbaum PJ. Homeostasis of extracellular ATP in human erythrocytes. J Biol Chem. 2011;286:38397–38407. doi: 10.1074/jbc.M111.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorman MW, Feigl FO, Buffington CW. Human plasma ATP concentration. Clin Chem. 2007;53:318–325. doi: 10.1373/clinchem.2006.076364. [DOI] [PubMed] [Google Scholar]
- 29.Garbharran U, Chinthapalli S, Hopper I, George M, Back DL, Dockery F. Red cell distribution width is an independent predictor of mortality in hip fracture. Age Ageing. 2013;42:258–261. doi: 10.1093/ageing/afs176. [DOI] [PubMed] [Google Scholar]
- 30.Yaginuma H, Kawai S, Tabata K, Tomiyama K, Kakizuka A, Komatsuzaki T, Noji H, Imamura H. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci Rep (Naturecom) 2014;4:6522. doi: 10.1038/srep06522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rapaport E, Fishman RF, Gercel C. Growth inhibition of human tumor cells in soft-agar cultures by treatment with low levels of adenosine 5-triphosphate. Cancer Res. 1983;43:4402–4406. [PubMed] [Google Scholar]
- 32.Elmaleh DR, Zamecnik PC, Castronovo F, Jr, Strauss HW, Rapaport E. Tc-99m labeled nucleotides as tumor-seeking radiodiagnostic agents. Proc Natl Acad Sci U S A. 1984;81:918–921. doi: 10.1073/pnas.81.3.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham EH, Sterling KM, Kim RJ, Salikhova AY, Huffman HB, Crockett MA, Johnston N, Parker HW, Boyle WE, Jr, Hartov A, Demidenko E, Efird J, Kahn J, Grubman SA, Jefferson DM, Robson SC, Thakar JH, Lorico A, Rappa G, Sartorelli AC, Okunieff P. Erythrocyte membrane ATP binding cassette (ABC) proteins: MRP1 and CFTR as well as CD39 (ecto-apyrase) involved in RBC ATP transport and elevated blood plasma ATP of cystic fibrosis. Blood Cells Mol Dis. 2001;27(1):165–180. doi: 10.1006/bcmd.2000.0357. [DOI] [PubMed] [Google Scholar]
- 34.Abraham EH, Vos P, Kahn J, Grubman SA, Jefferson DM, Ding I, Okunieff P. Cystic fibrosis hetero- and homozygosity is associated with inhibition of breast cancer growth. Nat Med. 1996;2:593–596. doi: 10.1038/nm0596-593. [DOI] [PubMed] [Google Scholar]
- 35.Stylianopoulosa T, Jain RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci U S A. 2013;110:18632–18637. doi: 10.1073/pnas.1318415110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]