

Abstract
Background
Activation of fibroblast growth factor receptor (FGFR)-dependent signalling by FGF23 may contribute to the complex pathogenesis of left ventricular hypertrophy (LVH) in chronic kidney disease (CKD). Pan FGFR blockade by PD173074 prevented development of LVH in the 5/6 nephrectomy rat model of CKD, but its ability to treat and reverse established LVH is unknown.
Methods
CKD was induced in rats by 5/6 nephrectomy. Two weeks later, rats began treatment with vehicle (0.9% NaCl) or PD173074, 1 mg/kg once-daily for 3 weeks. Renal function was determined by urine and blood analyses. Left ventricular (LV) structure and function were determined by echocardiography, histopathology, staining for myocardial fibrosis (Sirius-Red) and investigating cardiac gene expression profiles by real-time PCR.
Results
Two weeks after inducing CKD by 5/6 nephrectomy, rats manifested higher (mean ± SEM) systolic blood pressure (208 ± 4 versus 139 ± 3 mmHg; P < 0.01), serum FGF23 levels (1023 ± 225 versus 199 ± 9 pg/mL; P < 0.01) and LV mass (292 ± 9 versus 220 ± 3 mg; P < 0.01) when compared with sham-operated animals. Thereafter, 3 weeks of treatment with PD173074 compared with vehicle did not significantly change blood pressure, kidney function or metabolic parameters, but significantly reduced LV mass (230 ± 14 versus 341 ± 33 mg; P < 0.01), myocardial fibrosis (2.5 ± 0.7 versus 5.4 ± 0.95% staining/field; P < 0.01) and cardiac expression of genes associated with pathological LVH, while significantly increasing ejection fraction (18 versus 2.5% post-treatment increase; P < 0.05).
Conclusions
FGFR blockade improved cardiac structure and function in 5/6 nephrectomy rats with previously established LVH. These data support FGFR activation as a potentially modifiable, blood pressure-independent molecular mechanism of LVH in CKD.
Keywords: CKD, echocardiography, FGF receptor, FGF23, left ventricular hypertrophy
INTRODUCTION
Left ventricular hypertrophy (LVH) is a common cardiovascular complication of chronic kidney disease (CKD) that worsens with declining renal function and increases risks of congestive heart failure and death [1, 2]. The complex pathogenesis of LVH in CKD includes known mechanisms such as ventricular pressure and volume overload, but may also include novel emerging pathways, for example those mediated by fibroblast growth factors (FGF) [3].
FGF23 is one of 22 members of the FGF family of proteins that bind and activate alternatively spliced forms of four tyrosine kinase FGF receptors (FGFR1-4) [4]. Along with parathyroid hormone (PTH) and calcitriol, FGF23 is a central endocrine regulator of phosphate and calcium homeostasis. Through incompletely understood mechanisms, circulating FGF23 levels rise substantially and progressively in patients with advancing CKD. This compensatory increase in FGF23 levels helps maintain normal serum phosphate levels in CKD [5], but at a cost of calcitriol deficiency, secondary hyperparathyroidism and perhaps klotho deficiency [6].
Large epidemiological studies demonstrated that elevated FGF23 levels are strongly associated with higher risks of death and cardiovascular events in multiple CKD and even non-CKD populations [7–10]. As a potential underlying mechanism, epidemiological studies consistently identified independent associations between elevated FGF23 levels and greater left ventricular mass and higher prevalence and incidence of LVH [11, 12]. In support of the epidemiological data, we found that FGF23 can cause hypertrophic growth of individual cardiac myocytes in vitro and can induce LVH in mice via FGFR-dependent activation of the calcineurin–nuclear factor of activated T cells (NFAT) signalling cascade, which is known to mediate pathological cardiac hypertrophy in response to other pathogenic factors [3, 13].
These findings suggest that FGF23 excess may be a novel and potentially targetable mechanism that contributes to the pathogenesis of LVH in CKD. In a proof-of-concept experiment, we demonstrated that administering the pan-FGFR blocker, PD173074 [14], beginning at the time CKD was induced prevented development of LVH in the classic 5/6 nephrectomy rat model of CKD, independently of blood pressure and kidney function [3]. The purpose of this study was to extend these results from a prevention model to a treatment model. We tested the hypothesis that beginning treatment with PD173074 after LVH is already established would reverse or attenuate LVH in 5/6 nephrectomy rats.
