

Abstract
Metastasis is the ultimate cause of death for most cancer patients. While many mechanisms have been delineated for regulation of growth and tumor initiation of the primary tumor, very little is known about the process of metastasis. Metastasis requires dynamic alteration of cellular processes in order for cells to disseminate from the primary tumor to distant sites. These alterations often involve dramatic changes in the regulation of cytoskeletal and cell-environment interactions. Furthermore, controlled refinement of these interactions requires feedback to regulatory networks in the nucleus. MTA2 is a member of the metastasis tumor associated family of transcriptional regulators and is a central component of the nucleosome remodeling and histone deacetylation complex. MTA2 acts as a central hub for cytoskeletal organization and transcription and provides a link between nuclear and cytoskeletal organization. We will focus on MTA2 in this chapter, especially its role in breast cancer metastasis.
Keywords: Metastasis-associated protein, MTA2, Metastasis, Breast cancer, Tumor progression
1 MTA2 protein structure and function
MAT2 is a 668 amino acid protein and contains four distinct regions (Figure 1): a BAH domain from positions 1-144, an ELM2 domain from positions 145-256, a SANT domain from positions 263-315, and an atypical GATA Zinc finger domain from positions 367-394 [1]. Post-translational modifications are generally localized to the latter portion of the protein, and include four phosphorylation sites at S433[2], S435[2–4], T534[4], and S548[5]. An N6-acetyllysine modification has also been reported at position K152 [6] within the ELM domain. Thus, MTA2 has a complex domain structure with several potential regulatory regions.
Figure 1. Gene organization and mutational landscape of MTAs.

Each MTA family member is shown along the x axis, amino acid coordinates are indicated. BAH, ELM2, and GATA domains are indicated on each gene. Point mutations reported in cBioProtal are indicated and the frequency of mutations at those sites is shown on the y axis. Missense mutations are indicated in green and inactivating mutations (frame-shift and nonsense) are shown in red (purple is the combination of green and red).
The BAH, EML2, SANT, and zinc-finger domains are likely involved in protein-protein interactions, although these domains have been reported to also participate in DNA binding [7–10]. These domains are critical binding regions linking MTA2 to other members of the HDAC and NuRD complexes, as well as directing these complexes to target molecules [9, 11–13]. BAH, ELM2, SANT, and zinc-finger domains are shared by other MTA family members (MTA1 and MTA3) and are, therefore, likely critical for binding core components of the HDAC complex, though, positions 1-237 of MTA2 are sufficient for binding of estrogen receptor alpha (ERα) [12]. As MTAs act as core scaffolds for the NuRD complexes, these domains are critical for MTA family function.
MTA family members are central components of the Mi-2/NuRD complex [14, 15] [Figure 2] thereby regulating global gene expression networks. MTAs maintain the stability of the NuRD complex, as well as aiding in directing histone deacetylase activity from the associated HDAC proteins to target molecules. MTA proteins have been shown to interact with HIF1α [16, 17] and ERα [12], p53 [11], and regulate their activity via modulation of protein acetylation. Thus, MTAs exert their regulatory activity in two ways, one by controlling the global histone acetylation profile, and another by regulating the activity of key signaling pathways by acetylation of network hubs.
Figure 2. MTA2 in the NuRD complex.

NuRD complex components are shown with key MTA2 targets indicated, ER-alpha, p53, and transcriptional regulation.
Though all MTA family members participate in the NuRD complex, each member possesses distinct functions. MTA2 appears to regulate cytoskeletal reorganization via global gene expression modulation and activation of the Rho signaling pathway [18, 19]. MTA3 also plays a role in cytoskeletal regulation thru the master epithelial state via regulation of Snail [20]. Additionally, overall levels of MTA members appear to be regulated coordinately at the protein level. Fujita and colleagues [21] demonstrated that overexpression of MTA1 decreased protein levels of both MTA2 and MTA3, while increasing their level of mRNA expression. Similarly, Zhang and colleagues [22] showed that Polyoma middle-T antigen-overexpressing mammary tumors have increased expression of MTA1 and decreased expression of MTA3 with tumor progression. Thus, overall levels of MTAs may be limited by the availability of NuRD complex components with which to bind.
