

Abstract
Background. Untreated human immunodeficiency virus type 1 (HIV) infection is associated with persistent immune activation, which is an independent driver of disease progression in European and United States cohorts. In Uganda, HIV-1 subtypes A and D and recombinant AD viruses predominate and exhibit differential rates of disease progression.
Methods. HIV-1 seroconverters (n = 156) from rural Uganda were evaluated to assess the effects of T-cell activation, viral load, and viral subtype on disease progression during clinical follow-up.
Results. The frequency of activated T cells was increased in HIV-1–infected Ugandans, compared with community matched uninfected individuals, but did not differ significantly between viral subtypes. Higher HIV-1 load, subtype D, older age, and high T-cell activation levels were associated with faster disease progression to AIDS or death. In a multivariate Cox regression analysis, HIV-1 load was the strongest predictor of progression, with subtype also contributing. T-cell activation did not emerge an independent predictor of disease progression from this particular cohort.
Conclusions. These findings suggest that the independent contribution of T-cell activation on morbidity and mortality observed in European and North American cohorts may not be directly translated to the HIV epidemic in East Africa. In this setting, HIV-1 load appears to be the primary determinant of disease progression.
Keywords: HIV-1, AIDS, subtype D, immune activation, PD-1, viral load
Since the discovery of human immunodeficiency virus type 1 (HIV), we have improved our understanding of the complexities of viral transmission and subsequent disease progression. CD4 molecules on the surface of T-lymphocytes render these cells the primary targets of HIV-1 infection, and the absolute count of CD4 T cells in peripheral blood defines the extent of disease progression [1, 2]. HIV-1 load has a strong inverse relationship with CD4 T-cell absolute counts. While the introduction of combination antiretroviral therapy (cART) can reduce viremia to levels below detection, it does not eradicate the virus [3]. Strategies to achieve a cure require a deeper understanding of factors contributing to disease progression.
One key feature of HIV-1 immunopathogenesis is aberrant T-cell activation, and several studies indicate that chronic immune activation predicts the course of disease progression better than plasma viral load [4–6]. The prototypical marker of T-cell activation is CD38, which is used in combination with other phenotypic markers, such as HLA-DR, to improve the prognostic sensitivity and specificity of these biomarkers in HIV-1 disease [7]. Programmed death 1 (PD-1; also known as CD279), a receptor associated with decreased functionality and considered a negative regulator of T-cell activation, has been associated with disease progression [8–10]. Evidence shows that CD279 in conjunction with CD38 and HLA-DR identifies unique subsets of T cells, which are highly correlated with viral load and disease progression [11–13]. Driving forces behind T-cell activation may include persistent viral antigen, innate cytokines, and soluble factors such as type I interferon, microbial translocation, bystander activation, and coinfections [5, 11]. Cellular immune activation has been studied in greatest detail in US and European populations with HIV-1 subtype B infection. Comprehensive analyses of T-cell immune activation in sub-Saharan populations infected with non–subtype B viruses have been limited.
East Africa exhibits some of the greatest HIV-1 genetic diversity globally, which may account for the differential rates of transmission and disease progression observed in this region [14]. In Uganda, subtypes A and D predominate, with increasing proportions of intersubtype recombinants and dual infections [15–18]. HIV-1 subtype D, recombinant infection, and dual infections are associated with more-rapid decline in the CD4 T-cell count and faster progression to AIDS-defining events, compared with subtype A [19–22]. In this study, we evaluated the relative contribution of HIV-1 load, viral subtype, and T-cell immune activation to the progression to AIDS and death in a rural Ugandan setting. The well-characterized Rakai Community Cohort Study (RCCS), with 13 years of clinical follow-up, allowed assessment of the contributions of these virologic and immunologic parameters to disease outcome.
METHODS
Study Design and Participants
The study population was 156 HIV-1 seroconverters, identified by the RCCS during annual serosurveys between 1997 and 2002. Time of HIV-1 infection was estimated as the midpoint between the last seronegative and the first seropositive sample. HIV-1 load set point was estimated as the median viral load after the initial stage of infection. Seroconverters were followed longitudinally in a separate study, the Molecular Epidemiology Research (MER) study, with sample collection between 1999 and 2004 [19, 23]. Long-term follow-up generating outcome data was conducted annually through 2010, as previously described [24], and the median follow-up duration per participant was 10 years. The study population was chosen on the basis of availability of cryopreserved peripheral blood mononuclear cells (PBMCs) and HIV-1 subtype data, and 13 individuals were excluded from disease progression analysis because of loss to follow-up. cART was not available in Rakai at the time of this study [19]. Twenty-two HIV-1–uninfected individuals from RCCS were used as community matched controls. Baseline characteristics were similar between the subgroup of seroconverters in this study and the broader MER cohort for age (P = .209, by the χ2 test), sex (P = .387, by the χ2 test), and HIV subtype distribution (P = .876, by the χ2 test). The study was approved by institutional review boards of Uganda's National Council for Science and Technology, the Uganda Virus Research Institute's Science and Ethics Committee, the Human Subjects Protection Branch at the Walter Reed Army Institute of Research, and the Johns Hopkins Bloomberg School of Public Health. All study participants provided written informed consent.
Procedures and Laboratory Measurements
Venous blood was collected from participants at the first visit following detection of seroconversion and then 3, 6, and 12 months and annually thereafter. At each study visit, plasma and PBMCs were stored as previously described [25]. Viral RNA was quantified from plasma, using the Roche 1.5 Amplicor test (Roche Diagnostics, Manheim, Germany), with a lower detection limit of 400 copies/mL. HIV-1 subtype, recombinants, or dual infection was previously determined using a multiplex hybridization assay that was specially designed for this region [18, 26]. A total of 15.4% of the infections were subtype A, 62.2% were subtype D, and 22.4% were recombinant or dual infections. Absolute CD4 T-cell counts were determined at each study visit, using either the FACS MultiSET System or the FACSCount system (Becton Dickinson, San Jose, California). The time to AIDS was defined as the time between the estimated date of seroconversion to either a CD4 count <250 cells/µL or onset of AIDS defining diseases meeting World Health Organization clinical stage 4 criteria. Time to death was calculated as the interval between the estimated date of infection and the date of death.