MATERIALS AND METHODS
5/6 Nephrectomy model of CKD
Experiments were approved by a governmental committee on animal welfare ‘Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen’ and performed in accordance with national animal protection guidelines. We induced CKD in male Sprague Dawley rats by 5/6 nephrectomy, as described previously [13, 15]. In short, we removed the right kidney and selectively ligated two to three branches of the left renal artery through a mid-line incision. Sham operation consisted of decapsulation of the right kidney. Isoflurane inhalation was used for induction and maintenance of anaesthesia (4 and 1.5–2.5% isoflurane/oxygen mixture, respectively; Abbott GmbH & Co. KG, Wiesbaden, Germany). The analgesic buprenorphine (0.05 mg/kg) was administered for 3 days following the surgical procedure. We randomized rats into three groups: sham nephrectomy plus vehicle (NaCl 0.9%, n = 8), 5/6 nephrectomy plus vehicle (n = 8) and 5/6 nephrectomy plus PD1730714 (1 mg/kg once-daily; n = 7; R&D Systems, Minneapolis, MN, USA). Vehicle and PD173074 were applied intra-peritoneally, and the treatment lasted for 3 weeks.
Rats were kept in pairs at constant temperature (20°C) and humidity (25%) over the experimentation time. From the beginning of the experiment, they were fed a standard maintenance rat chow diet containing 0.7% phosphorus (diet #1320 Altromin, Lage, Germany). Rats were given ad libitum access to food and water. At the determined time points (just prior to starting treatment and 3 weeks after treatment), they were individually housed in metabolic cages for 24 h. Urine and blood samples were collected for further analysis.
Blood pressure in conscious rats was measured using a computerized rat-tail cuff technique (CODA, Kent Scientific Corporation, Torrington, CT, USA) averaging over 25 cycles of inflation/deflation of the cuff to obtain the mean systolic blood pressure. The rats were acclimatized to the system and pre-trained through 1 week before surgery. Blood pressure was measured every 2–3 days and mean results for each animal from the last three measurements were reported.
Serology
We analysed 24-h urine and blood samples for creatinine (enzymatic assay; Creatinine-Pap, Roche Diagnostics, Mannheim, Germany) and urea nitrogen (urease-GLDH method) [13]. Creatinine clearance (mL/min/100 g) was calculated using the following formula: {[(urine creatinine in mg/dL × urine volume in mL)/(serum creatinine in mg/dL × time in min)]/(body weight in grams/100)}. Time corresponds to the time in metabolic cage. In addition, urinary protein concentrations were determined with the Bradford protein assay following the manufacturer's instructions (Coomassie® Brilliant Blue G 250, SERVA Electrophoresis GmbH, Mannheim, Germany).
Plasma inorganic phosphate concentration was measured photometrically on a Modular P analyser (Roche, Mannheim, Germany) using the phosphomolybdate method. Circulating FGF23 was quantified in serum using a mouse/rat C-terminal ELISA Kit (Immutopics, San Clemente, CA, USA). PTH and 1,25-dihydroxyvitamin D [1,25(OH)2D] serum levels were measured using ELISA kits for rat bioactive intact PTH (Immutopics) and DHVD3 (USCN Life Science, Inc., Houston, TX, USA), respectively.
Assessment of left ventricular hypertrophy
We analysed left ventricular (LV) mass in vivo by high-resolution echocardiography at 2 weeks after surgery (baseline; i.e. just before initiation of treatment) and 3 weeks after treatment (end point) using an HDI 5000 Ultrasound system (Philips Medical Systems, Bothell, WA, USA) equipped with a 15-MHz transducer operating in harmonic imaging mode [13, 16]. Rats were maintained under inhalation anaesthesia with a 1.0–1.2% isoflurane/oxygen mixture (Abbott GmbH & Co. KG) minimizing cardiodepressive effects. We acquired and analysed at least three separate M-mode recordings to assess wall and chamber dimensions, LV mass and ejection fraction [16–18]. Each measurement was repeated at least three times, and mean values were analysed. LV mass was normalized to body weight [(LV mass/body weight) × 100]. After 3 weeks of treatment, we sacrificed the animals, and excised and weighted the hearts. We quantified LVH by measuring the ratio of heart weight to tibial length.
Assessment of cardiomyocyte hypertrophy, interstitial fibrosis and histology
At the end of the experiment, the animals were sacrificed and the hearts were excised, immersed in 4% ‘paraformaldehyde’ solution, dehydrated in a graded series of increasing alcohol concentrations, cleared in xylene and routinely embedded in paraffin. Five-micrometre-thick sections were cut. Sections were then deparaffinized, rehydrated and submitted to stainings and histological analyses.
The extent of cardiac myocyte hypertrophy, as measured by cross-sectional area, was determined on haematoxylin–eosin-stained sections. Multiple × 100 magnified pictures were captured from the left ventricle using a Carl Zeiss microscope and the AxionVisonLE Release 4.7.1 software (Carl Zeiss AG, Oberkochen, Germany). Myocytes (at least 20/heart) that showed round capillaries and clear membrane staining were included in the analysis. The image processing software ImageJ was used to calculate cross-sectional area.