MTA2 is not frequently mutated in human cancer (summarized in Figure 3). Howwever, mutations tend to cluster in the BAH and ELM2 domains (Figure 1), which may impact the protein-protein binding capabilities of these mutants. Furthermore, MTA2 is more frequently gene amplified in cancer than deleted, indicating that it's role in cancer and metastasis is likely via its upregulation as opposed to mutational activation.
Figure 3. MTA2 variation in cancer.

Mutations and CNV alterations are shown as reported from cBioPortal. Bar colors are indicated in the key.
2 Metastasis
The ultimate cause of death for most breast cancer patients is not the primary lesion, but rather complications from distant metastases [23]. In general the primary tumor may have been removed surgically years before signs of recurrence are evident. Metastasis is the process by which cells from the primary tumor are disseminated to distant sites in the body. These tumor cells may lay dormant for several years or even decades before “awakening” from dormancy to reappear as a clinically detectable metastatic lesion.
The process of metastasis involves complex orchestration of signaling pathways regulating survival, cytoskeletal alteration, and growth at a distant site [23]. Cells must first invade into the surrounding tissue of the tumor and gain access to the circulation. From there tumor cells or emboli must survive transit through the circulatory system and arrest at a distant metastatic site. Next the tumor cells must initiate the process of growth, which appears to require collaboration with the microenvironment at the metastatic site [23–25]. Pioneering experiments by Fidler and colleagues [26] demonstrated that less than 0.01% of B16 melanoma cells were capable of forming metastases in mice. Thus, competent metastasis is most probably a rare event.
While few cells may be capable of performing all of the required tasks to form metastases, genetic signatures and prognostic markers have been developed in specific cancer sites to predict the likelihood of distant metastasis. This is not surprising, as metastatic cells are thought to be selected for during the metastatic process, as opposed to being cells which adapt during the metastatic process [27]. Thus, while metastases may arise from one or a few cells, this process can be influenced by factors that are observable at the gross level and present in the primary tumor [28, 29]. Integration of these findings into a unified model is still controversial. The two models most often advanced are: 1) that all cells in the tumor are equally likely to form metastases. Since cancers shed millions of cells into the circulation, and the observation that a few metastases form is therefore not surprising. Different tumors may be primed to survive the metastatic process, but still metastasis formation is rare; alternatively 2) cancers are very heterogeneous and contain only few cells that are functionally specialized for metastasis. These metastatic cells may be present at higher or lower ratios within the primary tumor, and it is these ratios that molecular profiling is detecting allowing for useful prognostic signatures of metastatic behavior. Although the biological processes regulating metastatic dissemination are complex, several genes have been identified which increase the rate of metastasis formation in animal models and which predict metastasis in the clinical setting, including MTA proteins [13, 19].
3 Metastatic dissemination
Cancer cells must complete several steps in order to form a viable metastatic lesion [23]. In order to reach a distant site, metastatic cancer cells must first invade surrounding tissues. This process involves dramatic changes in the organization of the actin cytoskeleton and interactions with the extracellular environment. Invasion process invasion are required for both extravasation and intravasation, and it has been suggested that there are at least two means of invasion. The first involves proteolytic degradation of the extracellular matrix by proteinases. Proteolytic motion is associated with an epithelial to mesenchymal transition (EMT) phenotype and is observed in some breast cancer cell lines [30, 31]. The other type of migration involves protruding through the extracellular matrix without proteolytic degradation [32]. This type of amoeboid motion was first characterized in experiments of MDA-MB-231 breast cancer cells under conditions which blocked extracellular proteinases [33]. Both migration mechanisms rely on signaling through the Rho pathway, with RhoA signaling being dominant in amoeboid motility [33, 34] and Cdc42 and others involved in proteolytic migration [35]. These processes are intimately regulated by cellular machinery that actively engages the cytoskeleton, cell membrane, and extracellular matrix.
