

Summary
This review focuses on the current knowledge of signaling and regulation by NF-kB, including its debated pro- and anti-survival roles in cancer and highlights the significance of examining the exact role of NF-kB in bladder cancer.
Abstract
Signaling and regulation of transcription factor nuclear factor-kappaB (NF-κB) has been an area of extensive research since its first discovery nearly three decades ago. Members of the NF-κB family have been reported to critically mediate a multitude of responses in normal cells. Therefore, it is not surprising that NF-κB function can go awry and result in pathological conditions including cancer. Despite its critical importance, the functional role of NF-κB has not received the same attention in cancers of all tissue types. In the case of cancer of the urinary bladder, which is the second most common urologic cancer, the involvement of NF-κB in the development of superficial or muscle invasive disease and during cancer recurrence is rudimentary at best. Nuclear expression of p65/RelA is seen in bladder cancer patients and has been found to negatively affect survival of patients with superficial and muscle invasive disease. Despite these observations, the exact mechanism of NF-κB upregulation and function remains unknown. Furthermore, the emergence of a tumor suppressive role for NF-κB in recent years suggests that the family may play the role of a double-edged sword in cancer, which remains unexplored in bladder cancer. The challenge now is to delineate the increasing complexity of this pathway in the development and progression of bladder cancer. Here, we review key aspects of the current knowledge of signaling and regulation by the NF-κB family focusing on its controversial role in cancer and highlight the importance of studying NF-κB in bladder cancer in particular.
Introduction
Nuclear factor-kappaB (NF-κB) was identified as a regulator of the κB light chain in mature B cells and plasma cells (1). Following this initial discovery, NF-κB was found in almost all cell types and tissues where it regulates gene expression by binding to promoters/enhancers of a host of genes. Over the years, NF-κB has been found to regulate various responses to different stimuli and has been established as a critical mediator of physiological and pathological processes including many cancers. However, the role of NF-κB is context dependent and its tumor promoting and or tumor suppressing properties may depend to a large extent on the stage and type of cancer. Despite the critical importance of NF-κB in cancer, the function of NF-κB in urothelial cancer remains poorly defined. This review summarizes current knowledge of NF-κB-mediated transcriptional regulation and signaling in cancer and highlights the potential importance of NF-κB in bladder cancer and the existing gaps that should be investigated.
NF-κB family
The NF-κB family consists of five proteins, p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1) and p100/52 (NF-κB2) that form homo- and heterodimeric complexes by associating with each other to transcriptionally regulate target genes. All family members have a 300-amino acid long amino-terminal Rel homology domain (RHD (2)). The amino-terminus of RHD helps with DNA binding to the NF-κB consensus sequence present in regulatory elements of NF-κB target genes whereas the carboxy-terminus participates in dimerization and interaction with IκB (3–5) (Figure 1). RelA, RelB and c-Rel contain the carboxy-terminal transactivation domains (TAD) whereas p50 and p52, generated by processing of the precursor molecules p105 and p100, respectively, lack the TAD but have ankyrin (ANK) repeats, a characteristic of IκB proteins, the glycine-rich region and death domain (DD). The leucine zipper motif is present only in RelB. The complex structures of target promoters in conjunction with the different combination of NF-κB dimers, coactivators and corepressors regulate and initiate a variety of protein–protein interactions at the promoter, which makes NF-κB-mediated transcriptional control a key regulatory player.
Figure 1.

Diagrammatic representation of the functional domains of NF-κB family members. All members of the NF-κB proteins contain the Rel homology domain (RHD). RelA, RelB and c-Rel contain a transactivation domain (TAD) and Rel B is the only member with the Leucine zipper motif. Other structural features include the glycine-rich region (GRR), Ankyrin repeats (AR) and death domain (DD) that are only seen in the p52/p100 and p50/p105 members of the family. Modified from Oeckinghaus et al. (5).
Regulation of NF-κB signaling
NF-κB plays an important role in innate and adaptive immune responses and can be activated by bacterial and viral infections, inflammatory cytokines, UV- or γ-irradiation, ischemia, hyperosmotic shock and oxidative stresses (6). NF-κB activation generally occurs through either the classical or alternative pathways (7). In the classical pathway, stimulation by pro-inflammatory cytokines activates the inhibitor of nuclear factor kappa-B kinase (IKK) complex resulting in the phosphorylation of IκB proteins on two N-terminal serine residues leading to IκB ubiquitin-mediated degradation. In the alternative pathway, IKKα is phosphorylated by NF-κB inducing kinase, which phosphorylates p100 leading to polyubiquitination and degradation of the inhibitory molecules by the proteasome (8). The freed NF-κB dimers are translocated into the nucleus, where they transactivate target genes by binding to gene promoter/enhancer regions. In addition to the classical and alternative pathways, atypical pathways for activation of NF-κB also exist. While one of the atypical pathways involves UV-dependent activation of NF-κB by casein kinase 2 and calpain-mediated phosphorylation and degradation of IκB, the other occurs under genotoxic stress and involves the activation of the IKK complex though ATM kinase and the subsequent ubiquitination of nuclear factor kappa-B essential modulator (NEMO) that ultimately leads to activation of NF-κB (Figure 2) (9–12).