For the assessment of T-cell immune activation and exhaustion, cryopreserved specimens were thawed and washed with Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum, 2% HEPES, 2% l-glutamine, and 1% penicillin/streptomycin. Cell counts and viabilities were determined using Guava ViaCount and Guava PCA technology (Guava Technologies, Hayward, California), and the viability of all thawed samples was >80% as previously described for this cohort [25]. PBMCs were then distributed into a 96-well U-bottomed plate, washed, and stained for 30 minutes at 4°C in the dark. Monoclonal antibodies included anti-CD3 (clone SK7) conjugated to peridinin chlorophyll A protein (PerCP), anti-CD4 (clone SK3) and anti-CD8 (clone SK1) fluorescein isothiocyanate (FITC), anti-CD38 (clone HB7) R-phycoerythrin (PE), and anti-HLA-DR (clone L243) and anti-CD279 (PD-1, clone M1H4) allophycocyanin (APC), all from BD Biosciences (San Jose, California). Cells were washed with phosphate-buffered saline/bovine serum albumin/NaN3 buffer and fixed in 2% formaldehyde before data acquisition using a dual-laser FACSCalibur (BD Biosciences). Analysis was performed using FlowJo software, version 8.5 (Tree Star, Ashland, Oregon). Percentage of parent populations was used to quantify the frequency of T-cell populations expressing receptors alone or in combination, whereas the geometric mean was used to determine the mean fluorescent intensity (MFI) for individual proteins expressed on T-cell subsets, corresponding to the quantity of a receptor expressed on the surface of the cell.
Statistical Analyses
Statistical Analysis System (SAS) v9.2 was used for analysis. Two end points were considered—death or progression to AIDS. Nonparametric Spearman rank correlations were calculated between visit viral load/set point viral load and each of the T-cell immune activation measurements. Correlations between visit viral load and T-cell immune activation were considered statistically significant for P values of <.05. Kaplan–Meier survival analyses and univariate Cox proportional hazard models were used to explore the relationship between viral load, subtype, and immune activation on study end points. Hazard ratios (HRs) were adjusted for set point HIV-1 load. The log-rank test was used to detect the equality across strata. Quartiles were used to categorize continuous variables, such as viral load and all immune activation phenotypes, to select possible influential covariates and illustrate the relationship between variable and outcome. If the P value yielded by the log-rank test was <.2, then this variable was considered as a potential covariate for multivariate Cox proportional hazards regression models.
RESULTS
Population Characteristics
To examine the relative contribution of viral load, subtype, and T-cell immune activation on HIV-1 disease progression, we chose Rakai district, Uganda, to investigate these factors in a population with HIV-1 subtype A infection, HIV-1 subtype D infection, dual-subtype infection, or recombinant infection. Demographic characteristics for the 156 HIV-1–infected participants and 22 uninfected control subjects are shown in Table 1. No differences were observed in age and sex between HIV-infected and uninfected participants. A total of 64% of the HIV-1–infected participants were women, with a median age of 28 years at seroconversion. The time point chosen for virologic and immunologic assessment was a median of 584 days (interquartile range [IQR], 541–794 days) from the estimated day of seroconversion and was selected on the basis of the earliest cryopreserved PBMCs available. The median HIV-1 load and CD4 T-cell absolute counts, at the immunologically evaluated time point, were 35 351 copies/mL (IQR, 10 478–111 333 copies/mL) and 519 cells/µL (IQR, 374–761 cells/µL), respectively. There were no differences in CD4 T-cell absolute counts or time from seroconversion between men and women (data not shown). However, there was a trend toward higher viral loads in men, among whom the mean viral load (±standard deviation [SD]) was 61 718 ± 255 826 copies/mL, compared with 25 726 ± 146 244 copies/mL among women (P = .061). HIV-1 subtypes were similarly distributed between women and men (P = .897). There were no differences in CD4 T-cell count (P = .417), HIV-1 load (P = .240), or time from seroconversion (P = .395) across subtype A, subtype D, or dual and recombinant infections (Table 1).
Table 1.
Demographic and Human Immunodeficiency Virus Type 1 (HIV-1)–Associated Characteristics of Study Participants, by HIV-1 Status and Subtype
| Characteristic | HIV-1 Negative (n = 22) | HIV-1 Positive, by Subtype (n = 156) |
||
|---|---|---|---|---|
| A (n = 24) | AD (n = 35) | D (n = 97) | ||
| Sex, participants, no. (%) | ||||
| Female | 13 (59) | 16 (67) | 19 (54) | 65 (67) |
| Male | 9 (41) | 8 (33) | 16 (46) | 32 (33) |
| Age, y, mean ± SD | 31 ± 8 | 26 ± 8 | 27 ± 7 | 28 ± 7 |
| Time from seroconversion, d | NA | 564 (327–1356) | 581 (322–1270) | 588 (209–2054) |
| CD4 T-cell absolute count, cells/μLa | 731 (708–938) | 694 (245–1281) | 509 (270–1492) | 495 (98–1484) |
| HIV-1 load, copies/mL | NA | 25 692 (2148–427 096) | 28 555 (839–761 664) | 41 713 (262–1 330 850) |
| Study end point reached, participants, no. | ||||
| AIDS | NA | 7 | 14 | 48 |
| Death | NA | 3 | 12 | 25 |
Data are median value (range), unless otherwise indicated.
Abbreviations: NA, not applicable; SD, standard deviation.
a No. of individuals used for CD4 T-cell absolute counts: HIV-1 subtype A, 12; subtype AD (recombinant/dual infection), 16; and subtype D, 46.