After entering the circulation or accessing an organ plain, cancer cells must survive the stresses of migration. Primary obstacles include anoikis, immune surveillance, and shear stress [23, 36–38]. Despite these barriers, tumor cells can be detected in the circulation several decades after mastectomy with no evidence of clinical metastases [39]. Altered signaling through cytoskeletal regulators has been associated with anoikis resistance through Rho/ROCK [40]. Again, regulation of this process is intimately linked to the cytoskeleton. MTA proteins, primarily MTA2, control the gene expression networks of these pathways, thereby augmenting the metastatic potential of cancer cells [19, 41].
The final obstacle to metastasis formation is the process of colonizing distant sites. This process may take several years, and may involve a period of dormancy where there is little if any growth at the metastatic site. Meng and colleagues [39] demonstrated that circulating tumor cells could be detected months after definitive surgery for breast cancer without the presence of an overt metastatic lesion, therefore, early or dormant metastatic lesions may be in equilibrium between cell proliferation and sheading. In breast cancer, recurrences can present over twenty years after initial diagnosis and removal of the primary tumor [42]. Dormant cells represent an opportunity for therapeutic intervention to reduce the incidence of metastasis and recurrence. The biology of these dormant cells is not well understood, but is the subject of active research [39, 43–45]. Current adjuvant therapies must be able to target dormant micro-metastases because adjuvant therapies have significant clinical benefit though the primary tumor has been removed.
Many steps in the metastatic process require signaling from components of the cytoskeleton. The cytoskeleton is maintained via a complex network of interconnected pathways which integrate signaling from the cell surface (from molecules including focal adhesion kinase [FAK], integrins, and cadherins) and the nucleus (from gene regulators including Twist, Snail, Slug, and the MTA's) to regulate actin depolymerization and actin-myosin contraction [35]. These processes directly facilitate motility by orchestrating actin polymerization and focal adhesion formation at the leading front, primarily driven by Cdc42, and actin-myosin contraction and actin de-polymerization in the rear, primarily driven by Rho/ROCK [32, 35, 46].
MTA2 has been linked in several ways to regulation of cytoskeletal organization. An analysis of data published by Wang and colleagues [47] of MTA2 DNA bindingsites revealed several pathways to be enriched from the Molecular Signatures database [48] in genes associated with and potentally regulated by MTA2 (Table 1). These include the actin cytoskeleton, adherens junction, tight junction, and focal adhesion pathways. Covington and colleagues also found that MTA2 regulated expression of cytoskeletal pathways at the transcriptional level [19]. Gene expression analysis revealed that MTA2-overexpressing cells and clinical samples had differential regulation of focal adhesion, cytoskeletal, and MAPK signaling pathways relative to MTA2-low counterparts.
Table 1.
Molecular signatures associated with MTA2-chromatic binding interactions.