Figure 2.
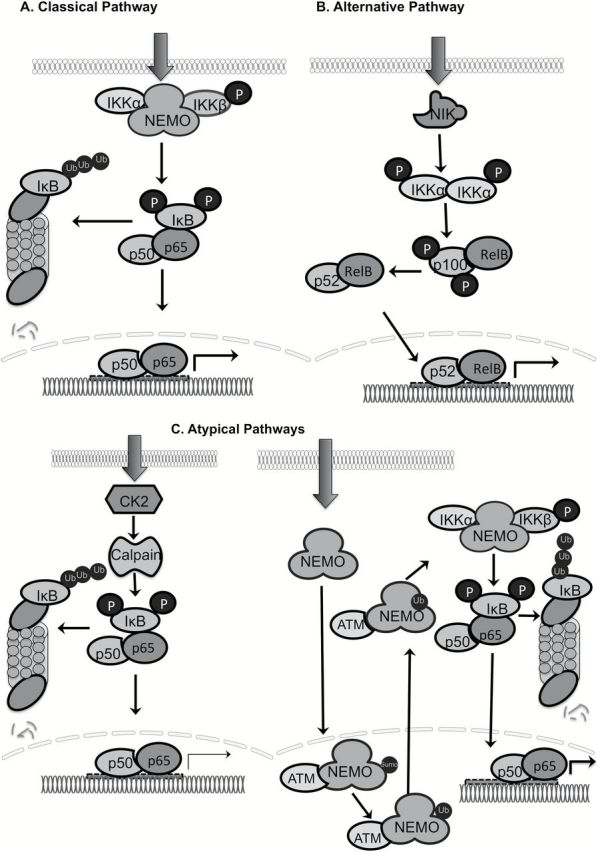
Different pathways of NF-κB activation. Classical NF-κB pathway is activated through phosphorylation of IκB by IKKβ. Proteasomal degradation of IκB after ubiquitination allows translocation of the p65/p50 dimer to the nucleus and transactivation of target genes. The alternative pathway utilizes NF-κB-inducing kinase (NIK) to activate IKKα, which induces proteolytic processing of p100 to p52 followed by translocation of RelB/p52 to the nucleus and transactivation of target genes. The atypical pathways include IκB degradation either through casein kinase 2/calpain- or through ATM and subsequent ubiquitination of NEMO to activate NF-κB-mediated transcription. Adapted from Hoesel and Schmid (11) and Godwin et al. (12).
There are several mechanisms by which activated NF-κB can be downregulated. The most well studied mechanism is the feedback loop where IκB binds to nuclear NF-κB and facilitates its cytosolic export. The IκB family members including IκBα, IκBβ, BCL-3, IκBε, IκBλ and the NF-κB precursor proteins, p100 and p105, share the seven ankyrin repeats that facilitates the binding of these proteins to the dimerization domain of NF-κB dimers (13). Crystallographic studies show that IκB proteins mask only the nuclear localization sequence (NLS) of p65 but not that of p50 (14). Nevertheless, constant shuttling of IκB/NF-κB between the nucleus and cytoplasm occurs due to the NLS of p50 and nuclear export sequences present on IκB and p65 (15). As a result, the nuclear localization of NF-κB is brought about by IκB degradation and unmasking of the NLS of p65 that helps its nuclear localization. The degradation of IκB is a highly regulated process. Phosphorylation of IκBα takes place on serine 32 and 36 by a large protein complex of 700–900kDa consisting of two catalytically active kinases, IKKα and IKKβ, and the regulatory subunit IKKγ (NEMO) (16–19). IKKα and IKKβ have an amino-terminal kinase domain, a leucine zipper and a carboxy terminal domain. The leucine zipper helps in dimerization, whereas the carboxy-terminus contains the hexapeptide sequence (LDWSWL) referred to as the NEMO-binding domain that facilitates their interaction with the regulatory subunit IKKγ (20,21). The IKK complex is activated by phosphorylation of two serines in the SLCTS sequence motif of the T-loop regions (17,22). The mechanism of this phosphorylation is controversial. It is suggested that it may occur either through trans-auto phosphorylation or through phosphorylation by an upstream kinase (23,24). The role of IKKα in the classical NF-κB signaling is not well defined. However, in the alternative pathway IKKα is activated by NF-κB inducing kinase, serving a dual role as an IKKα-activating kinase and a scaffold protein bringing IKKα and p100 together (25).
Post-translational modifications of NF-κB
Posttranslational modifications of NF-κB have been shown to add an additional layer of complexity to the regulation of NF-κB signaling (26) (Figure 3).
Figure 3.
