

Abstract
GM3 synthase (ST3GAL5) is the first biosynthetic enzyme of a- and b-series gangliosides. Patients with GM3 synthase deficiency suffer severe neurological disability and deafness. Eight children (ages 4.1 ± 2.3 years) homozygous for ST3GAL5 c.694C>T had no detectable GM3 (a-series) or GD3 (b-series) in plasma. Their auditory function was characterized by the absence of middle ear muscle reflexes, distortion product otoacoustic emissions and cochlear microphonics, as well as abnormal auditory brainstem responses and cortical auditory-evoked potentials. In St3gal5−/− mice, stereocilia of outer hair cells showed signs of degeneration as early as postnatal Day 3 (P3); thereafter, blebs devoid of actin or tubulin appeared at the region of vestigial kinocilia, suggesting impaired vesicular trafficking. Stereocilia of St3gal5−/− inner hair cells were fused by P17, and protein tyrosine phosphatase receptor Q, normally linked to myosin VI at the tapered base of stereocilia, was maldistributed along the cell membrane. B4galnt1−/− (GM2 synthase-deficient) mice expressing only GM3 and GD3 gangliosides had normal auditory structure and function. Thus, GM3-dependent membrane microdomains might be essential for the proper organization and maintenance of stereocilia in auditory hair cells.
Introduction
The organ of Corti is the auditory sensory organ of the cochlea, comprised of hair cells, supporting cells and the tectorial membrane. Inner hair cells (IHCs) are directly involved in sound transduction, whereas outer hair cells (OHCs) amplify the vibrations. Cochlear fluids in scala tympani cause to drive the deflection of the stereocilia on apical surfaces of IHCs and OHCs. Movement of stereocilia opens mechanotransduction potassium channels located at their tips and triggers hair cell depolarization (1,2).
Sialic acid-containing glycosphingolipids (GSLs), also called gangliosides, are enriched in the central nervous system and vital to normal brain development and function (3–5). Gangliosides localize to the outer leaflet of membranes to form microdomains called lipid rafts, which regulate membrane organization, transmembrane signaling and cell adhesion (6). GM3 synthase (a.k.a lactosylceramide sialyltransferase; ST3GAL5) is the first enzyme mediating biosynthesis of complex a- and b-series gangliosides. The second enzyme, B4GALNT1, transfers GalNAc to LacCer, GM3 or GD3 to form GA2, GM2 or GD2, respectively (Fig. 1A).
Figure 1.
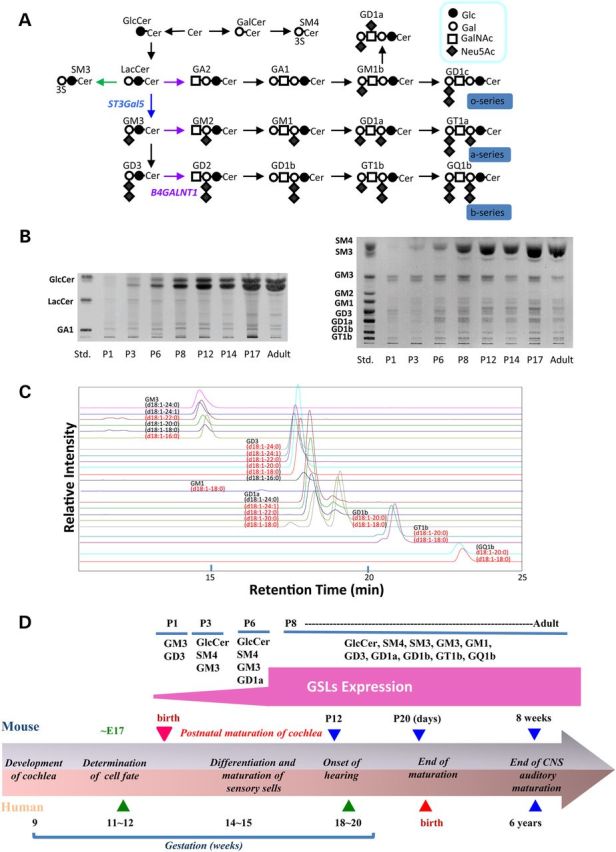
Marked increase of GSLs in cochlea during postnatal maturation period. (A) Schematic presentation of GSL biosynthesis. (B) High performance-thin-layer chromatograms (HPTLC analysis of gangliosides in cochlea of wild-type mice during the postnatal maturation period. Left panel is neutral glycolipids and right panel is acidic glycolipids. Cochlea were isolated and subjected for the purification of GSL analysis as described in Materials and Methods. Each purified fraction was spotted as 2 mg protein per lane. (C) Mass chromatograms of gangliosides prepared from cochlea of the wild mice at P17. The acidic fraction was analyzed by liquid chromatography-ion trap-mass spectrometry with a NH2 column under the conditions described in the Materials and Methods. The MS1 chromatogram was monitored with theoretical values of [M – H]−1 or [M − 2H]−2 for each ganglioside. The structures indicated in the figure is tentative, and based on information with only molecular related ions indicated with black and, in addition, carbohydrate-related fragment ions indicated with red, and requires the consideration of possibility of d20:1 sphingosine. (D) Summary of stage-specific expression of GSLs during postnatal maturation of cochlea based on the information of Figure 1B and C. Comparative development of the cochlea of both humans and mice are also shown.