T-Cell Activation and Exhaustion in HIV-1 Subtype A, Subtype D, and Recombinant AD Infection
T-cell expression of CD38 and HLA-DR, biomarkers associated with T-cell activation, or CD38 and CD279, corresponding to deregulated activated T cells with potentially impaired function, were assessed by flow cytometry (Figure 1). CD38, HLA-DR, and CD279 expression alone or in combination were elevated in HIV-1–infected subjects, compared with controls (Supplementary Table 1). Despite dynamic ranges, most T-cell phenotypic measures did not differ statistically between the 3 HIV-1 subtype categories (Table 2). The exception to this was CD38 mean fluorescence intensity (MFI) on CD8 T cells, which was modestly higher in HIV-1 subtype D and recombinant or dual infections, compared to HIV-1 subtype A infection (P = .030). HIV-1 load was positively correlated with the frequency of CD4 T cells coexpressing CD38 and HLA-DR (ρ = 0.433; P < .001; Figure 1B), as well as CD38 and CD279 (ρ = 0.350; P < .001; Figure 1C). Additionally, HIV-1 load was positively correlated with the frequency of CD8 T cells coexpressing CD38 and HLA-DR (ρ = 0.421; P < .001; Figure 1E) or CD38 and CD279 (ρ = 0.280; P < .001; Figure 1F). Similar positive correlations were observed between HIV-1 set point viral load and the frequency of T cells with an activated or exhausted phenotype (data not shown). These data indicate that T-cell activation levels in this cohort show similar cross-sectional relationships with viral load, as observed in cohorts from developed countries. Furthermore, viral subtypes appear to be only modestly, if at all, associated with differences in activation levels.
Figure 1.
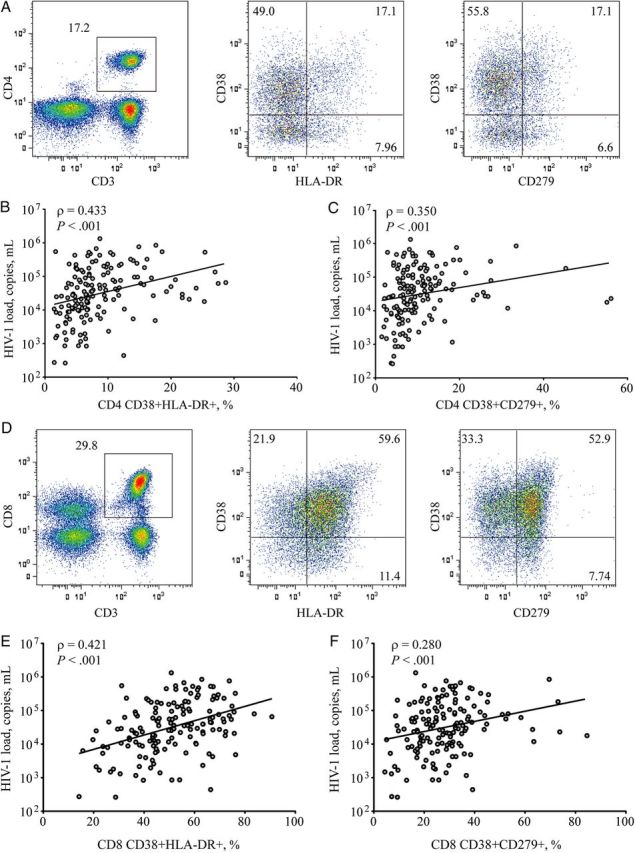
Characterization of immune activation and relationship to human immunodeficiency virus type 1 (HIV-1) load. Phenotypic markers of immune activation were enumerated from cryopreserved peripheral blood mononuclear cells, using flow cytometry. Lymphocytes were identified on the basis of size and granularity scatter profiles. A, Pseudo-color histogram of CD3-PerCP expression in combination with CD4-FITC identified helper T cells on a representative HIV-infected donor. CD38-PE and HLA-DR-APC, associated with activated phenotypes, or CD38-PE and CD279-APC, corresponding to activated T cells with potentially impaired function, were calculated. B and C, Relationship to HIV-1 load was determined for CD38-PE and HLA-DR-APC (B) and CD38-PE and CD279-APC (C), where positive Spearman correlations were observed. D, Pseudo-color histogram of cytotoxic T cells identified from CD3-PerCP expression in combination with CD8-FITC from a representative HIV-1–infected donor. CD38 and HLA-DR or CD279 in combination were evaluated. E and F, CD38-PE and HLA-DR-APC (E) and CD38-PE and CD279-APC (F) showed a positive relationship to HIV-1 load using a spearman correlation.
Table 2.
T-Cell Phenotypic Changes in Subtype A, D, and Recombinant AD Human Immunodeficiency Virus Type 1 (HIV-1) Infection
| Variablea | Mean Value ± SD, By Subtype |
P Valuesb | ||
|---|---|---|---|---|
| A (n = 24) | AD (n = 35) | D (n = 97) | ||
| CD8 T cells | ||||
| CD38+, % | 72.3 ± 12.5 | 74.8 ± 14.6 | 76.4 ± 13.7 | .313 |
| CD38+, MFI | 123.2 ± 46.1 | 131.9 ± 56.9 | 153.0 ± 60.4 | .030 |
| CD279+, % | 30.3 ± 15.3 | 29.8 ± 17.1 | 31.3 ± 13.6 | .541 |
| HLA-DR+, % | 57.0 ± 13.4 | 59.4 ± 13.5 | 58.6 ± 14.4 | .794 |
| CD38+HLA-DR+, % | 47.3 ± 13.3 | 50.6 ± 15.1 | 51.2 ± 15.6 | .426 |
| CD38+CD279+, % | 26.9 ± 13.7 | 27.2 ± 15.9 | 28.4 ± 12.8 | .555 |
| CD4 T cells | ||||
| CD38+, % | 52.2 ± 16.4 | 51.7 ± 16.9 | 56.0 ± 14.3 | .243 |
| CD38+, MFI | 112.5 ± 32.5 | 119.5 ± 48.1 | 132.3 ± 44.3 | .058 |
| CD279+, % | 14.2 ± 13.4 | 15.5 ± 15.2 | 14.3 ± 9.0 | .393 |
| HLADR+, % | 13.0 ± 8.1 | 14.5 ± 8.4 | 13.7 ± 7.8 | .666 |
| CD38+HLA-DR+, % | 7.6 ± 5.1 | 9.0 ± 6.2 | 9.0 ± 5.8 | .359 |
| CD38+CD279+, % | 9.9 ± 9.9 | 11.7 ± 12.2 | 10.4 ± 6.2 | .156 |
Data are mean value ± SD.
Abbreviations: MFI, mean fluorescence intensity; SD, standard deviation.
a Percentages denote the percentage of the T-cell subset expressing the specified marker.
b By the Kruskal–Wallis test.