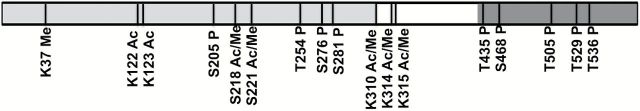
Post-translational modifications of RelA. RelA undergoes multiple post-translation modifications such as phosphorylation, acetylation, methylation and ubiquitination, which regulate its activation, function and degradation. Phosphorylation (P), acetylation (Ac), methylation (Me) at amino acid Serine (S), Threonine (T), Lysine (K) residues. Modified from Huang et al. (26).
Phosphorylation
RelA is phosphorylated in the cytoplasm and the nucleus in response to multiple stimuli. Most phosphorylation sites lie within the N-terminal RHD and the C-terminal TAD and modulate its transcriptional activation. Phosphorylation of p65 at S276 within the RHD by protein kinase A may alter the conformation and affect the binding of RelA with different cofactors and increase its transactivation function (27,28). RelA phosphorylation at S536 occurs within the TAD and is mediated by different kinases including IKKs, ribosomal subunit kinase-1 (RSK1), and TANK binding kinase (TBK1) in response to various stimuli resulting in different biological output (29). RelA phosphorylation can also negatively regulate transcriptional activity of NF-κB. For example, phosphorylation of S468 of RelA within the TAD by three kinases (GSK3β, IKKβ and IKKε) decreases NF-κB activity (30). However, phosphorylation of S468 by IKKε in T cells increases transcription function of NF-κB showing that functional regulation of NF-κB by phosphorylation can be context dependent (31).
Acetylation
RelA can be acetylated at seven lysine residues, including, K122, 123, 218, 221, 310, 314 and 315. Although most acetylation is carried out by p300/CBP, PCAF, has also been found to be involved (32). Specific acetylation is time-dependent with differential functional output. For example acetylation at K221 increases DNA binding of NF-κB, K218 acetylation disrupts its binding with IκBα whereas acetylation at K122 and K123 by p300/CBP and PCAF decreases RelA binding to the κB enhancer (33). Histone deacetylases (HDACs) such as HDAC1, HDAC3 and SIRT1 deacetylate RelA thus regulating NF-κB activity (32,34).
Methylation
Recently regulation of NF-κB activity by methylation has been reported. Nuclear receptor-binding SET domain-containing protein1 (NSD1) monomethylates K218 and dimethylates K221 thereby increasing the transcriptional activity of NF-κB at its target genes (35). Arginine methylation mediated by arginine methyltransferases, such as PRMT1 and PRMT2, have been found to regulate NF-κB-dependent transcription (36). SETD6, a protein lysine methyltransferase, has also been found to monomethylate RelA at Lys310 recruiting GLP (G9A-like protein) to the NF-κB target genes resulting in transcriptional repression (37).
Ubiquitination
The strength and duration of NF-κB is controlled by ubiquitination. After activation, RelA is ubiquitinated and degraded by the proteasome (38). SOCS1 (suppressor of cytokine signaling 1) is an E3 ligase and has been found to ubiquitinate RelA in a LPS-dependent manner (39). The murine herpesvirus-4 protein, ORF73, also ubiquitinates RelA thereby mediating its proteasomal degradation (40). There is evidence of crosstalk among various post-translational modifications in that prior phosphorylation can facilitate acetylation at nearby residues making these modifications critical and complicated regulators of NF-κB signaling.
Conflicting roles of NF-κB in cancer
Protumorigenic role of NF-κB
A major focus of the role of NF-κB in cancer biology has been on its tumor facilitating function and there are many excellent reviews on the subject so we only discuss a few key points regarding its protumorigenic function. The idea that NF-κB family might promote carcinogenesis came from the fact that NF-κB1 displayed sequence homology to v-Rel, an avian reticuloendotheliosis viral oncogene, and the protooncogene c-Rel (41). The hypothesis was further validated when in some B- and T-cell lymphomas p100 was found to undergo genetic rearrangement, leading to truncated and constitutively active forms of the protein (42). Rearrangements, amplifications and missense mutations in the NF-κB subunits have been found in solid and hematopoietic tumors (42). Though genetic alterations in the NF-κB family are not as common as other oncogenes, NF-κB can be deregulated by multiple ways. There are numerous cellular processes that are regulated by NF-κB and deregulation of these genes and pathways are mechanisms by which NF-κB promotes tumorigenesis. For example, aberrant activation and nuclear localization of NF-κB, alterations in proteins involved in pathways regulating NF-κB, constitutive activation of upstream molecules, defects in negative feedback mechanisms and changes in tumor microenvironment can lead to NF-κB-mediated tumorigenesis (43–45).