Various patterns of ganglioside deficiency have been modeled in genetically engineered mice (7). GD3 synthase null (Siat8−/−) mice lack b-series gangliosides and have thermal hyperalgesia and mechanical allodynia (8). B4galnt1−/− mice, which express only two ganglioside species (GM3 and GD3), have reduced neural conduction velocity, ataxia and progressive axonal degeneration (9,10). Mice that express only GM3 (B4galnt1−/−; Siat8−/− double knockout) have audiogenic seizures and sudden death (5,11). Mice lacking all a- and b-series ganglioside species, generated by double knockout of both St3gal5 and B4galnt1, have a severe neurodegenerative disease characterized by reduced brain weight, aberrant axon-glial interactions, axonal degeneration and decreased survival (12).
In humans, homozygous or compound heterozygous loss-of-function mutations of ST3GAL5 cause systemic a- and b-series ganglioside deficiency and are associated with infantile-onset epileptic encephalopathy, slow brain growth, stagnant psychomotor development, growth failure, blindness, dyspigmentation and deafness (13–17). St3gal5−/− mice produce no GM3 and also have severe hearing loss manifest at the time of normal hearing onset accompanied by degeneration of cochlear hair cells (18). Based on the auditory phenotype of human and murine GM3 synthase-deficiency, we hypothesized that GM3 plays an essential role in the postnatal maturation and function of the organ of Corti (18). Here, we study auditory function of human patients with homozygous ST3GAL5 c.694C>T mutations and explore mechanisms of hearing loss using St3gal5−/− and B4galnt1−/− transgenic mice. Our results implicate a specific and indispensable role for GM3 ganglioside in development and viability of cochlear hair cells.
Results
GM3 synthase (ST3GAL5) deficiency in humans
Eight children (ages 4.1 ± 2.3 years, 4 female) from the Old Order Amish community of Pennsylvania were homozygous for ST3GAL5 c.694C>T mutations. As reported by Simpson et al. (13), we confirmed that plasma of affected children had undetectable GM3 and its downstream biosynthetic derivatives as well as elevated lactosylceramide (LacCer; the proximate), globoside and paragloboside metabolites (data not shown). All eight children had the characteristic phenotype of systemic a- and b-series ganglioside deficiency, including slow postnatal head growth, intractable epileptic encephalopathy, severe psychomotor delay, visual impairment and hearing loss. Patient electroencephalograms (EEGs) revealed slow (2–4 Hz), high voltage (>300 µV), chaotic background activity. Sleep–wake variation, posterior rhythm, and photic driving response were consistently absent. Multifocal spike-slow wave discharges at a frequency of 1.5–3 Hz lasting 3–10 s were seen in all children, and electrographic seizures of similar frequency and duration were captured on 29% of EEG (Fig. 2). Magnetic resonance images from a subgroup of patients showed normal brain size and structure at birth, but evidence of hypomyelination and delayed brain maturation with advancing age (Fig. 2).
Figure 2.

Upper panel from left to right, T2, fluid-attenuated inversion recovery, diffusion-weighted, and 1H-MR spectroscopy at 1.5 T show normal cortical and subcortical structure, but severe hypomyelination and increased water diffusion throughout the corona radiata, as well as elevated choline (Cho) relative to creatine–phosphocreatine (Cr) and N-acetylaspartate 1H signals (single voxel PRESS, TE 144/TR 1500). (Lower panel) EEG of a 2-year-old child homozygous for ST3GAL5 c.694C>T shows a slow, chaotic background, absent posterior rhythm and no discernable change through a behavioral sleep–wake cycle. Beginning at the dashed red line, there is a burst of generalized, frontal predominant, spike-slow wave discharges at 3 Hz in excess of 450 µV (note voltage sensitivity).