T-Cell Activation, HIV-1 Subtype, Viral Load, and HIV-1 Infection Progression to AIDS or Death
Kaplan–Meier survival analyses were performed to determine relationships between T-cell activation, viral load and subtype, and clinical progression to AIDS or death (Figure 2). The frequency of CD4 T cells coexpressing CD38 and HLA-DR was associated with faster progression to death, but this relationship did not reach statistical significance for CD38 and CD279 (P = .004 and P = .081, respectively, by the log rank test). The frequency of CD4 T cells coexpressing CD38 and HLA-DR or CD38 and CD279 was associated with faster progression to AIDS (P < .001 and P = .005, respectively, by the log rank test). The frequency of CD8 T cells coexpressing CD38 and HLA-DR (Figure 2A) but not CD38 and CD279 was associated with a shorter time to AIDS-defining events (P = .014 and P = .191, respectively, by the log rank test). CD8 T-cell coexpression of CD38 and HLA-DR (Figure 2B) but not CD38 and CD279 were associated with a shorter time to death (P = .001 and P = .156 respectively, by the log rank test). With regard to HIV-1 subtype, we observed a trend toward faster progression to AIDS (P = .051, by the log rank test; Figure 2C) and death (P = .078, by the log rank test; Figure 2D) for subtype D or recombinant/dual infections, compared with HIV-1 subtype A infection. Analysis of quartiles of viral load revealed that higher quartiles of viremia were associated with faster progression to AIDS (P < .001, by the log rank test; Figure 2E) and death (P < .001, by the log rank test; Figure 2F).
Figure 2.

Kaplan–Meier estimates to AIDS or death by T-cell activation, human immunodeficiency virus type 1 (HIV-1) subtype, or viral load. Kaplan–Meier estimates are presented, comparing the time to AIDS, as defined by CD4 T-cell counts of <250 cells/µL of whole blood, or the time to death for T-cell activation, HIV-1 subtype, and viral load. A and B, Individuals were stratified into quartiles on the basis of the percentage of CD8 T cells coexpressing CD38 and HLA-DR from lowest frequency (first quartile; blue) to the highest frequency (fourth quartile; brown) for time to AIDS (A) or time to death (B). C and D, Kaplan–Meier estimates for HIV-1 subtype A (blue), recombinant/dual infections (red), and subtype D (green) are displayed for time to AIDS (C) or time to death (D). E and F, Individuals were stratified into quartiles on the basis of HIV-1 load from lowest frequency (first quartile; blue) to highest frequency (fourth quartile; brown) for time to AIDS (E) or time to death (F). All P values were calculated with the log-rank test.
We next analyzed possible associations between T-cell activation, viral subtype and viral load, and the clinical follow-up data (Table 3). Examining quartiles of CD38 and HLA-DR coexpression as a percentage of CD4 T cells demonstrated a HR of 5.85 (95% CI, 1.68–20.32), adjusted for set point HIV-1 load, for expression levels of >10.5%, compared with levels of <4.8%, for disease progression to death and an adjusted HR of 2.58 (95% CI, 1.16–5.74) for progression to AIDS. Similarly, for the frequency of CD38 and HLA-DR coexpression of >61.6%, compared with levels of <39.3%, on CD8 T cells the adjusted HR was 4.82 (95% CI, 1.62–14.31) for disease progression to death. However, despite a trend, there was no significant association between CD8 T-cell activation (CD38 and HLA-DR coexpression) and progression to AIDS. The highest quartile of CD38 and CD279 coexpression on CD4 T cells was associated with an adjusted HR of 2.66 (95% CI, 1.28–5.51) for progression to AIDS but was not significantly associated with time to death. CD8 T-cell simultaneous expression of CD38 and CD279 was not significantly associated with time to AIDS or death. These data indicate that an increased frequency of T cells expressing an activated phenotype was associated with faster progression to AIDS or death.
Table 3.
T-Cell Phenotype Survival Statistics
| T-Cell Subset, Phenotype, Event, Quartilea | Events, No. | Events/1000 Person-Years (95% CI) | Adjustedb HR (95%CI) | P Values |
|---|---|---|---|---|
| CD4 T cells | ||||
| CD38+HLA-DR+ | ||||
| Death | ||||
| 1st (1.47–4.76) | 3 | 11.0 (2.3–32.2) | Reference | .030 |
| 2nd (4.76–6.7) | 10 | 36.6 (17.5–67.2) | 2.85 (.78–10.38) | |
| 3rd (6.7–10.5) | 11 | 53.7 (26.8–96.2) | 4.20 (1.15–15.41) | |
| 4th (10.5–28.7) | 16 | 79.4 (45.4–128.9) | 5.85 (1.68–20.32) | |
| AIDS | ||||
| 1st (1.47–4.76) | 10 | 39.9 (19.2–73.5) | Reference | .020 |
| 2nd (4.76–6.7) | 22 | 87.7 (54.9–132.7) | 2.03 (.96–4.31) | |
| 3rd (6.7–10.5) | 21 | 135.6 (83.9–207.2) | 3.39 (1.56–7.39) | |
| 4th (10.5–28.7) | 16 | 111.0 (63.4–180.2) | 2.58 (1.16–5.74) | |
| CD38+CD279+ | ||||
| Death | ||||
| 1st (1.49–5.61) | 6 | 22.4 (8.2–48.8) | Reference | .101 |
| 2nd (5.61–8.27) | 11 | 46.2 (23.1–82.7) | 2.06 (.76–5.58) | |
| 3rd (8.27–12.6) | 8 | 35.8 (15.5–70.6) | 1.27 (.43–3.77) | |
| 4th (12.6–56.1) | 15 | 67.2 (37.6–110.9) | 2.88 (1.11–7.44) | |
| AIDS | ||||
| 1st (1.49–5.61) | 12 | 47.4 (24.5–82.7) | Reference | .013 |
| 2nd (5.61–8.27) | 16 | 75.5 (43.2–122.6) | 1.65 (.77–3.50) | |
| 3rd (8.27–12.6) | 22 | 124.8 (78.2–189.0) | 2.89 (1.42–5.86) | |
| 4th (12.6–56.1) | 19 | 119.5 (72.0–186.7) | 2.66 (1.28–5.51) | |
| CD8 T cells | ||||
| CD38+HLA-DR+ | ||||