| Description$ | k/K | p-value | FDR q-value# |
|---|---|---|---|
| MAPK signaling pathway | 0.1124 | 4.82E-13 | 8.97E-11 |
| Calcium signaling pathway | 0.1292 | 1.43E-11 | 1.33E-09 |
| Cytokine-cytokine receptor interaction | 0.1011 | 8.05E-11 | 4.99E-09 |
| Regulation of actin cytoskeleton | 0.1111 | 1.29E-10 | 5.99E-09 |
| Adherens junction | 0.1867 | 1.04E-09 | 3.88E-08 |
| Pathways in cancer | 0.0854 | 1.77E-09 | 5.50E-08 |
| Purine metabolism | 0.1195 | 3.17E-09 | 8.42E-08 |
| Fc gamma R-mediated phagocytosis | 0.1546 | 4.07E-09 | 9.46E-08 |
| Focal adhesion | 0.1045 | 5.54E-09 | 1.15E-07 |
| Leukocyte transendothelial migration | 0.1356 | 8.81E-09 | 1.64E-07 |
| Chemokine signaling pathway | 0.1053 | 1.12E-08 | 1.89E-07 |
| Axon guidance | 0.124 | 3.21E-08 | 4.98E-07 |
| Parkinson's disease | 0.1203 | 4.97E-08 | 7.11E-07 |
| TGF-beta signaling pathway | 0.1512 | 5.80E-08 | 7.71E-07 |
| Huntington's disease | 0.0973 | 1.99E-07 | 2.46E-06 |
| Adipocytokine signaling pathway | 0.1642 | 2.55E-07 | 2.92E-06 |
| Alzheimer's disease | 0.1006 | 2.67E-07 | 2.92E-06 |
| Tight junction | 0.1119 | 3.35E-07 | 3.47E-06 |
| Oxidative phosphorylation | 0.1111 | 3.70E-07 | 3.62E-06 |
|
| |||
| Insulin signaling pathway | 0.1095 | 4.48E-07 | 4.16E-06 |
Selection of top 20 signatures by FDR q-value
Description provided by Molecular Signatures database
Specifically, MTA2 was associated with down-regulation of Rho GTP-dissociation inhibitor alpha (Rh oGDIα), and increased activation (GTP-bound) Rho [19]. MTA2-enhanced migration and anchorage-independent growth was dependent on Rho signaling as blockade either by Rho GDIα re-expression or ROCK inhibitors which were able to revert the MTA2-overexpression phenotype. MTA2 overexpression resulted in increased lamellipodia formation in breast cancer cell lines (Figure 4), consistent with activation of Rho signaling and indicating that MTA2 enhances an amoeboid phenotype [46, 49]. Lamellipodia formation was inhibited by blockade of ROCK signaling. These data establish that MTA2 regulates cytoskeletal and motility pathways dependent on Rho/ROCK signaling.
Figure 4. Cytoskeletal changes associated with MTA2 overexpression and ROCK inhibition.
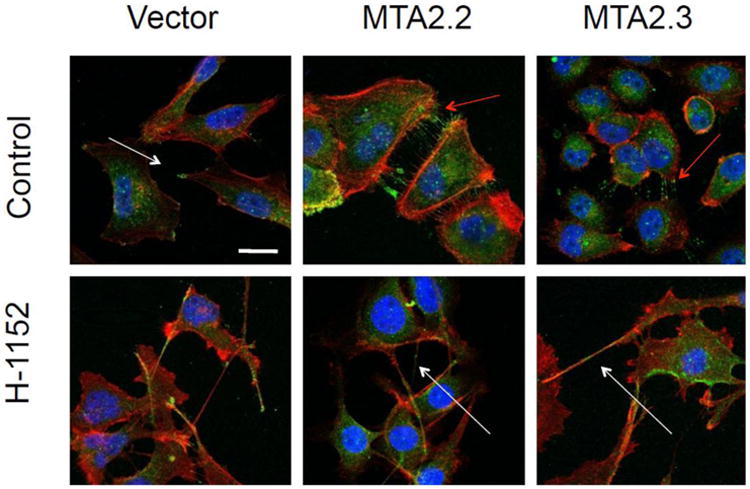
MTA2-overexpressing or vector control cells are shown treated with either vehicle or H-1152, a potent ROCK inhibitor. Cells were stained for p-LIMK1/2 (green), phaloidin (red), and DAPI (blue). Pseudopodia are indicated with white arrows, lamelopodia are indicated with red arrows.