Auditory phenotype of humans with ST3GAL5 deficiency
Children homozygous for ST3GAL5 c.694C>T had absent middle ear muscle reflexes (MEMRs) at all frequencies in most ears tested (Table 1). Thresholds were normal in only one ear of one patient, but responses were absent for two out of four frequencies in that subject's other ear. Another patient had elevated responses in one ear, but absent responses at all frequencies in the other ear. Distortion product otoacoustic emissions (DPOAEs) were absent bilaterally in all subjects, consistent with the observation that all ST3GAL5 c.694C>T homozygotes who were tested failed the newborn hearing screen.
Table 1.
Middle ear muscle reflexes in children homozygous for ST3GAL5 c.694C>T
| Age (years) | Middle ear muscle reflexes |
|||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 kHz | 1 kHz |
2 kHz |
4 kHz |
|||||
| Left | Right | Left | Right | Left | Right | Left | Right | |
| 1.6 | NR | 90 | NR | 95 | 95 | 95 | 95 | 90 |
| 2.0 | NR | NR | NR | NR | 100 | NR | 100 | NR |
| 2.8 | NR | NR | NR | NR | NR | 100 | NR | NR |
| 3.1 | NR | 95 | NR | NR | NR | NR | NR | NR |
| 3.6 | 100 | 100 | 100 | 95 | 100 | NR | NR | NR |
| 4.5 | 85 | NR | NR | NR | NR | NR | NR | NR |
| 7.2 | 100 | NR | NR | NR | 100 | NR | NR | NR |
| 7.9 | 95 | NR | NR | NR | NR | NR | NR | NR |
NR, no response; 85, 90, 95 or 100 dB hearing level.
Auditory brainstem responses (ABRs) had abnormal thresholds in all ears and cochlear microphonics (CM) was consistently absent (Table 2). One child had no reproducible ABR waves. Wave I was observed in one subject, questionable in another, and absent in remaining patients. In six children, only Waves III and V could be clearly defined. Wave latencies were normal (80 dB) in the majority of patients. In most ears, comparison of condensation and rarefaction responses revealed waveform phase reversals (Fig. 2). Cortical auditory-evoked potentials (CAEPs) were detectable in all ST3GAL5 c.694C>T homozygotes (Table 2), but their morphology was abnormal in 7 (88%) and P2/N2 latency was delayed in 6 (75%) patients.
Table 2.
Auditory brainstem responses and CAEPs in children homozygous for ST3GAL5 c.694C>T
| Age (years) | Auditory brainstem responses |
CAEPs |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CM |
Morphology |
Waves (80 dB) | Thresholds |
Latency (80 dB) | Phase reversal |
Morphology |
Latency |
|||||||||
| Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | Left | Right | |
| 1.6 | Abs | Abs | – | – | Abs | Abs | – | – | – | – | – | – | Abn | Abn | D | D |
| 2.0 | Abs | Abs | Abn | Abn | I, III, V | I, III,V | 50 | 50 | N | N | No | Yes | Abn | Abn | D | D |
| 2.8 | Abs | Abs | Abn | Abn | III, V | III, V | 70 | 70 | D | D | Yes | Yes | Abn | Abn | D | D |
| 3.1 | Abs | Abs | Abn | Abn | III, V | III, V | 80 | 80 | N | N | No | Yes | Abn | Abn | D | D |
| 3.6 | Abs | Abs | Abn | Abn | III, V | III, V | 80 | 80 | N | N | Yes | Yes | Abn | Abn | D | D |
| 4.5 | Abs | Abs | Abn | Abn | Ia, III, V | Ia, III, V | DNT | DNT | N | N | Yes | Yes | Abn | Abn | N | N |
| 7.2 | Abs | Abs | Abn | Abn | III, V | III, V | 60 | 50 | N | N | No | Yes | N | N | N | N |
| 7.9 | Abs | Abs | Abn | Abn | IIIa, Va | IIIa, Va | 80 | 80 | N | D | No | a | Abn | Abn | D | D |
Abn, abnormal; Abs, absent; CM, cochlear microphonic; D, delayed, N, normal; DNT, did not test.
aEquivocal result.
Postnatal GSL expression in wild-type murine cochlea
Healthy mice begin to recognize sound by postnatal Day 12 (P12), designated the ‘onset of hearing’ (Fig. 1D). We investigated the expression of GSLs in wild-type murine cochlea during the postnatal maturation period from P1 to P17. As shown in Figure 1B, GM3 is the dominant cochlear GSL at P1. After P3, there is marked increase of GM3 as well as other GSLs, including glucosylceramide, complex gangliosides (GM1, GD1a, GD3, GD1b, GT1b) and sulfatides (SM3 and SM4) (Fig. 1B). The structures of each ganglioside species were examined by liquid chromatography mass spectrometry (LC-MS) (Fig. 1C).