To assess interstitial fibrosis, the nuclei were stained with Weigert's haematoxylin (Merck, Darmstadt, Germany) followed by staining in Sirius-Red for 1 h (Waldeck GmbH, Münster, Germany). Digitalized pictures were taken using a Carl Zeiss microscope and the AxionVisonLE Release 4.7.1 software (Carl Zeiss AG). The fibrosis area was calculated using the image processing software ImageJ (developed by NIH) according to ImageJ Tutorial. The percentage of area within the threshold in the whole image was measured as the fibrosis area [13]. We analysed kidney and heart histology by H&E (Roth, Karslruhe, Germany) and von Kossa stainings (Merck) according to manufacturers' instructions.
Gene expression analysis
We analysed gene expression by real-time PCR using the SYBR Green PCR Master Mix with the ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Darmstadt, Germany) [13]. All measurements were performed with support of the Integrated Functional Genomics facilities of the IZKF, University of Münster, Germany. Relative gene expression values were evaluated with the method using gapdh as housekeeping gene. Primer sequences are: GAPDH reverse 5′-actccacgacatactcagcca-3′ and forward 5′-catcaacgaccccttcatt-3′; α-myosin heavy chain (αMHC) reverse 5′-gagcaggagctgatcgagac-3′ and forward 5′-cctctgcgttcctacactcc-3′; β-myosin heavy chain (βMHC) reverse 5′-tgtccaagttccgcaaggt-3′ and forward 5′-caagatctactcttcattcaggcc-3′; atrial natriuretic peptide (ANP) reverse 5′-cggaagctgttgcagccta-3′ and forward 5′-gccctgagcgagcagaccga-3′; BNP reverse 5′-tcgaagtctctcctggatcc-3′ and forward 5′-ccagaacaatccacgatgc-3′. Primers annealing temperature was 60°C.
Statistical analysis
Data are presented as mean ± SEM or median (25th–75th percentiles). Kolmogorov–Smirnov test was used to test normality. Non-normally distributed data were analysed after log-transformation by ANOVA. We compared groups by ANOVA along with post hoc Tukey test. The t-test was used when appropriate. A level of P < 0.05 was considered statistically significant. Analyses were performed using GraphPad Prism version 4.0 for windows.
RESULTS
We treated 5/6 nephrectomy rats with 1 mg/kg of PD173074 or vehicle by once-daily intra-peritoneal injection for 3 weeks beginning 2 weeks after surgery (Figure 1A). We used the same dose of PD173074 in this study as in our previous LVH prevention study [3]. Sham-operated rats served as an additional control group. Renal and metabolic parameters measured 2 weeks after sub-total nephrectomy and prior to administration of the study drug are presented in Table 1. Compared with the sham-operated rats (n = 8), both 5/6 nephrectomy groups (n = 15 total) demonstrated significantly reduced kidney function, and significantly increased blood pressure, proteinuria and serum FGF23 levels. After 3 weeks of treatment with PD173074 or vehicle, there were no significant changes within treatment groups in kidney function, blood pressure, serum phosphate or FGF23 levels (Table 1).
FIGURE 1:

FGFR blockade reverses LVH in an animal model of CKD. (A) Experiment outline. CKD was induced by 5/6 nephrectomy (Nx). After 2 weeks (pre-treatment), LV dimensions and function were determined by echocardiography. Rats were then treated for 3 weeks with vehicle (NaCl 0.9%) or PD173074, 1 mg/kg once-daily. A second echocardiography was repeated at the end of the experiment (3 weeks post-treatment). (B) Echocardiographic measurements of LV mass pre- and post-treatment. (C) At the end of the experiment, LVH was determined as the ratio of heart weight to tibia length. (D) Quantitative analysis of cardiomyocyte cross-sectional area. (E) Expression profile of adult cardiac genes (αMHC), fetal genes (βMHC) and markers of cardiac stress and remodelling (ANP, BNP). Relative gene expression was determined by real-time PCR using gapdh as housekeeping gene. Sham-operated rats were set as control. (F) Representative images of interstitial cardiac fibrosis of 5/6 nephrectomy rats treated with vehicle (left panel) or PD173074 (right panel) for 3 weeks. Staining was performed with Sirius-Red (dark red) (original magnification, ×20; scale bar: 50 µm). The bottom graphic represents quantification of the extent of cardiac fibrosis after 3 weeks of treatment. (G) Echocardiographic measurements of LV ejection fraction pre- and post-treatment. All data are mean ± SEM, n = 8 for Sham–Vehicle, 4 for Sham–PD173074, n = 8 for 5/6 nephrectomy rats receiving vehicle (Nx–Vehicle) and n = 7 for PD173074 (Nx–PD173074). †P < 0.05 for NaCl- and PD173074-treated rats in comparison with Sham–NaCl pre-treatment, §P < 0.05 for PD173074-treated rats in comparison with pre-treatment, &P < 0.05 for NaCl-treated rats in comparison with PD173074 post-treatment, *P < 0.05 in comparison with Sham–NaCl, **P < 0.05 in comparison with Sham–NaCl and Sham–PD173074; and #P < 0.05 in comparison with 5/6 nephrectomy rats receiving NaCl (vehicle).