Conversely the Rho signaling pathway regulated MTA2 levels in ERα-negative breast cancer cells. Rho GDIα-overexpression decreased levels of MTA2 expression and resulted in further upregulation of endogenous Rho GDIα levels [19]. As both MTA2 and Rho GDIα were exogenously expressed, these findings imply that Rho GDIα regulates MTA2 at the protein level since endogenous and exogenous MTA2 were similarly decreased. Barone and colleagues found that knockdown of Rho GDIα was associated with upregulation of MTA2 levels in ERα-positive breast cancer cells [18]. In this setting, Rho GDIα knockdown lead to a tamoxifen-resistant phenotype and increased anchorage-independent growth. In this context, Rho GDIα knockdown increased the rate and frequency of distant lung metastases in orthotopic xenograft models. These data highlight the bi-directional signaling between the cytoskeleton and the nucleus that regulates cytoskeletal organization, which is coordinated by the MTA2/Rho GDIα axis.
MTA2 also regulates the activity of the master cell state regulator Twist [41, 50]. Twist is essential for mesoderm induction and the process of epithelial to mesenchymal transition [50, 51]. Fu and colleagues found that Twist binds to the MTA2-containing Mi2 NuRD complex and that this complex was required for E-cadherin suppression, migration, invasion, and metastasis [41]. Further work is required to more fully define the complex relationship between MTA2, nuclear coregulators, and the Rho signaling pathway.
4 Targeting MTA2
MTA2 is a component of the NuRD complex, which relies on associated HDAC activity to mediate post-translational modification, specifically deacetylation, of target proteins. HDAC activity can be blocked by HDAC inhibitors. The HDAC inhibitor Vorinostat (Zolinza) has been tested in clinical breast cancer trials [52], however, no clinical responses and many adverse events were observed. Future studies attempting to target MTA2 specifically may be required. Possible therapies include MTA2-containing NuRD inhibitors, or specific inhibitors of MTA2 itself, potentially in the form of siRNA targeted by nanoparticles [53] or peptides that can block MTA2 interactions with NuRD components [18].
Current work suggests that MTA2 is intimately linked with cytoskeletal and cell motility regulation. Covington and colleagues [19] demonstrated that MTA2's activities were dependent upon ROCK activity and that ROCK targeting agents could block motility and anchorage-independent growth in an MTA2-dependent manner. As ROCK is essential for amoeboid dependent cell motility, these data make ROCK an attractive clinical target.
Studies in the clinical setting will also require specifically determining if MTA2 enhances the dissemination of breast cancer cells to distant sites and/or is required for the formation of clinically-detectable metastases after dissemination. These studies will necessitate the development of inducible systems to express MTA2 during each stage of metastatic spread and potentially targeting MTA2 with inhibitors during these stages.
5 Conclusions
The current literature highlights MTA2 as a central regulator of key gene expression pathways central to metastatic dissemination. MTA2 was shown in several studies to regulate cytoskeletal and motility pathways which are essential processes in the metastatic cascade. Metastasis is intimately tied to dynamic changes in cell-environment interactions as metastatic cells spread to distant sites. While cell survival and oncogenic signaling networks are still important for cancer cell survival, activation of these pathways is also important for primary tumor growth, and therefore are not additional changes required for metastasis. However, traversal through the circulation and establishment of the metastatic niche in a distant organ site are much different stresses than a pre-metastatic cancer cell would previously have been exposed to (and thus selected for). Activation of cell motility pathways allows a single step to gain many of the abilities that a metastatic cell requires to reach and colonize a distant site.
Several therapeutic agents are currently being developed which can affect either MTA2 or the Rho/ROCK pathways. Specifically, Vorinostat [52] targets HDAC complexes and is approved for use in cutaneous T-cell lymphoma and has been tested in other cancers. Fasudil, a potent ROCK inhibitor, [54, 55] has been approved for clinical use in Japan and is being tested in cancer studies. Perhaps combination therapies targeting these two pathways would be beneficial in the metastatic setting.