Tumor suppressive role of NF-κB
The most convincing data that NF-κB is required for p53-mediated apoptosis came from the work of Ryan et al. (46). Induction of p53 increased NF-κB DNA-binding activity and increased apoptosis that is distinct from TNFα-induced NF-κB activation. Resistance to p53-mediated apoptosis but not TNFα-induced apoptosis in RelA-deficient cells challenged the previous dogma that NF-κB only facilitates cell survival. Further evidence for proapoptotic function of NF-κB comes from observations that in the proliferative human basal epithelial layer, NF-κB is cytoplasmic while in the nonproliferative supra basal layer it is localized in the nucleus (47). Transgenic animals carrying dominant-negative IκBα (no NF-κB nuclear DNA binding activity) or overexpressing nuclear NF-κB showed epidermal hyperplasia or hypoplasia, respectively. Human keratinocytes overexpressing either nuclear p50 or IκBα grafted on SCID mice, respectively, show atrophic epidermis or hyperplastic epithelium that penetrates deep into the dermis (47). These lines of evidence show that in the epidermal tissue NF-κB activity results in growth inhibition, whereas loss of NF-κB increases proliferation. A recent study by Jacque et al. (48) showed that RelB also decreases proliferation, and xenograft tumors through a p53-dependent manner. A RelB knockdown mouse fibroblast cell line significantly promoted tumor growth most probably due to lack of p53 transcriptional activation in the absence of RelB. Although overexpression of RelB increased p53 levels and knocking it down decreased p53 levels no direct interaction was observed between the two proteins. In vitro studies with p53−/− and p53wt MEFs show that RelB overexpression does not inhibit proliferation without the presence of p53. Further, p53-mediated Doxorubicin response was reduced in RelB knockdown cells. This observation in conjunction with the work of Ryan and colleagues, suggests that both RelA and RelB may work together possibly forming a heterodimer to induce p53 activation and apoptosis. These published data show that RelA/p65 subunit of NF-κB and the p53 tumor suppressor can be interdependent as cells respond to death-inducing stimuli. Taken together, it is evident that NF-κB can potentially act as a tumor suppressor through a variety of mechanisms, which may be context-dependent (Table 1) (49).
Table 1.
Mechanisms of tumor promotion and tumor suppression by NF-κB. Modified from Perkins et al. (49)
| Mechanisms of tumor promotion by NF-κβ | Mechanisms of tumor suppression by NF-κβ |
|---|---|
| Antiapoptotic (induction of antiapoptotic genes such as Bcl-xL, XIAP, cIAP1 and 2) | Proapoptotic (induction of pro-apoptotic genes such as FAS, DR4 and 5) |
| Metastasis (regulation of genes involved in cell adhesion and migration such as ICAM-1. VCAM-1, ELAM-1, MMP-9, uPA) | Tumor suppressor (aids in p53 induced apoptosis) |
| Angiogenesis (regulation of VEGF, IL1, IL8, TNF) | Antiproliferative role (JNK inhibition, induction of p21WAF1/CIP1 expression) |
| Proliferation (induction of oncogenes such as cyclin D1, c-Myc) | Suppression of transformation |
| Immortalization (induction of telomerase) | |
| Inflammation (regulation of inflammatory gene expression such as cyclooxygenase 2 and iNOS) |
NF-κB in urothelial tumorigenesis
Histologically, bladder cancer is classified either as urothelial (~95%) or non-urothelial (~5%) (50). In this review we focus on urothelial carcinoma. Multifocal development of urothelial cancer in the urinary tract can occur both synchronously (two or more in number, detected within 6 months after the first resection) and metachronously (detected ~6 months post removal of the primary tumor, and located in a different area), which leads to recurrence after the initial resection of tumor.
Knowledge of molecular changes including NF-κB associated with these disease stages is still rudimentary. Immunohistochemical analysis of tumor samples from 140 patients (94 males and 46 females) showed a strong correlation between cyclooxygenase 2 and nuclear NF-κB immunoreactivity (51,52). A novel T to G polymorphism at −1186 of the NF-κB binding site of the cyclooxygenase 2 promoter was shown to be significantly associated with an increased risk of urothelial carcinoma development (53). Notably, polymorphisms located in the human NF-κB promoter, located on chromosome 4q24, have also been reported in the bladder (54). Specifically, an insertion/deletion ATTG functional polymorphism at -94 has been shown to be involved in carcinoma risk and recurrence. Three insertion/deletion allelotypes (ins/ins, ins/del, or del/del) have differential regulatory effects of the NF-κB (p50) gene and the susceptibility of individuals to malignancy (55,56). The risk of recurrence is higher in patients with homozygous deletions compared with homozygous insertions in the promoter. There is also increased risk for non-muscle invasive, grade I, single and small tumor carcinoma in patients with homozygous deletions compared with ins/ins and ins/del. Among urothelial cancer patients, individuals with homozygous insertions have twice as much NF-κB promoter activity compared with single allelotypes. Although the −94 polymorphisms are associated with age, male gender, family history of cancer and smoking habitus, there is no association with tumor grade and stage. Additionally, quantitative data of immunohistochemical levels of p65/RelA expression in paraffin-embedded tissue from 116 patients shows that the majority of patients display concurrent cytoplasmic and nuclear expression, with only three patients being negative for p65/RelA (57). Univariate and multivariate analysis of superficial and muscle invasive carcinomas show nuclear p65/RelA expression increases while cytosolic expression decreases with increasing tumor grade and T-category. The nuclear expression of p65/RelA negatively affects survival in both cases. This suggests that in high-grade bladder cancer, p65 may have an oncogenic role helping in progression and invasion. Expression was also determined using in situ hybridization of twenty patient samples, and these results indicate that NF-κB plays a role in the pathogenesis of transitional cell carcinoma (58). Examination of the TCGA database shows that loss of NF-κB expression is associated with bladder cancer (59). Oncomine data from Sanchez-Carbayo et al. show that there may be a difference between superficial and infiltrating bladder cancers with NF-κB increasing in superficial bladder cancer and decreasing in infiltrating bladder cancer (60,61).