Table 1.
Renal and metabolic effects of 3 weeks of treatment with PD173074 or vehicle in 5/6 nephrectomy (Nx) rats
| Sham (reference values) | Sham + PD173074 |
5/6 Nx + Vehicle |
5/6 Nx + PD173074 |
ANOVA | ||||
|---|---|---|---|---|---|---|---|---|
| Pre-treatment | Post-treatment | Pre-treatment | Post-treatment | Pre-treatment | Post-treatment | P-value | ||
| (n = 8) | (n = 4) | (n = 4) | (n = 8) | (n = 8) | (n = 7) | (n = 7) | ||
| Body weight (g) | 353 ± 13 | 196 ± 6*,***,****,***** | 335 ± 21 | 311 ± 13 | 378 ± 20 | 292 ± 20** | 356 ± 19 | <0.0001 |
| Weight gain over 3 weeks (g) | 163 ± 10 | 140 ± 15 | 67 ± 11* | 76 ± 27* | 0.0007 | |||
| Serum creatinine (mg/dL) | 0.19 ± 0.02 | 0.18 ± 0.01 | 0.21 ± 0.01 | 0.52 ± 0.04*,**,*** | 0.56 ± 0.04*,**,*** | 0.46 ± 0.06*,**,*** | 0.52 ± 0.05*,**,*** | <0.0001 |
| Blood urea nitrogen (mg/dL) | 17 ± 0.1 | 14 ± 1.7 | 15 ± 0.7 | 37 ± 3.3*,**,*** | 35 ± 1.6*,**,*** | 41 ± 7.4*,**,*** | 33 ± 3.4*,**,*** | <0.0001 |
| Creatinine clearance (mL/min/100 g) | 0.99 ± 0.07 | 0.90 ± 0.09 | 0.94 ± 0.06 | 0.40 ± 0.03*,**,*** | 0.40 ± 0.04*,**,*** | 0.46 ± 0.06*,**,*** | 0.45 ± 0.05*,**,*** | <0.0001 |
| Urine protein (mg/mg creatinine) | 0.3 ± 0.03 | 0.3 ± 0.05 | 0.36 ± 0.02 | 5.8 ± 1.9 | 3.9 ± 1.4 | 5.4 ± 1.5 | 4.2 ± 1.6 | 0.0185 |
| Systolic blood pressure (mmHg) | 139 ± 2.5 | 137 ± 6.3 | 138 ± 4.1 | 213 ± 4.7*,**,*** | 215 ± 6.4*,**,*** | 203 ± 6.2*,**,*** | 203 ± 5.2*,**,*** | <0.0001 |
| Serum phosphate (mg/dL) | 6.9 ± 0.3 | 6.4 ± 0.2 | 7.1 ± 0.6 | 6.1 ± 0.5 | 6.0 ± 0.3 | 5.9 ± 0.6 | 5.7 ± 0.5 | 0.3297 |
| Serum FGF23 (pg/mL) | 194 (179–225) | 190 (171–208) | 292 (230–351) | 947 (676–1159)*,**,*** | 1315 (345–1900)*,**,*** | 548 (402–1111)*,**,*** | 865 (649–1434)*,**,*** | <0.0001 |
| PTH (pg/mL) | 73 ± 18 | nd | nd | 177 ± 26 | 118 ± 40 | 127 ± 29 | 60 ± 39 | 0.0848 |
| 1,25(OH)2D | 143 ± 9 | nd | nd | 144 ± 10 | 125 ± 11 | 144 ± 9 | 131 ± 8 | 0.4932 |
Results are mean ± SEM or median (25–75th percentiles). Reference values: data from sham rats after 3 weeks of treatment with vehicle. Sham animals receiving NaCl pre-treatment: 2 weeks after 5/6 nephrectomy surgery; post-treatment: after 3 weeks of treatment with vehicle or PD173074; weight gain: difference (in g) between weight from pre- to post-treatment; FGF, fibroblast growth factor; PTH, parathyroid hormone; 1,25(OH)2D, 1,25-dihydroxyvitamin D. nd, not determined.
*P < 0.001 in comparison with Sham.
**P < 0.05 in comparison with Sham + PD173074 pre-treatment.
***P < 0.05 in comparison with Sham + PD173074 post-treatment.
****P < 0.05 in comparison with 5/6 Nx + vehicle post-treatment.
*****P < 0.05 in comparison to 5/6 Nx + PD173074 post-treatment.
Compared with the sham-operated rats, 5/6 nephrectomy rats developed a significant increase in LV mass before initiation of therapy, as assessed by echocardiography (Figure 1B). Mean LV mass increased by 28 ± 6% in the seven rats assigned to PD173074 and by 33 ± 6% in the eight rats assigned to vehicle (P < 0.05 for each versus sham). Three weeks of treatment with PD173074 reduced LV mass significantly in 5/6 nephrectomy rats by ∼20%, nearly to the same levels as the healthy sham-operated animals (Figure 1B). In contrast, 5/6 nephrectomy rats treated with vehicle experienced a further 16% increase in LV mass (Figure 1B). After 3 weeks of treatment, neither PD173074 nor vehicle had any significant effects on LV mass or other cardiac parameters presented below in sham-operated rats (Figure 1 and Table 1).