6 Key Unanswered Questions
While the MTA2 and Rho pathways were shown to form a feed-forward regulatory loop [19], the molecular mechanisms of this regulatory pathway have yet to be fully explored. Future work may reveal additional targets within this pathway and yield increased understanding of cell motility regulation. Published studies indicate that MTA2 has specific transcriptional regulatory effects, however, MTAs are components of the core HDAC complex and thus regulate global gene expression patterns. Future work needs to focus on the functions specific to MTA2 within the HDAC complex, and delineate how these effects serve to finely regulate cellular processes. Finally, current data indicates that MTA2, along with Rho GDIα, are involved in treatment resistance and metastasis in breast cancer. However, it has not been shown that MTA2 is involved in tumor initiation of primary tumors. These findings may establish if MTA2 activation is an early or late event in cancer formation.
Acknowledgments
SAWF is supported by NIH/NCI R01 CA72038 and P30 CA125123-05, CPRIT RP120732-P7, and the Susan G. Komen Foundation PG12221410.
References
- 1.The UniProt Consortium. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Research. 2014;42(D1):D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(31):10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayya V, Lundgren DH, Hwang SI, Rezaul K, Wu L, Eng JK, et al. Han DK. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Science signaling. 2009;2(84):ra46. doi: 10.1126/scisignal.2000007. [DOI] [PubMed] [Google Scholar]
- 4.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, et al. Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Science signaling. 2010;3(104):ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 5.Rigbolt KTG, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, et al. Blagoev B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Science signaling. 2011;4(164):rs3. doi: 10.1126/scisignal.2001570. [DOI] [PubMed] [Google Scholar]
- 6.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science (New York, NY) 2009;325(5942):834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 7.Callebaut I, Courvalin JC, Mornon JP. The BAH (bromo-adjacent homology) domain: a link between DNA methylation, replication and transcriptional regulation. FEBS letters. 1999;446(1):189–193. doi: 10.1016/s0014-5793(99)00132-5. [DOI] [PubMed] [Google Scholar]
- 8.Boyer LA, Latek RR, Peterson CL. The SANT domain: a unique histone-tail-binding module? Nature reviews Molecular cell biology. 2004;5(2):158–163. doi: 10.1038/nrm1314. [DOI] [PubMed] [Google Scholar]
- 9.Aasland R, Stewart AF, Gibson T. The SANT domain: a putative DNA-binding domain in the SWI-SNF and ADA complexes, the transcriptional co-repressor N-CoR and TFIIIB. Trends in biochemical sciences. 1996;21(3):87–88. [PubMed] [Google Scholar]
- 10.Solari F, Bateman A, Ahringer J. The Caenorhabditis elegans genes egl-27 and egr-1 are similar to MTA1, a member of a chromatin regulatory complex, and are redundantly required for embryonic patterning. Development (Cambridge, England) 1999;126(11):2483–2494. doi: 10.1242/dev.126.11.2483. [DOI] [PubMed] [Google Scholar]
- 11.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408(6810):377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 12.Cui Y, Niu A, Pestell R, Kumar R, Curran EM, Liu Y, Fuqua SAW. Metastasis-associated protein 2 is a repressor of estrogen receptor alpha whose overexpression leads to estrogen-independent growth of human breast cancer cells. Mol Endocrinol. 2006;20(9):2020–2035. doi: 10.1210/me.2005-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar R, Wang RA, Bagheri-Yarmand R. Emerging roles of MTA family members in human cancers. Semin Oncol. 2003;30(5 Suppl 16):30–37. doi: 10.1053/j.seminoncol.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Bowen NJ, Fujita N, Kajita M, Wade PA. Mi-2/NuRD: multiple complexes for many purposes. Biochim Biophys Acta. 2004;1677(1-3):52–57. doi: 10.1016/j.bbaexp.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11(8):588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon HE, Cheon H, Chun KH, Lee SK, Kim YS, Jung BK, et al. Lee MS. Metastasis-associated protein 1 enhances angiogenesis by stabilization of HIF-1alpha. Oncology reports. 2006;16(4):929–935. [PubMed] [Google Scholar]