| Death | ||||
| 1st (14.2–39.3) | 4 | 14.9 (4.1–38.2) | Reference | .012 |
| 2nd (39.3–50.6) | 6 | 24.1 (8.9–52.5) | 1.62 (.46–5.75) | |
| 3rd (50.6–61.6) | 11 | 53.7 (26.8–96.1) | 3.24 (1.03–10.20) | |
| 4th (61.6–90.7) | 19 | 82.4 (49.6–128.7) | 4.82 (1.62–14.31) | |
| AIDS | ||||
| 1st (14.2–39.3) | 11 | 44.2 (22.1–79.2) | Reference | .062 |
| 2nd (39.3–50.6) | 23 | 106.1 (67.3–159.2) | 2.36 (1.14–4.86) | |
| 3rd (50.6–61.6) | 17 | 100.3 (58.4–160.6) | 2.23 (1.03–4.83) | |
| 4th (61.6–90.7) | 18 | 108.7 (64.4–171.9) | 2.66 (1.25–5.67) | |
| CD38+CD279+ | ||||
| Death | ||||
| 1st (4.39–18.7) | 7 | 28.3 (11.4–58.2) | Reference | .248 |
| 2nd (18.7–27.4) | 9 | 42.3 (19.3–80.2) | 1.33 (.48–3.68) | |
| 3rd (27.4–34.4) | 8 | 32.6 (14.1–64.2) | 0.90 (.33–2.51) | |
| 4th (34.4–84.6) | 16 | 65.1 (37.2–105.6) | 1.96 (.80–4.78) | |
| AIDS | ||||
| 1st (4.39–18.7) | 15 | 66.1 (37–109) | Reference | .598 |
| 2nd (18.7–27.4) | 12 | 71.6 (37–125.1) | 1.47 (.68–3.17) | |
| 3rd (27.4–34.4) | 26 | 125.2 (81.8–183.5) | 1.51 (.79–2.87) | |
| 4th (34.4–84.6) | 16 | 80.7 (46.1–131.0) | 1.19 (.58–2.41) |
Abbreviations: CI, confidence interval; HIV, human immunodeficiency virus; HR, hazard ratio.
a Values in parentheses denote the percentage of the T-cell subset with the specified phenotype.
b Adjusted for set point HIV-1 load.
Relative Effect of HIV-1 Load, HIV-1 Subtype, and T-Cell Activation on Disease Progression
We first compared the individual contribution of demographic, immunologic, and virologic parameters to the rate of disease progression (Table 4), to select possible influential covariates and down-select parameters for multivariate Cox proportional hazards regression models (Table 5). The full multivariate analysis is shown in Supplementary Table 2 for progression to AIDS and in Supplementary Table 3 for death. In the final multivariate model, HIV-1 AD recombinants were associated with shorter time to death (adjusted HR, 3.77; P = .044). Log10 viral load was a strong predictor of death (adjusted HR, 3.09; P < .0001), and older age at seroconversion showed a trend toward predicting death due to HIV-1 infection (adjusted HR, 1.04; P = .0522). Use of progression to AIDS as an end point revealed that HIV-1 subtype AD recombinant (adjusted HR, 3.11; P = .016) and log10 viral load at visit (adjusted HR, 2.75; P < .0001) were significant factors, but age at seroconversion was not. The Cox models did not significantly violate the assumption of proportionality, using AIDS or death as an end point (P = .113 and P = .523, respectively). These data indicate that in this rural Ugandan cohort, HIV-1 load is an independent strong predictor of disease progression, as is infection with AD recombinant virus. Surprisingly, however, the frequency of T cells expressing an activated or exhausted phenotype did not emerge as an independent predictor of disease progression from this particular cohort.
Table 4.
Univariate Analysis for Modeling
| Parameter | P Valuesa | HR (95% CI) |
|---|---|---|
| Univariate analysis results for AIDS | ||
| CD8 T cells | ||
| CD38+ (10% increment) | .0018 | 10.33 (10.12–10.54) |
| CD38+ (MFI) | <.0001 | 1.009 (1.005–1.013) |
| CD279+ (10% increment) | .5155 | 10.05 (9.89–10.22) |
| HLADR+ (10% increment) | .0655 | 10.16 (9.99–10.33) |
| CD38+HLA-DR+ (10% increment) | .0047 | 10.22 (10.07–10.38) |
| CD38+CD279+ (10% increment) | .1492 | 10.13 (9.95–10.32) |
| CD4 T cells | ||
| CD38+ (10% increment) | .0485 | 10.16 (10.00–10.33) |
| CD38+ (MFI) | .0007 | 1.009 (1.004–1.014) |
| CD279+ (10% increment) | .1660 | 10.13 (9.95–10.32) |
| HLADR+ (10% increment) | .0034 | 10.46 (10.15–10.79) |
| CD38+HLA-DR+ (10% increment) | .0012 | 10.65 (10.25–11.06) |
| CD38+CD279+ (10% increment) | .0170 | 10.30 (10.05–10.55) |
| Age at SC (10 year increment) | .5333 | 10.10 (9.79–10.42) |
| Set point VL (log10) | <.0001 | 2.062 (1.478–2.878) |
| Visit VL (log10) | <.0001 | 2.791 (1.944–4.008) |
| HIV-1 Subtype (AD) | .0222 | 2.916 (1.165–7.300) |
| HIV-1 Subtype (D) | .0342 | 2.362 (1.066–5.231) |
| Sex (female) | .4978 | 1.194 (0.715–1.992) |
| Univariate analysis results for death | ||
| CD8 T cells | ||
| CD38+ (10% increment) | .0049 | 10.42 (10.13–10.73) |
| CD38+ (MFI) | <.0001 | 1.01 (1.006–1.014) |
| CD279+ (10% increment) | .0369 | 10.18 (10.01–10.35) |
| HLADR+ (10% increment) | .0016 | 10.38 (10.14–10.62) |
| CD38+HLA-DR+ (10% increment) | .0003 | 10.38 (10.17–10.60) |
| CD38+CD279+ (10% increment) | .0043 | 10.28 (10.09–10.47) |
| CD4 T cells | ||
| CD38+ (10% increment) | .0685 | 10.2 (9.98–10.42) |
| CD38+ (MFI) | .003 | 1.009 (1.003–1.015) |
| CD279+ (10% increment) | .0933 | 10.16 (9.97–10.34) |
| HLADR+ (10% increment) | .0006 | 10.59 (10.25–10.93) |
| CD38+HLA-DR+ (10% increment) | .0001 | 10.85 (10.41–11.31) |
| CD38+CD279+ (10% increment) | .012 | 10.31 (10.07–10.55) |
| Age at SC (10 year increment) | .077 | 10.34 (9.96–10.74) |
| Set point VL (log10) | .0049 | 1.863 (1.208–2.871) |
| Visit VL (log10) | <.0001 | 3.117 (1.887–5.149) |
| HIV-1 Subtype (AD) | .032 | 4.058 (1.128–14.593) |
| HIV-1 Subtype (D) | .0866 | 2.854 (0.86–9.469) |
| Sex (female) | .2108 | 0.667 (0.354–1.258) |
Abbreviations: CI, confidence interval; HIV, human immunodeficiency virus; MFI, mean fluorescence intensity; SC, seroconversion; VL, HIV-1 viral load.
a By χ2 analysis.