Aberrant signaling and transcription networks through activation of NF-κB ultimately promote epithelial-to-mesenchymal transition, migration, invasion and stem cell characteristics. Urothelial cancer is more often associated with environmental exposure versus genetic origin. Exposure to noxious stimuli from the environment has the capacity to disrupt signaling and transcription networks and induce an inflammatory response. Interleukins have a profound effect on bladder cancer progression. Many of the interleukins including IL-28A, IL-20, IL-8 and IL-5 have been reported to be highly expressed in muscle invasive bladder cancer, enhance migration of bladder cancer cells through NF-κB-mediated activation of various signaling pathways. In particular, IL-8 mediates angiogenesis, tumorigenicity and metastasis through the NF-κB-mediated transcription upregulation. Targeting IL-8 in an orthotopic model of bladder cancer significantly inhibits tumor growth by decreasing matrix metalloproteinase (MMP)-2 and MMP-9 due to decreased NF-κB transcriptional activity (62). Work by Karashima et al. (63) show that NF-κB increases under hypoxic and acidic conditions in multiple urothelial carcinoma cell lines. There is also significant increase in the secretion of IL-8, a known target of NF-κB. Furthermore, they report that mutant IκBα inhibits NF-κB from shuttling into the nucleus, and decreases NF-κB DNA binding activity (63). In addition, transient nicotine exposure, which is a key component of cigarette smoke and a major bladder cancer risk factor has been shown to stimulate the activation of NF-κB DNA binding activity affecting cyclin D1 expression and cell proliferation in bladder cancer cells (64).
Future directions
Increased inflammation and tumor infiltration with activated inflammatory cells that serve as sources of chemokines/cytokines to perpetuate inflammation and immune dysfunction have been described in bladder cancer (65). Although the role of NF-κB in the inflammatory response and its effects on increased expression of downstream targets, such as IL-8, are key to understanding the functional roles of NF-κB, the observed decrease of NF-κB in advanced disease on one hand and increase in IL-8 on the other appears to be counterintuitive and needs further exploration. On the basis of the pro- and antisurvival functions of NF-κB we propose that NF-κB plays differential roles in the two phenotypic pathways implicated in bladder cancer (59) (Figure 4). Although conflicting data exist on NF-κB expression in bladder cancer, a large number of studies show decreased expression and/or copy number in bladder cancer (66–69) (and Figure 5). Analysis of 131 urothelial carcinomas in the TCGA database shows loss of NF-κB in infiltrating bladder cancer compared with normal bladder urothelium, suggesting that NF-κB may play a tumor suppressive role in this form of invasive bladder cancer. As discussed in this review, NF-κB levels increase in superficial bladder cancer and decrease in infiltrating bladder cancer (compared with normal urothelium) suggesting that its oncogenic or tumor-suppressive role in bladder cancer cannot be argued without the context of stage of disease. In this regard, analyses of expression from multiple data sets point to an oncogenic role in progression of early disease yet suppressive role in invasive disease.
Figure 4.
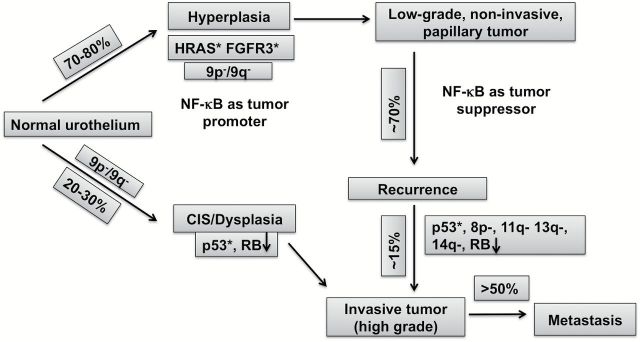
Hypothetical model of NF-κB in bladder tumorigenesis. Urothelial tumors arise via two divergent phenotypic pathways. Some transform from urothelial hyperplasia to low-grade noninvasive superficial papillary tumors whereas the more aggressive form evolves from flat, high-grade carcinoma in situ to highly invasive tumors. On the basis of the pro- and antisurvival functions of NF-κB, differential roles have been attributed to NF-κB in the two phenotypic pathways implicated in bladder cancer. Modified from Netto et al. (59).
Figure 5.