- 17.Singh RR, Kumar R. MTA family of transcriptional metaregulators in mammary gland morphogenesis and breast cancer. J Mammary Gland Biol Neoplasia. 2007;12(2-3):115–125. doi: 10.1007/s10911-007-9043-7. [DOI] [PubMed] [Google Scholar]
- 18.Barone I, Brusco L, Gu G, Selever J, Beyer A, Covington KR, et al. Fuqua SAW. Loss of Rho GDIα and resistance to tamoxifen via effects on estrogen receptor α. J Natl Cancer Inst. 2011;103(7):538–552. doi: 10.1093/jnci/djr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Covington KR, Brusco L, Barone I, Tsimelzon A, Selever J, Corona-Rodriguez A, et al. Fuqua SAW. Metastasis tumor-associated protein 2 enhances metastatic behavior and is associated with poor outcomes in estrogen receptor-negative breast cancer. Breast cancer research and treatment. 2013 doi: 10.1007/s10549-013-2709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113(2):207–219. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 21.Fujita N, Kajita M, Taysavang P, Wade PA. Hormonal regulation of metastasis-associated protein 3 transcription in breast cancer cells. Mol Endocrinol. 2004;18(12):2937–2949. doi: 10.1210/me.2004-0258. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Stephens LC, Kumar R. Metastasis tumor antigen family proteins during breast cancer progression and metastasis in a reliable mouse model for human breast cancer. Clin Cancer Res. 2006;12(5):1479–1486. doi: 10.1158/1078-0432.CCR-05-1519. [DOI] [PubMed] [Google Scholar]
- 23.Fidler IJ. Critical factors in the biology of human cancer metastasis: twenty-eighth G.H.A. Clowes memorial award lecture. Cancer Res. 1990;50(19):6130–6138. [PubMed] [Google Scholar]
- 24.Psaila B, Kaplan RN, Port ER, Lyden D. Priming the “soil” for breast cancer metastasis: the pre-metastatic niche. Breast Dis. 2006;26:65–74. doi: 10.3233/bd-2007-26106. [DOI] [PubMed] [Google Scholar]
- 25.Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the “pre-metastatic niche”: within bone and beyond. Cancer Metastasis Rev. 2006;25(4):521–529. doi: 10.1007/s10555-006-9036-9. [DOI] [PubMed] [Google Scholar]
- 26.Fidler IJ. Metastasis: guantitative analysis of distribution and fate of tumor embolilabeled with 125 I-5-iodo-2′-deoxyuridine. J Natl Cancer Inst. 1970;45(4):773–782. [PubMed] [Google Scholar]
- 27.Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197(4306):893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- 28.Buyse M, Loi S, van't Veer L, Viale G, Delorenzi M, Glas AM, et al. Consortium, T. R. A. N. S. B. I. G. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006;98(17):1183–1192. doi: 10.1093/jnci/djj329. [DOI] [PubMed] [Google Scholar]
- 29.Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, et al. Bertucci F. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res Treat. 2011;126(2):407–420. doi: 10.1007/s10549-010-0897-9. [DOI] [PubMed] [Google Scholar]
- 30.Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, et al. Thompson EW. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. 2010;15(2):235–252. doi: 10.1007/s10911-010-9175-z. [DOI] [PubMed] [Google Scholar]
- 31.Alves CC, Carneiro F, Hoefler H, Becker KF. Role of the epithelial-mesenchymal transition regulator Slug in primary human cancers. Front Biosci. 2009;14:3035–3050. doi: 10.2741/3433. [DOI] [PubMed] [Google Scholar]
- 32.Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005;96(7):379–386. doi: 10.1111/j.1349-7006.2005.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, et al. Friedl P. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. The Journal of cell biology. 2003;160(2):267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carr HS, Zuo Y, Oh W, Frost JA. Regulation of focal adhesion kinase activation, breast cancer cell motility, and amoeboid invasion by the RhoA guanine nucleotide exchange factor Net1. Molecular and cellular biology. 2013;33(14):2773–2786. doi: 10.1128/MCB.00175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009;28(1-2):65–76. doi: 10.1007/s10555-008-9170-7. [DOI] [PubMed] [Google Scholar]