Table 5.
Multiparameter Regression Analysis to AIDS or Death
| Parameter | HR (95% CI) | P Valuesa |
|---|---|---|
| Maximum likelihood estimates (AIDS) | ||
| HIV-1 Subtype AD | 3.11 (1.23–7.82) | .016 |
| HIV-1 Subtype D | 1.91 (0.85–4.26) | .117 |
| Visit VL (log10) | 2.75 (1.91–3.94) | <.0001 |
| Maximum likelihood estimates (death) | ||
| HIV-1 Subtype (AD) | 3.77 (1.04–13.73) | .044 |
| HIV-1 Subtype (D) | 2.36 (0.71–7.89) | .163 |
| Visit VL (log10) | 3.09 (1.87–5.10) | <.0001 |
| Age at SC | 1.04 (1.00–1.09) | .052 |
Abbreviations: CI, confidence interval; HIV, human immunodeficiency virus; HR, hazard ratio; SC, seroconversion; VL, HIV-1 viral load.
a By χ2 analysis.
DISCUSSION
HIV/AIDS-related mortality ranked as the sixth leading cause of death globally in 2010, accounting for >10% of deaths in individuals aged 15–49 years, and remains the leading cause for years of life lost in areas of sub-Saharan Africa [27]. Factors contributing to HIV-1 disease progression include host genetics, viral diversity, and coinfections endemic to resource-limited tropical settings. We used multivariate regression to assess the complex associations between immunologic and virologic factors contributing to AIDS and death. Viral load and subtype were the strongest predictors of HIV-1 disease progression. Despite significant associations in univariate analysis, a number of immunologic variables, including T-cell activation, did not emerge as independent predictors of disease progression and mortality in the subgroup of seroconverters studied from this particular cohort.
Cellular immune activation, commonly characterized by receptors on the surface of T cells, is considered to be of great importance to the prognosis of untreated HIV-1 infection. Treatments that prevent or limit immune activation are being explored [28]. Chloroquine administered in chronic untreated HIV-1 infection lowered T-cell activation levels with no clear effect on HIV-1 load [29]. Contrary to this report, hydroxychloroquine, an analogue of chloroquine used in treatment of systemic lupus erythematosus, showed no changes in T-cell activation and, in fact, suggested advanced disease progression in the treatment arm of this study [30]. Another study examining an HMG-CoA reductase inhibitor with antiinflammatory properties showed modest reductions in T-cell immune activation but had no effect on viral load [31]. In Ethiopia, successful treatment of intestinal parasitic infections was associated with a minor reduction in T-cell activation in HIV-1–negative individuals but not in HIV-1–positive individuals [32]. It is clear that patients initiating cART experience a lowering of HIV-1 load and subsequent reductions in T-cell immune activation during immune reconstitution [33, 34]. Interestingly, persistent CD8 T-cell immune activation has been associated with mortality after initiation of cART in virologically suppressed Ugandans [35]. In addition, cART initiated during acute HIV-1 infection does not resolve T-cell activation to levels observed in HIV-1–uninfected healthy individuals [36, 37]. Together, these data may suggest that while T-cell immune activation is integrally linked to HIV-1 load, different treatment regimens may reduce levels of immune activation without corresponding reductions in viral load.
Our data support a model in which reduction in HIV-1 replication remains the primary rationale for treatment. It is important to note that our data represent heterosexually transmitted infection in a rural sub-Saharan setting and may not extend to other regions or populations. It is also important to note that the current study may be confounded by patient follow-up. In African cohort studies, it has been shown that mortality is underestimated because of loss to follow-up [38]. We conducted a sensitivity analysis and found that if all participant loss to follow-up was due to mortality, higher CD38 expression on CD8 T cells (as measured by MFI) emerged as a weak independent predictor of HIV-associated death. However, in this simulated model, viral load and subtype remained the strongest predictors of HIV-1 disease progression.
The observation that T-cell immune activation was similar between subtype A, subtype D, and subtype D–containing recombinants was surprising because of literature that independently link both subtype and activation to HIV-1 disease progression [4, 6, 19, 22]. There are limited data examining T-cell immune activation in HIV-1 subtypes circulating in Africa, compared with HIV-1 subtype B infection, and those data that exist are conflicting [34, 39]. This is most likely because of difficulties in controlling for biological and technical differences. Uganda presents a unique opportunity to compare activation across HIV-1 subtypes, within a single cohort. We have previously shown a differential loss of T-cell subsets with regulatory functions between subtype A and D infection [40]. Bousheri et al have shown that HIV-1 subtype D infection is associated with increased CD4 T-cell apoptosis, compared with HIV-1 subtype A, despite similar T-cell activation levels [12]. Although our data is consistent with regard to T-cell activation, we did not see measurable differences in PD-1, which could be due to differences in reagents, urban and rural differences between Kampala and Rakai, or HIV-1 disease state. In addition, in our study, enumeration of subsets expressing both PD-1 and CD38 were predictive of disease progression to AIDS and death in univariate but not multivariate analyses. Thus, our data suggest that HIV-1 disease progression to AIDS or death in this East African cohort is primarily dependent on viral load, rather than T-cell activation.