It has been reported that ganglioside GM1 is present on the surface of the cochlea of the chinchilla using FITC-labeled cholera toxin B subunits (19). Table 3 summarizes the distinctive membrane distributions of GM1 and GM3 in wild-type organ of Corti. In OHC stereocilia, GM3 was high but GM1 was undetectable (Fig. 3C and D), and neither ganglioside species was expressed on the basolateral cell body (Fig. 3E). In IHC stereocilia, both GM1 and GM3 were expressed from the taper region to the top (Fig. 3C and D), but GM3 was absent from the IHC body. Expression of GM1 (but not GM3) was especially high on the surface of supporting Deiters cells and pillar cells expressed only GM1 (Fig. 3E).
Table 3.
Comparison of GM3 and GM1 expression of specific regions in the organ of Corti at P14
| OHC |
IHC |
DC |
PC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Stereocilia | Cuticular plate | Cell body | Stereocilia | Cuticular plate | Cell body | Cell surface | Cell body | Cell body | |
| GM3 | ++ | + | − | ++ | + | − | − | + | − |
| GM1 | − | − | − | ++ | + | + | ++ | + | + |
OHC, outer hair cell; IHC, inner hair cell; DC, Deiters cell; PC, pillar cell.
Figure 3.
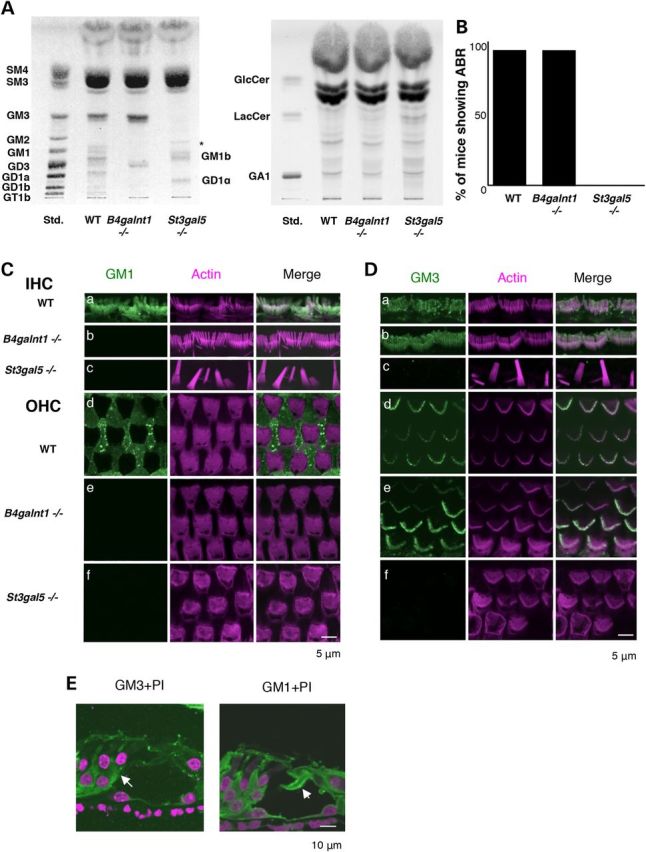
Identification of GM3 as an essential molecule for hair cell function. (A) HP-TLC of acidic (left) and neutral (right) GSLs from the cochlea of 4-week-old wild-type (WT), B4galnt1−/− and St3gal5−/− mice. The cochlea of B4galnt1−/− mice were mainly expressed GM3 and GD3. Each purified fraction was spotted 2 mg protein per lane. (B) Auditory-evoked brainstem response (ABR) of wild-type, B4galnt1−/− and St3gal5−/− mice at 4 weeks old. (C and D) Comparison of gangliosides expression in hair cells among wild-type, B4galnt1−/− and St3gal5−/− mice aged 4 weeks. Whole-mount immunostaining was performed. GM1 (C, green) and GM3 (D, green) of IHC (a–c) and OHC (d–f) stereocilia in wild-type (a and d), B4galnt1−/− (b and e) and St3gal5−/− (c and f) mice. In IHC, both GM1 and GM3 were expressed along the stereocilium from the taper region to top (Ca and Da). On the other hand, only GM3 was highly enriched in OHC stereocilia (Dd) without staining of GM1 (Cd). The stereocilia of IHC and OHC in B4galnt1−/− mice were expressed GM3 but not GM1 (Cb and Db). In St3gal5−/− mice, fused stereocilia was observed in IHC (Cc and Dc) and irregular phalloidin staining was observed in OHC (Cf and Df). Notably, the expression levels of GM1 but not GM3 was especially high on the surface of supporting cells (Deiters cell) (Cd and Dd). (E) Confocal images of cross sections of the cochleae in wild-type mice stained for GM3 (green, left panel), GM1 (green, right panel) and propidium iodide to label cell nuclei. (Arrow) Deiters cell, (arrow head) pillar cell. The red color of phalloidin staining (C and D) and propidium iodide (E) has been converted to magenta using Adobe Photoshop CC2014 software.