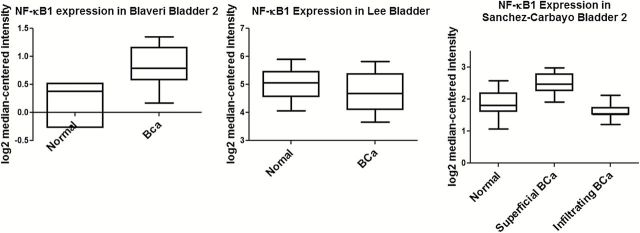
Differential expression of NF-κB in bladder cancer. Oncomine datasets show that NF-κB expression is variable between normal and bladder cancer samples (panels A and B) (55,66). Comparison between superficial and infiltrating bladder cancer samples show decreased NF-κB expression in the latter suggesting an association with disease stage (57).
About 50–70% of invasive bladder cancers have p53 mutations and there is evidence of crosstalk between p53 and NF-κB during hypoxic, genotoxic, metabolic and oxidative stress in other tumor models. Recently, we reported that inhibition of cellular antioxidant defense systems induced p53 degradation and led to induction of NF-κB due to increased access to transcription cofactor, CBP/p300. This led to increased pro-inflammatory gene expression and promoted survival of prostate cancer cells (70). It remains to be seen whether p53 and NF-κB exhibit this competition for transcription cofactors and how mutant p53 fits into this puzzle in the urothelial context. Research efforts should also be directed in identifying if and when during papillary bladder tumor recurrence there is a switch in NF-κB from a protumorigenic to an antitumorigenic function. Another area that begs investigation in bladder cancer development is post-translational modifications that are critical in regulating NF-κB activity.
The first line of therapy for noninvasive bladder cancer is transurethral resection of bladder tumor. Following transurethral resection of bladder tumor different forms of adjuvant therapy are used to prevent recurrence and progression of bladder cancer. The most frequently used therapies are administration of Bacillus Calmette Guerin, intravesical instillation of chemotherapeutic drugs such as Mitomycin C, Doxorubicin and hyperthermia (71). Despite these strategies recurrence is as high as 70% with about 15% of recurrent cancers progressing to muscle invasive disease. The mechanism through which BCG, functions is still not clearly understood. Whether BCG affects gene expression changes other than the well-known proinflammatory network remains unresolved. Interestingly, increased expression of NF-κB1 is associated with BCG treatment, reflecting a role for NF-κB in BCG-mediated immune response (72). BCG-treated patients with NF-κB del/del genotype were reported to have a 2.5-fold increased risk of recurrence compared with ins/ins genotype (73). The association between NF-κB genotype and BCG treatment response underscores the importance of personalized medicine in bladder cancer therapy. Our model has implications for the use of NF-κB as a therapeutic target in bladder cancer. According to our model it would not suffice to only know whether NF-κB is present or absent in tumors. Rather it would be critical to understand whether NF-κB functions as a growth promoter or inhibitor in specific tumors before embarking on a treatment strategy that affects the various signaling networks influenced by NF-κB.
Funding
R01CA149516 (R.G.), NIHT32 CA136058 (E.C.) and Greehey Fellowship (T.J.H.).
Acknowledgements
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- IKK
inhibitor of nuclear factor kappa-B kinase
- NF-κB
nuclear factor-kappaB
- RHD
Rel homology domain
- TAD
transactivation domains.
References
- 1. Sen R., et al. (1986). Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell, 47, 921–928. [DOI] [PubMed] [Google Scholar]
- 2. Perkins N. (2012). The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer, 12, 121–132. [DOI] [PubMed] [Google Scholar]
- 3. Ghosh G., et al. (1995). Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature, 373, 303–310. [DOI] [PubMed] [Google Scholar]
- 4. Baldwin A. (1996). The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol., 14, 649–683. [DOI] [PubMed] [Google Scholar]
- 5. Oeckinghaus A., et al. (2009). The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol., 1, a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baeuerle P., et al. (1994). Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol., 12, 141–179. [DOI] [PubMed] [Google Scholar]
- 7. Bonizzi G., et al. (2004). The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol., 25, 280–288. [DOI] [PubMed] [Google Scholar]
- 8. Karin M., et al. (2004). The IKK NF-kappa B system: a treasure trove for drug development. Nat. Rev. Drug Discov., 3, 17–26. [DOI] [PubMed] [Google Scholar]
- 9. Kato T., et al. (2003). CK2 Is a C-Terminal IkappaB kinase responsible for NF-kappaB activation during the UV response. Mol. Cell, 12, 829–839. [DOI] [PubMed] [Google Scholar]
- 10. Huang T.T., et al. (2003). Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell, 115, 565–576. [DOI] [PubMed] [Google Scholar]
- 11. Hoesel B., et al. (2013). The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer, 12, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Godwin P., et al. (2013). Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol., 3, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hatada E., et al. (1992). The ankyrin repeat domains of the NF-kappa B precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-kappa B DNA binding. Proc. Natl. Acad, Sci. USA, 89, 2489–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huxford T., et al. (1998). The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell, 95, 759–770. [DOI] [PubMed] [Google Scholar]