Several factors may influence the differential associations with disease progression observed in the present study and in previous studies based on cohorts from developed countries. The present study evaluated a heterosexual transmission group, predominantly female, whereas previous studies reporting the independent association of T-cell activation with disease progression were based on primarily male cohorts of homosexual transmission or injection drug use [4, 6]. In HIV-1 subtype B infection, risk factors associated with disease progression vary on the basis of sex and race [41–43]. Moreover, HIV-infected women maintain lower HIV-1 loads than men but experience accelerated rates of disease progression [44–46]. Furthermore, sex differences in innate signaling pathways may impact HIV-1 disease progression rates [47]. Additionally, host immunogenetics are probably different between the Rakai, Uganda, cohort and participants from previous studies characterizing HIV-1 subtype B. Two gene families, KIR and HLA, are critically involved in immunologic processes, highly polymorphic, and associated with HIV-1 disease outcome [48, 49]. There is considerable variability in KIR/HLA loci between African populations and cohorts in the United States and Europe [50]. Other factors may also contribute to immunological differences between East Africa and Europe and the United States, including climate, diet, and endemic diseases, such as malaria, tuberculosis, and helminthic infections. The extent to which all of these factors combine to influence the HIV-1 disease course remains unclear.
Although immune activation is strongly associated with disease progression in HIV-1 subtype B infections, our data suggest that the rate of viral replication is the primary driver of HIV-1 disease in Uganda. This striking observation strongly suggests that renewed studies of HIV-1 immunopathogenesis are needed and furthermore indicates that immunopathogenesis may be critically influenced by the local conditions under which an infected individual is living.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the Rakai District community, leadership, and study participants, for their valuable contribution; and the Rakai District Health Science Program and the Makerere University Walter Reed Project, for their tireless work to combat the HIV/AIDS epidemic at the front lines.
M. A. E., M. S. O., A. D. R., and J. K. S. wrote the manuscript. M. A. E., M. S. O., C. K., and L. A. E. contributed to study design, experimentation, and collection of data. M. A. E., M. S. O., M. L., C. K., J. K., and M. M. performed data and statistical analysis. M. A. E., A. D. R., O. L., T. C. Q., J. K. S., and M. L. R. contributed to interpretation of the data. O. L., M. J. W., N. K., R. H. G., D. S., N. K. S., T. C. Q., N. L. M., F. W.-M., and M. L. R. contributed to the design, execution, collection and analysis of primary cohort data. All authors reviewed the final manuscript.
Disclaimer. The views expressed are those of the authors and should not be construed to represent the positions of the US Army or the Department of Defense. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Financial support. This work was supported by a cooperative agreement (W81XWH-11-2-0174) between the Henry M. Jackson Foundation for the Advancement of Military Medicine and the US Army Medical Research and Materiel Command; and the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Cao Y, Qin L, Zhang L, Safrit J, Ho DD. Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N Eng J Med 1995; 332:201–8. [DOI] [PubMed] [Google Scholar]
- 2.Pantaleo G, Menzo S, Vaccarezza M, et al. Studies in subjects with long-term nonprogressive human immunodeficiency virus infection. N Eng J Med 1995; 332:209–16. [DOI] [PubMed] [Google Scholar]
- 3.Palella FJ, Jr, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Eng J Med 1998; 338:853–60. [DOI] [PubMed] [Google Scholar]
- 4.Giorgi JV, Hultin LE, McKeating JA, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis 1999; 179:859–70. [DOI] [PubMed] [Google Scholar]
- 5.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 2006; 12:1365–71. [DOI] [PubMed] [Google Scholar]
- 6.Deeks SG, Kitchen CM, Liu L, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 2004; 104:942–7. [DOI] [PubMed] [Google Scholar]
- 7.John L, Lutwama F. A review of the use of activation markers in Africa. J HIV Ther 2010; 15:11–4. [PubMed] [Google Scholar]
- 8.Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 2006; 12:1198–202. [DOI] [PubMed] [Google Scholar]
- 9.Petrovas C, Casazza JP, Brenchley JM, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med 2006; 203:2281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006; 443:350–4. [DOI] [PubMed] [Google Scholar]
- 11.Eller MA, Blom KG, Gonzalez VD, et al. Innate and adaptive immune responses both contribute to pathological CD4 T cell activation in HIV-1 infected Ugandans. PLoS One 2011; 6:e18779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bousheri S, Burke C, Ssewanyana I, et al. Infection with different hiv subtypes is associated with CD4 activation-associated dysfunction and apoptosis. J Acquir Immune Defic Syndr 2009; 52:548–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatano H, Jain V, Hunt PW, et al. Cell-Based Measures of Viral Persistence Are Associated With Immune Activation and Programmed Cell Death Protein 1 (PD-1)-Expressing CD4+ T cells. J Infect Dis 2013; 208:50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor BS, Hammer SM. The challenge of HIV-1 subtype diversity. N Engl J Med 2008; 359:1965–6. [DOI] [PubMed] [Google Scholar]
- 15.Harris ME, Serwadda D, Sewankambo N, et al. Among 46 near full length HIV type 1 genome sequences from Rakai District, Uganda, subtype D and AD recombinants predominate. AIDS Res Hum Retroviruses 2002; 18:1281–90. [DOI] [PubMed] [Google Scholar]
- 16.Peeters M, Toure-Kane C, Nkengasong JN. Genetic diversity of HIV in Africa: impact on diagnosis, treatment, vaccine development and trials. AIDS 2003; 17:2547–60. [DOI] [PubMed] [Google Scholar]
- 17.Hemelaar J, Gouws E, Ghys PD, Osmanov S. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS 2011; 25:679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arroyo MA, Sateren WB, Serwadda D, et al. Higher HIV-1 incidence and genetic complexity along main roads in Rakai District, Uganda. J Acquir Immune Defic Syndr 2006; 43:440–5. [DOI] [PubMed] [Google Scholar]
- 19.Kiwanuka N, Laeyendecker O, Robb M, et al. Effect of human immunodeficiency virus Type 1 (HIV-1) subtype on disease progression in persons from Rakai, Uganda, with incident HIV-1 infection. J Infect Dis 2008; 197:707–13. [DOI] [PubMed] [Google Scholar]