Treatment with PD173074 significantly decreased LV inter-ventricular septal and posterior wall thickness, heart weight-to-tibia length ratio and cardiomyocyte cross-sectional area (Table 2 and Figure 1C and D). Gene expression profile analyses demonstrated reduced expression of atrial natriuretic peptides (ANP) and βMHC, and increased αMHC expression in response to PD173074, consistent with reversal of the reactivation of fetal gene programmes associated with pathological LVH (Figure 1E).
Table 2.
Echocardiographic effects of 3 weeks of treatment with PD173074 or vehicle in 5/6 nephrectomy (Nx) rats
| Sham (reference values) | Sham + PD173074 |
5/6 Nx + Vehicle |
5/6 Nx + PD173074 |
ANOVA | ||||
|---|---|---|---|---|---|---|---|---|
| Pre-treatment | Post-treatment | Pre-treatment | Post-treatment | Pre-treatment | Post-treatment | P-value | ||
| (n = 8) | (n = 4) | (n = 4) | (n = 8) | (n = 8) | (n = 7) | (n = 7) | ||
| IVS, d (mm) | 1.3 ± 0.03 | 1.3 ± 0.04 | 1.4 ± 0.06 | 1.5 ± 0.05 | 1.8 ± 0.09*,**,***,****,*****,****** | 1.5 ± 0.09 | 1.4 ± 0.06 | <0.0001 |
| IVS, s (mm) | 2.3 ± 0.03 | 2.3 ± 0.03 | 2.4 ± 0.05 | 2.6 ± 0.08 | 3.0 ± 0.14*,**,***,****,*****,****** | 2.3 ± 0.12 | 2.4 ± 0.10 | <0.0001 |
| LVPW, d (mm) | 1.3 ± 0.03 | 1.3 ± 0.04 | 1.4 ± 0.05 | 1.5 ± 0.06 | 1.8 ± 0.1*,**,***,****,****** | 1.5 ± 0.10 | 1.4 ± 0.06 | 0.0002 |
| LVPW, s (mm) | 2.3 ± 0.05 | 2.3 ± 0.04 | 2.5 ± 0.1 | 2.6 ± 0.08 | 3.0 ± 0.14*,**,***,****,*****,****** | 2.4 ± 0.09 | 2.4 ± 0.10 | <0.0001 |
Results are mean ± SEM. Reference values: data from sham rats after 3 weeks of treatment with vehicle. Pre-treatment: 2 weeks after 5/6 nephrectomy surgery; post-treatment: after 3 weeks of treatment with vehicle or PD173074; IVS: ‘inter-ventricular septal’ wall thickness in ‘diastole’ (d) and in ‘systole’ (s); LVPW: left ventricular posterior wall thickness.
*P < 0.001 in comparison with Sham.
**P < 0.05 in comparison with Sham + PD173074 pre-treatment.
***P < 0.05 in comparison with 5/6 Nx + PD173074 post-treatment.
****P < 0.05 in comparison with 5/6 Nx + NaCl pre-treatment.
*****P < 0.05 in comparison with 5/6 Nx + PD173074 pre-treatment.
******P < 0.05 in comparison with 5/6 Nx + PD173074 post-treatment.
Interstitial myocardial fibrosis is a hallmark of LVH in CKD that likely contributes to its arrhythmogenicity [19, 20]. Compared with vehicle, treatment with PD173074 significantly ameliorated the extent of myocardial fibrosis in 5/6 nephrectomy rats (Figure 1F). Histological sections of heart and kidney from 5/6 nephrectomy rats treated for 3 weeks with PD173074 or vehicle showed no evidence of mineralization by H&E or von Kossa staining (Figure 2).