- 15. Johnson C., et al. (1999). An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J., 18, 6682–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Z., et al. (1996). Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell, 84, 853–862. [DOI] [PubMed] [Google Scholar]
- 17. Mercurio F., et al. (1997). IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science (New York, N.Y.), 278, 860–866. [DOI] [PubMed] [Google Scholar]
- 18. Ozes O., et al. (1999). NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature, 401, 82–85. [DOI] [PubMed] [Google Scholar]
- 19. Zandi E., et al. (1997). The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell, 91, 243–252. [DOI] [PubMed] [Google Scholar]
- 20. May M., et al. (2002). Characterization of the Ikappa B-kinase NEMO binding domain. J. Biol. Chem., 277, 45992–46000. [DOI] [PubMed] [Google Scholar]
- 21. May M., et al. (2000). Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science (New York, N.Y.), 289, 1550–1554. [DOI] [PubMed] [Google Scholar]
- 22. Ling L., et al. (1998). NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. USA, 95, 3792–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scheidereit C. (2006). IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene, 25, 6685–6705. [DOI] [PubMed] [Google Scholar]
- 24. Hayden M.S., et al. (2004). Signaling to NF-kappaB. Genes Dev., 18, 2195–224. [DOI] [PubMed] [Google Scholar]
- 25. Xiao G., et al. (2004). Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J. Biol. Chem., 279, 30099–30105. [DOI] [PubMed] [Google Scholar]
- 26. Huang B., et al. (2010). Posttranslational modifications of NF-kappaB: another layer of regulation for NF-kappaB signaling pathway. Cell. Signal., 22, 1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhong H., et al. (1997). The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell, 89, 413–424. [DOI] [PubMed] [Google Scholar]
- 28. Jamaluddin M., et al. (2007). TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell. Signal., 19, 1419–1433. [DOI] [PubMed] [Google Scholar]
- 29. Sakurai H., et al. (1999). IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem., 274, 30353–30356. [DOI] [PubMed] [Google Scholar]
- 30. Buss H., et al. (2004). Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J. Biol. Chem., 279, 49571–49574. [DOI] [PubMed] [Google Scholar]
- 31. Mattioli I., et al. (2006). Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J. Biol. Chem., 281, 6175–6183. [DOI] [PubMed] [Google Scholar]
- 32. Kiernan R., et al. (2003). Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem., 278, 2758–2766. [DOI] [PubMed] [Google Scholar]
- 33. Chen L.-f., et al. (2002). Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J., 21, 6539–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yeung F., et al. (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J., 23, 2369–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu T., et al. (2010). Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. USA, 107, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ganesh L., et al. ( 2006. ) Protein methyltransferase 2 inhibits NF-kappaB function and promotes apoptosis. Mol. Cell. Biol., 26, 3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levy D., et al. (2011). Lysine methylation of the NF-κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat. Immunol., 12, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Natoli G., et al. (2008). Nuclear ubiquitin ligases, NF-kappaB degradation, and the control of inflammation. Sci. Signal., 1, pe1. [DOI] [PubMed] [Google Scholar]
- 39. Ryo A., et al. (2003). Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell, 12, 1413–1426. [DOI] [PubMed] [Google Scholar]
- 40. Rodrigues L., et al. (2009). Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J., 28, 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gilmore T.D. (1999). Multiple mutations contribute to the oncogenicity of the retroviral oncoprotein v-Rel. Oncogene, 18, 6925–6937. [DOI] [PubMed] [Google Scholar]
- 42. Rayet B., et al. (1999). Aberrant rel/nfkb genes and activity in human cancer. Oncogene, 18, 6938–6947. [DOI] [PubMed] [Google Scholar]
- 43. Garg A., et al. (2002). Nuclear transcription factor-kappaB as a target for cancer drug development. Leukemia, 16, 1053–1068. [DOI] [PubMed] [Google Scholar]
- 44. Baldwin A. (2001). Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J. Clin. Invest., 107, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karin M., et al. (2002). NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer, 2, 301–310. [DOI] [PubMed] [Google Scholar]
- 46. Ryan K.M., et al. (2000). Role of NF-kappaB in p53-mediated programmed cell death. Nature, 404, 892–897. [DOI] [PubMed] [Google Scholar]
- 47. Seitz C.S., et al. (1998). Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc. Natl. Acad. Sci. USA, 95, 2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jacque E., et al. (2013). RelB inhibits cell proliferation and tumor growth through p53 transcriptional activation. Oncogene, 32, 2661–2669. [DOI] [PubMed] [Google Scholar]