- 36.Cavallo F, De Giovanni C, Nanni P, Forni G, Lollini PL. 2011: the immune hallmarks of cancer. Cancer immunology, immunotherapy: CII. 2011;60(3):319–326. doi: 10.1007/s00262-010-0968-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 38.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 39.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Uhr JW. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10(24):8152–8162. doi: 10.1158/1078-0432.CCR-04-1110. [DOI] [PubMed] [Google Scholar]
- 40.Schackmann RCJ, van Amersfoort M, Haarhuis JHI, Vlug EJ, Halim VA, Roodhart JML, et al. Derksen PWB. Cytosolic p120-catenin regulates growth of metastatic lobular carcinoma through Rock1-mediated anoikis resistance. J Clin Invest. 2011;121(8):3176–3188. doi: 10.1172/JCI41695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu J, Qin L, He T, Qin J, Hong J, Wong J, et al. Xu J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21(2):275–289. doi: 10.1038/cr.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(EBCTCG), E. B. C. T. C. G. Davies C, Godwin J, Gray R, Clarke M, Cutter D, et al. Peto R. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–784. doi: 10.1016/S0140-6736(11)60993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer. 2010;10(12):871–877. doi: 10.1038/nrc2933. [DOI] [PubMed] [Google Scholar]
- 44.Heyn C, Ronald JA, Ramadan SS, Snir JA, Barry AM, MacKenzie LT, et al. Foster PJ. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn Reson Med. 2006;56(5):1001–1010. doi: 10.1002/mrm.21029. [DOI] [PubMed] [Google Scholar]
- 45.Riethdorf S, Pantel K. Disseminated tumor cells in bone marrow and circulating tumor cells in blood of breast cancer patients: current state of detection and characterization. Pathobiology. 2008;75(2):140–148. doi: 10.1159/000123852. [DOI] [PubMed] [Google Scholar]
- 46.Kedrin D, van Rheenen J, Hernandez L, Condeelis J, Segall JE. Cell motility and cytoskeletal regulation in invasion and metastasis. J Mammary Gland Biol Neoplasia. 2007;12(2-3):143–152. doi: 10.1007/s10911-007-9046-4. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, et al. Shang Y. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138(4):660–672. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 48.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67(9):545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7) doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 51.Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71(1):245–254. doi: 10.1158/0008-5472.CAN-10-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vansteenkiste J, Cutsem EV, Dumez H, Chen C, Ricker JL, Randolph SS, Schöffski P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs. 2008;26(5):483–488. doi: 10.1007/s10637-008-9131-6. [DOI] [PubMed] [Google Scholar]
- 53.Pulford B, Reim N, Bell A, Veatch J, Forster G, Bender H, et al. Zabel MD. Liposome-siRNA-peptide complexes cross the blood-brain barrier and significantly decrease PrP on neuronal cells and PrP in infected cell cultures. PLoS One. 2010;5(6):e11085. doi: 10.1371/journal.pone.0011085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu GJ, Wang ZJ, Wang YF, Xu LL, Wang XL, Liu Y, et al. Zeng YJ. Systematic assessment and meta-analysis of the efficacy and safety of fasudil in the treatment of cerebral vasospasm in patients with subarachnoid hemorrhage. Eur J Clin Pharmacol. 2012;68(2):131–139. doi: 10.1007/s00228-011-1100-x. [DOI] [PubMed] [Google Scholar]
- 55.Ying H, Biroc SL, Li WW, Alicke B, Xuan JA, Pagila R, et al. Dinter H. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol Cancer Ther. 2006;5(9):2158–2164. doi: 10.1158/1535-7163.MCT-05-0440. [DOI] [PubMed] [Google Scholar]