FIGURE 2:
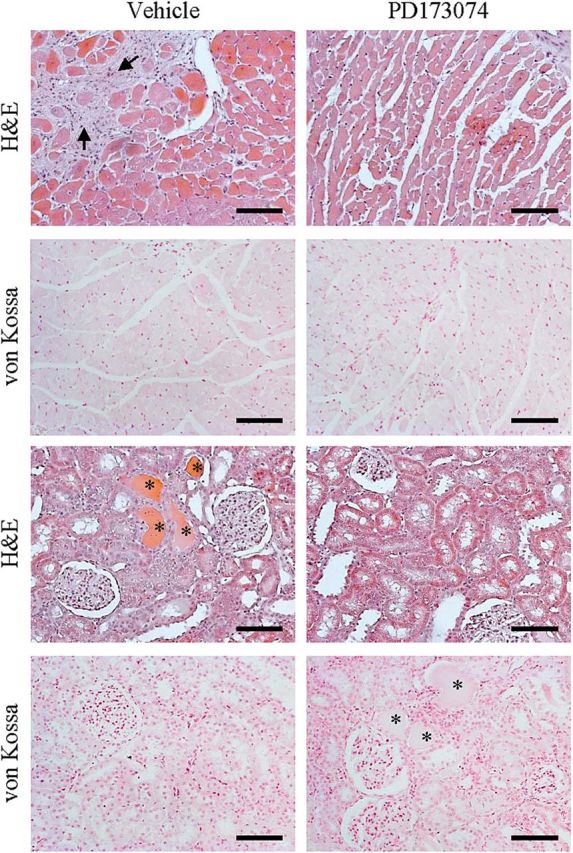
Histological effects of FGFR blockade on the hearts and kidneys of CKD animals. CKD was induced by 5/6 nephrectomy (Nx). Representative histological sections of the myocardium (upper panels) and kidneys (lower panels) from 5/6 nephrectomy rats treated for 3 weeks with vehicle (NaCl 0.9%) or PD173074 (1 mg/kg/day). Tissues show no evidence of mineralization by H&E or von Kossa staining, suggesting no adverse, toxic effects associated with the use of PD173074 at the dose used for up to 3 weeks. Magnification, ×20; scale bar: 100 µm. Interstitial fibrosis (arrows); tubular proteinaceous casts (asterisks).
Ultimately, pathological ventricular remodelling results in progressive cardiac systolic dysfunction and congestive heart failure. Although systolic function remained within the normal range throughout the study period, PD173074 treatment significantly increased ejection fraction by 18% (P < 0.05) compared with the vehicle-treated 5/6 nephrectomy rats which demonstrated no significant change (Figure 1G). Taken together, these findings indicate that FGFR blockade improved cardiac structure and function in 5/6 nephrectomy rats with previously established LVH.
DISCUSSION
The development, severity and persistence of LVH and cardiac fibrosis are strongly associated with mortality and cardiovascular events in patients with CKD and end-stage renal disease. As a potentially modifiable mechanism of disease, London et al. [2] reported that a 10% decrease in left ventricular mass translated into a 28% decrease in cardiovascular mortality during 5 years of follow-up in a cohort of patients undergoing haemodialysis treatment. However, current therapeutic approaches in CKD and end-stage renal disease inadequately control pathological cardiac remodelling as evidenced by increasing prevalence of LVH as CKD progresses and worsening severity of LVH in most patients undergoing conventional haemodialysis [2, 19, 21, 22]. Novel therapeutic targets are needed.
We show here that FGFR blockade can partially reverse components of LVH and cardiac fibrosis in a classic animal model of CKD despite sustained renal impairment and severe hypertension. Systolic function was maintained in the normal range across all studied groups, but improved significantly after treatment with PD173074. This could be attributed to the fact that ejection fraction was markedly lower in CKD rats randomized into this group, or to a reduction in their left ventricular remodelling.
These findings emphasize the importance of additional non-haemodynamic factors in the development and progression of LVH in CKD and suggest a novel pathway that could be targeted to reduce the burden of cardiovascular disease in this high-risk population. Furthermore, we demonstrated cardioprotective effects of FGFR blockade in the setting of no changes in serum phosphate or tissue mineralization (Table 1 and Figure 2, respectively). This suggests the presence of a therapeutic window within which toxic effects of FGFR activation can theoretically be mitigated without precipitating severe hyperphosphatemia or soft tissue calcification that occurs in response to much higher doses of pan-FGFR inhibitors or neutralizing anti-FGF23 antibodies that globally block all FGFR-dependent effects of FGF23 [23–25].
The primary physiological function of FGF23 is to regulate phosphate homeostasis [26]. As renal function declines, FGF23 levels become elevated in the vast majority of patients with CKD, beginning early in the course of disease [5]. High FGF23 levels promote renal phosphate excretion by binding FGFR1-klotho complexes, which activates the mitogen-activated protein kinase (MAPK) signalling cascade leading to down-regulation of sodium-dependent phosphate co-transport in the proximal tubule [27]. In contrast, when klotho is not present, for example in cardiac myocytes, we previously demonstrated that FGF23 is capable of activating FGFRs but instead, stimulates calcineurin-dependent NFAT signalling [3], which is a known mediator of pathological cardiac hypertrophy [28]. In support of these findings, we also previously reported that cyclosporine, which blocks calcineurin, can attenuate LVH in uraemic rats [13]. Furthermore, a recent report validated klotho-independent activation of NFAT signalling by FGF23, this time in the parathyroid glands, which were previously thought to be classical, klotho-dependent targets of FGF23 [29].