- 20.Kaleebu P, French N, Mahe C, et al. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J Infect Dis 2002; 185:1244–50. [DOI] [PubMed] [Google Scholar]
- 21.Kiwanuka N, Robb M, Laeyendecker O, et al. HIV-1 viral subtype differences in the rate of CD4+ T-cell decline among HIV seroincident antiretroviral naive persons in Rakai district, Uganda. J Acquir Immune Defic Syndr 2010; 54:180–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amornkul PN, Karita E, Kamali A, et al. Disease progression by infecting HIV-1 subtype in a seroconverter cohort in sub-Saharan Africa. AIDS 2013; 27:2775–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiwanuka N, Robb M, Kigozi G, et al. Knowledge about vaccines and willingness to participate in preventive HIV vaccine trials: a population-based study, Rakai, Uganda. J Acquir Immune Defic Syndr 2004; 36:721–5. [DOI] [PubMed] [Google Scholar]
- 24.Wawer MJ, Gray RH, Sewankambo NK, et al. A randomized, community trial of intensive sexually transmitted disease control for AIDS prevention, Rakai, Uganda. AIDS 1998; 12:1211–25. [DOI] [PubMed] [Google Scholar]
- 25.Olemukan RE, Eller LA, Ouma BJ, et al. Quality monitoring of HIV-1-infected and uninfected peripheral blood mononuclear cell samples in a resource-limited setting. Clin Vaccine Immunol 2010; 17:910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoelscher M, Dowling WE, Sanders-Buell E, et al. Detection of HIV-1 subtypes, recombinants, and dual infections in east Africa by a multi-region hybridization assay. AIDS 2002; 16:2055–64. [DOI] [PubMed] [Google Scholar]
- 27.Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380:2095–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plaeger SF, Collins BS, Musib R, Deeks SG, Read S, Embry A. Immune activation in the pathogenesis of treated chronic HIV disease: a workshop summary. AIDS Res Hum Retroviruses 2012; 28:469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murray SM, Down CM, Boulware DR, et al. Reduction of immune activation with chloroquine therapy during chronic HIV infection. J Virol 2010; 84:12082–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paton NI, Goodall RL, Dunn DT, et al. Effects of hydroxychloroquine on immune activation and disease progression among HIV-infected patients not receiving antiretroviral therapy: a randomized controlled trial. JAMA 2012; 308:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganesan A, Crum-Cianflone N, Higgins J, et al. High dose atorvastatin decreases cellular markers of immune activation without affecting HIV-1 RNA levels: results of a double-blind randomized placebo controlled clinical trial. J Infect Dis 2011; 203:756–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kassu A, Tsegaye A, Wolday D, et al. Role of incidental and/or cured intestinal parasitic infections on profile of CD4+ and CD8+ T cell subsets and activation status in HIV-1 infected and uninfected adult Ethiopians. Clin Exp Immunol 2003; 132:113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koblavi-Deme S, Maran M, Kabran N, et al. Changes in levels of immune activation and reconstitution markers among HIV-1-infected Africans receiving antiretroviral therapy. AIDS 2003; 17(suppl 3):S17–22. [DOI] [PubMed] [Google Scholar]
- 34.Eggena MP, Barugahare B, Okello M, et al. T cell activation in HIV-seropositive Ugandans: differential associations with viral load, CD4+ T cell depletion, and coinfection. J Infect Dis 2005; 191:694–701. [DOI] [PubMed] [Google Scholar]
- 35.Hunt PW, Cao HL, Muzoora C, et al. Impact of CD8+ T-cell activation on CD4+ T-cell recovery and mortality in HIV-infected Ugandans initiating antiretroviral therapy. AIDS 2011; 25:2123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gay CL, Mayo AJ, Mfalila CK, et al. Efficacy of NNRTI-based antiretroviral therapy initiated during acute HIV infection. AIDS 2011; 25:941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinikoor MJ, Cope A, Gay CL, et al. Antiretroviral therapy initiated during acute HIV infection fails to prevent persistent T-cell activation. J Acquir Immune Defic Syndr 2013; 62:505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng EH, Emenyonu N, Bwana MB, Glidden DV, Martin JN. Sampling-based approach to determining outcomes of patients lost to follow-up in antiretroviral therapy scale-up programs in Africa. JAMA 2008; 300:506–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weisman Z, Kalinkovich A, Borkow G, Stein M, Greenberg Z, Bentwich Z. Infection by different HIV-1 subtypes (B and C) results in a similar immune activation profile despite distinct immune backgrounds. J Acquir Immune Defic Syndr 1999; 21:157–63. [PubMed] [Google Scholar]
- 40.Flach B, Naluyima P, Blom K, et al. Differential loss of invariant natural killer T cells and FoxP3(+) regulatory T cells in HIV-1 subtype A and subtype D infections. J Acquir Immune Defic Syndr 2013; 63:289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hessol NA, Kalinowski A, Benning L, et al. Mortality among participants in the Multicenter AIDS Cohort Study and the Women's Interagency HIV Study. Clin Infect Dis 2007; 44:287–94. [DOI] [PubMed] [Google Scholar]
- 42.Hariri S, McKenna MT. Epidemiology of human immunodeficiency virus in the United States. Clin Microbiol Rev 2007; 20:478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemly DC, Shepherd BE, Hulgan T, et al. Race and sex differences in antiretroviral therapy use and mortality among HIV-infected persons in care. J Infect Dis 2009; 199:991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farzadegan H, Hoover DR, Astemborski J, et al. Sex differences in HIV-1 viral load and progression to AIDS. Lancet 1998; 352:1510–4. [DOI] [PubMed] [Google Scholar]
- 45.Evans JS, Nims T, Cooley J, et al. Serum levels of virus burden in early-stage human immunodeficiency virus type 1 disease in women. J Infect Dis 1997; 175:795–800. [DOI] [PubMed] [Google Scholar]
- 46.Sterling TR, Lyles CM, Vlahov D, Astemborski J, Margolick JB, Quinn TC. Sex differences in longitudinal human immunodeficiency virus type 1 RNA levels among seroconverters. J Infect Dis 1999; 180:666–72. [DOI] [PubMed] [Google Scholar]
- 47.Meier A, Chang JJ, Chan ES, et al. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med 2009; 15:955–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bashirova AA, Thomas R, Carrington M. HLA/KIR restraint of HIV: surviving the fittest. Annu Rev Immunol 2011; 29:295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carrington M, Martin MP, van Bergen J. KIR-HLA intercourse in HIV disease. Trends Microbiol 2008; 16:620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Single RM, Martin MP, Gao X, et al. Global diversity and evidence for coevolution of KIR and HLA. Nat Genet 2007; 39:1114–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.