- 49. Perkins N., et al. (2006). Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ., 13, 759–772. [DOI] [PubMed] [Google Scholar]
- 50. Dahm P., et al. (2003). Malignant non-urothelial neoplasms of the urinary bladder: a review. Eur. Urol., 44, 672–681. [DOI] [PubMed] [Google Scholar]
- 51. Kontos S., et al. (2010). Inverse expression of estrogen receptor-beta and nuclear factor-kappaB in urinary bladder carcinogenesis. Int. J. Urol. 17, 801–809. [DOI] [PubMed] [Google Scholar]
- 52. Kontos S., et al. (2010). Coordinated increased expression of Cyclooxygenase2 and nuclear factor κB is a steady feature of urinary bladder carcinogenesis. Adv. Urol., 2010. 10.1155/2010/871356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kang S., et al. (2005). Polymorphism in the nuclear factor kappa-B binding promoter region of cyclooxygenase-2 is associated with an increased risk of bladder cancer. Cancer Lett., 217, 11–16. [DOI] [PubMed] [Google Scholar]
- 54. Riemann K., et al. (2007). Insertion/deletion polymorphism in the promoter of NFKB1 as a potential molecular marker for the risk of recurrence in superficial bladder cancer. Int. J. Clin. Pharmacol.Ther., 45, 423–430. [DOI] [PubMed] [Google Scholar]
- 55. Tang T., et al. (2010). Insertion/deletion polymorphism in the promoter region of NFKB1 gene increases susceptibility for superficial bladder cancer in Chinese. DNA Cell Biol., 29, 9–12. [DOI] [PubMed] [Google Scholar]
- 56. Li P., et al. (2013). Functional promoter -94 ins/del ATTG polymorphism in NFKB1 gene is associated with bladder cancer risk in a Chinese population. PloS One, 8, e71604. doi: 10.1371/journal.pone.0071604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Levidou G., et al. (2008). Clinical significance of nuclear factor (NF)-kappaB levels in urothelial carcinoma of the urinary bladder. Virchows Archiv., 452, 295–304. [DOI] [PubMed] [Google Scholar]
- 58. Kadhim H., et al. (2006). Possible role of nuclear factor kappaB detected by in situ hybridization in the pathogenesis of transitional cell carcinoma of the bladder. J. Med. Liban., 54, 196–199. [PubMed] [Google Scholar]
- 59. Netto G. (2013). Clinical applications of recent molecular advances in urologic malignancies: no longer chasing a “mirage”? Adv. Anat. Pathol.,. 20, 175–203. [DOI] [PubMed] [Google Scholar]
- 60. Cancer Genome Atlas Research Network. (2014). Comprehensive molecular characterization of urothelial bladder carcinoma. Nature, 507, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sanchez-Carbayo M., et al. (2003). Gene discovery in bladder cancer progression using cDNA microarrays. Am. J. Pathol., 163, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mian B., et al. (2003). Fully human anti-interleukin 8 antibody inhibits tumor growth in orthotopic bladder cancer xenografts via down-regulation of matrix metalloproteases and nuclear factor-kappaB. Clin. Cancer Res., 9, 3167–3175. [PubMed] [Google Scholar]
- 63. Karashima T., et al. (2003). Nuclear factor-kappaB mediates angiogenesis and metastasis of human bladder cancer through the regulation of interleukin-8. Clin. Cancer Res., 9, 2786–2797. [PubMed] [Google Scholar]
- 64. Chen R.-J., et al. (2008). Rapid activation of Stat3 and ERK1/2 by nicotine modulates cell proliferation in human bladder cancer cells. Toxicol. Sci., 104, 283–293. [DOI] [PubMed] [Google Scholar]
- 65. Zhu Z., et al. (2012). Inflammatory pathways as promising targets to increase chemotherapy response in bladder cancer. Mediators Inflamm., 2012 , 528690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dyrskjøt L., et al. (2009). Genomic profiling of microRNAs in bladder cancer: miR-129 is associated with poor outcome and promotes cell death in vitro . Cancer Res., 69, 4851–4860. [DOI] [PubMed] [Google Scholar]
- 67. Modlich O., et al. (2004). Identifying superficial, muscle-invasive, and metastasizing transitional cell carcinoma of the bladder: use of cDNA array analysis of gene expression profiles. Clin. Cancer Res., 10, 3410–3421. [DOI] [PubMed] [Google Scholar]
- 68. Blaveri E., et al. (2005). Bladder cancer outcome and subtype classification by gene expression. Clin. Cancer Res., 11, 4044–4055. [DOI] [PubMed] [Google Scholar]
- 69. Lee J.-S., et al. (2010). Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol., 28, 2660–2667. [DOI] [PubMed] [Google Scholar]
- 70. Thapa D., et al. (2014). NQO1 suppresses NF-κB-p300 interaction to regulate inflammatory mediators associated with prostate tumorigenesis. Cancer Res., 74, 5644–5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Holmäng S., et al. (1995). The relationship among multiple recurrences, progression and prognosis of patients with stages Ta and T1 transitional cell cancer of the bladder followed for at least 20 years. J. Urol., 153, –1823. [PubMed] [Google Scholar]
- 72. Kamat A.M., et al. (2009). Curcumin potentiates the antitumor effects of Bacillus Calmette-Guerin against bladder cancer through the downregulation of NF-kappaB and upregulation of TRAIL receptors. Cancer Res., 69, 8958–8966. [DOI] [PubMed] [Google Scholar]
- 73. Ahirwar D., et al. (2010). IL-8 -251 T > A polymorphism is associated with bladder cancer susceptibility and outcome after BCG immunotherapy in a northern Indian cohort. Arch. Med. Res., 41, 97–103. [DOI] [PubMed] [Google Scholar]