Putative cardiac effects of FGF23 are supported by epidemiological findings of strong independent associations between higher FGF23 levels and greater left ventricular mass and higher prevalence and incidence of LVH in a variety of populations [5, 7, 9, 11, 12]. In support of the potential clinical relevance of adverse effects of FGF23 on the heart, high FGF23 levels are also independently predictive of higher risk of congestive heart failure events in CKD and also non-CKD patients [30, 31]. Given the strong relationships between LVH, congestive heart failure and death [7–9, 11, 12, 30, 31], it is intriguing to speculate that activation of FGFRs by FGF23 may contribute causally to adverse cardiovascular outcomes in CKD.
Although we previously demonstrated that interfering with FGFR activation in the heart can prevent LVH and now show that this treatment strategy can ameliorate established LVH in animals, a limitation of these experiments is that neither proves that FGF23 is the primary FGFR ligand underlying LVH in 5/6 nephrectomy rats. However, even though we cannot exclude contributions of other FGFs known to contribute to LVH, such as FGF2 [3, 32, 33], the collective results nonetheless support FGFR activation as a novel and perhaps targetable mechanism of LVH in CKD. Further support for the importance of this pathway is derived from a cardiac-specific, transgenic mouse model engineered to overexpress a constitutively active FGFR mutant [34]. The animals developed a severe cardiac phenotype including LVH and fibrosis that began only a few days after inducing expression of the transgene, but was also partially reversible when overexpression was discontinued, even after 42 days. This suggests potent but treatable effects of FGFR signalling in the heart. Interestingly, the Ras/MAPK pathway was not activated in the myocardium of the transgenic mice, but there was evidence of aberrant calcium signalling suggesting possible calcineurin–NFAT activation, as we observed previously [3, 13].
A second limitation is that we could not pinpoint which one or more of the known FGFR isoforms mediate the pro-hypertrophic effects that we successfully blocked. From a clinical therapeutic and drug development perspective, it would be ideal if the FGFR responsible for the adverse cardiac effects was distinct from FGFR1, which mediates the phosphaturic effects of FGF23 that are critical for maintaining normal serum phosphate levels in CKD [23]. Additional research is needed to catalogue the particular FGFRs and their ligands that mediate specific effects in different end-organs.
Our data add to the growing body of research implicating FGFR activation as an important blood pressure- and volume-independent molecular mechanism in the pathogenesis of LVH in CKD. These data support the need for additional investigation into the role of disordered mineral metabolism in the pathogenesis of LVH in CKD to determine whether FGF23 excess and FGFR activation could be targets for novel therapeutic agents aimed at treating LVH and preventing its morbid consequences.
ACKNOWLEDGEMENTS
The authors thank Katrin Beul and Richard Holtmeier for their excellent technical assistance. MB and this study was supported by Stifterverband für die Deutsche Wissenschaft und der Simon-Claussen-Stiftung (Project H1405409999915626). S.R., D.K. and A.G. were supported by the Deutsche Forschungsgemeinschaft, CRC 656, Project C7.
CONFLICT OF INTEREST STATEMENT
The results presented in this paper have not been previously published, in whole or part.
REFERENCES
- 1.Aurigemma GP, Gottdiener JS, Shemanski L, et al. Predictive value of systolic and diastolic function for incident congestive heart failure in the elderly: the cardiovascular health study. J Am Coll Cardiol. 2001;37:1042–1048. doi: 10.1016/s0735-1097(01)01110-x. [DOI] [PubMed] [Google Scholar]
- 2.London GM, Pannier B, Guerin AP, et al. Alterations of left ventricular hypertrophy in and survival of patients receiving hemodialysis: follow-up of an interventional study. J Am Soc Nephrol. 2001;12:2759–2767. doi: 10.1681/ASN.V12122759. [DOI] [PubMed] [Google Scholar]
- 3.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem. 2011;149:121–130. doi: 10.1093/jb/mvq121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–1378. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutierrez O, Isakova T, Rhee E, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205–2215. doi: 10.1681/ASN.2005010052. [DOI] [PubMed] [Google Scholar]
- 7.Gutierrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305:2432–2439. doi: 10.1001/jama.2011.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jean G, Terrat JC, Vanel T, et al. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol Dial Transplant. 2009;24:2792–2796. doi: 10.1093/ndt/gfp191. [DOI] [PubMed] [Google Scholar]
- 10.Parker BD, Schurgers LJ, Brandenburg VM, et al. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Ann Intern Med. 2010;152:640–648. doi: 10.1059/0003-4819-152-10-201005180-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez OM, Januzzi JL, Isakova T, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119:2545–2552. doi: 10.1161/CIRCULATIONAHA.108.844506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mirza MA, Larsson A, Melhus H, et al. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis. 2009;207:546–551. doi: 10.1016/j.atherosclerosis.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Di Marco GS, Reuter S, Kentrup D, et al. Cardioprotective effect of calcineurin inhibition in an animal model of renal disease. Eur Heart J. 2011;32:1935–1945. doi: 10.1093/eurheartj/ehq436. [DOI] [PubMed] [Google Scholar]
- 14.Mohammadi M, Froum S, Hamby JM, et al. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 1998;17:5896–5904. doi: 10.1093/emboj/17.20.5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Marco GS, Reuter S, Hillebrand U, et al. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J Am Soc Nephrol. 2009;20:2235–2245. doi: 10.1681/ASN.2009010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghanem A, Troatz C, Elhafi N, et al. Quantitation of myocardial borderzone using reconstructive 3-D echocardiography after chronic infarction in rats: incremental value of low-dose dobutamine. Ultrasound Med Biol. 2008;34:559–566. doi: 10.1016/j.ultrasmedbio.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Ghanem A, Roll W, Hashemi T, et al. Echocardiographic assessment of left ventricular mass in neonatal and adult mice: accuracy of different echocardiographic methods. Echocardiography. 2006;23:900–907. doi: 10.1111/j.1540-8175.2006.00323.x. [DOI] [PubMed] [Google Scholar]
- 18.Stypmann J, Engelen MA, Troatz C, et al. Echocardiographic assessment of global left ventricular function in mice. Lab Anim. 2009;43:127–137. doi: 10.1258/la.2007.06001e. [DOI] [PubMed] [Google Scholar]
- 19.Glassock RJ, Pecoits-Filho R, Barberato SH. Left ventricular mass in chronic kidney disease and ESRD. Clin J Am Soc Nephrol. 2009;4(Suppl 1):S79–S91. doi: 10.2215/CJN.04860709. [DOI] [PubMed] [Google Scholar]
- 20.Ritz E, Wanner C. The challenge of sudden death in dialysis patients. Clin J Am Soc Nephrol. 2008;3:920–929. doi: 10.2215/CJN.04571007. [DOI] [PubMed] [Google Scholar]
- 21.Schmid H, Schiffl H, Lederer SR. Erythropoiesis-stimulating agents, hypertension and left ventricular hypertrophy in the chronic kidney disease patient. Curr Opin Nephrol Hypertens. 2011;20:465–470. doi: 10.1097/MNH.0b013e3283497057. [DOI] [PubMed] [Google Scholar]
- 22.Zoccali C, Benedetto FA, Mallamaci F, et al. Left ventricular mass monitoring in the follow-up of dialysis patients: prognostic value of left ventricular hypertrophy progression. Kidney Int. 2004;65:1492–1498. doi: 10.1111/j.1523-1755.2004.00530.x. [DOI] [PubMed] [Google Scholar]
- 23.Hasegawa H, Nagano N, Urakawa I, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010;78:975–980. doi: 10.1038/ki.2010.313. [DOI] [PubMed] [Google Scholar]
- 24.Yanochko GM, Vitsky A, Heyen JR, et al. Pan-FGFR inhibition leads to blockade of FGF23 signaling, soft tissue mineralization, and cardiovascular dysfunction. Toxicol Sci. 2013;135:451–464. doi: 10.1093/toxsci/kft161. [DOI] [PubMed] [Google Scholar]
- 25.Shalhoub V, Shatzen EM, Ward SC, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest. 2012;122:2543–2553. doi: 10.1172/JCI61405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012;82:737–747. doi: 10.1038/ki.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prie D, Urena TP, Friedlander G. Latest findings in phosphate homeostasis. Kidney Int. 2009;75:882–889. doi: 10.1038/ki.2008.643. [DOI] [PubMed] [Google Scholar]
- 28.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 29.Olauson H, Lindberg K, Amin R, et al. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013;9:e1003975. doi: 10.1371/journal.pgen.1003975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ix JH, Katz R, Kestenbaum BR, et al. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study) J Am Coll Cardiol. 2012;60:200–207. doi: 10.1016/j.jacc.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scialla JJ, Xie H, Rahman M, et al. Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol. 2014;25:349–360. doi: 10.1681/ASN.2013050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kardami E, Jiang ZS, Jimenez SK, et al. Fibroblast growth factor 2 isoforms and cardiac hypertrophy. Cardiovasc Res. 2004;63:458–466. doi: 10.1016/j.cardiores.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 33.House SL, House BE, Glascock B, et al. Fibroblast growth factor 2 mediates isoproterenol-induced cardiac hypertrophy through activation of the extracellular regulated kinase. Mol Cell Pharmacol. 2010;2:143–154. doi: 10.4255/mcpharmacol.10.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cilvik SN, Wang JI, Lavine KJ, et al. Fibroblast growth factor receptor 1 signaling in adult cardiomyocytes increases contractility and results in a hypertrophic cardiomyopathy. PLoS One. 2013;8:e82979. doi: 10.1371/journal.pone.0082979. [DOI] [PMC free article] [PubMed] [Google Scholar]