GSL expression in cochlea of St3gal5 and B4galnt1 null mice
Cochlea of St3gal5−/− mice were devoid of a- and b-series gangliosides, replaced by compensatory o-series gangliosides (GM1b and GD1α) (Fig. 3A, left panel) and a high content of LacCer, the proximate substrate of GM3 synthase (Fig. 3A right panel). In B4galnt1−/− mice, the only other ganglioside detected was GD3 (Fig. 3A). Neutral glycolipid composition of B4galnt1−/− cochlea was similar to wild type.
Auditory system morphology and function in St3gal5 and B4galnt1 null mice
To better define the role of various ganglioside species in hearing, we compared auditory function between St3gal5−/− and B4galnt1−/− mice. At age 4 weeks, hearing ability of B4galnt1−/− mice was comparable to that of wild-type mice at all frequencies tested (Fig. 3B). In contrast, ABRs were absent in 4-week-old St3gal5−/− mice (Fig. 3B).
We used confocal laser microscopy with phalloidin staining to examine hair cell morphology in transgenic animals. In B4galnt1−/− mice, OHC and IHC morphology were normal at 4 weeks-old (Fig. 3C and D). In contrast, St3gal5−/− mice had structural derangements of both OHCs and IHCs that progressed over time (Figs 4 and 5). At P1, hair bundles of St3gal5−/− mice resembled those of wild-type mice, but by P3 abnormal OHC hair bundle morphology was evident in most regions of the basal cochlea (Fig. 4A). By P8, scanning electron microscopy (SEM) revealed blebs along both apical and basal coils of vestigial kinocilia (Fig. 4B, left panel), but these disappeared by P17 (Fig. 4B, right panel). As depicted in Figure 4C, OHC blebs in P8 St3gal5−/− mice did not stain with anti-acetylated tubulin, suggesting they did not derive from tubulin-based kinocilia. Negative phalloidin staining indicated that these blebs contained neither cytoplasmic nor cortical actin. Blebs generally indicate cellular injury and subsequent necrosis or apoptosis (20–22), but TUNEL staining showed no signs of apoptosis in cochlea of P8 St3gal5−/− mice (Fig. 4D). In this vestigial kinocilia region of the cuticular plate, actin filaments were depleted and connections between the actin cortical layer and plasma membrane were weak (23), but membrane recycling, endocytosis, and exocytosis appeared active (24). Transmission electron microscopy (TEM) revealed the appearance of intracellular vesicles in OHCs of St3gal5−/− mice (Fig. 4E). Magnified view clearly showed that these vesicles contained the intracellular membranous structures. Taken together, these observations suggest that blebs and intracellular vesicles seen in OHCs of St3gal5−/− mice might reflect an imbalance of endocytosis and exocytosis.
Figure 4.

Degeneration of OHC in St3gal5 null mice. (A) Confocal images of the basal coil of phalloidin-stained cochlear whole mounts from St3gal5+/+ and −/− mice at P1 and P3. Defects in structure of hair bundle are first in the outer hair cells (arrows) at P3. (B) SEM images of OHC of St3gal5+/+ and St3gal5−/− mice at P8 and 17. At P8, the blebs at the vestigial kinocilium location were observed (arrows), but disappeared at P17. (C) Expression of acetylated tubulin. Confocal images from showing stereocilia of St3gal5+/+ and −/− mice stained for acetylated tubulin (green) and F-actin (phalloidin, magenta) at P3 and P8. At P3, hair cells of St3gal5+/+ and −/− mice were expressed kinocilia with or without degeneration of stereocilia. At P8, kinocilia were disappeared from OHC in both St3gal5+/+ and −/− mice. Three rows of OHCs are indicated by the white arrowheads. (D) TUNEL staining. Apoptotic cell was not detected in both St3gal5+/+ and −/− mice at P8. (E) TEM images of OHC in St3gal5+/+ and −/− mice at P17. The red color of phalloidin staining (C) and propidium iodide (D) has been converted to magenta using Adobe Photoshop CC2014 software.
Figure 5.

Fused stereocilia formation of IHC in St3gal5 null mice. (A)The stereocilia of the IHCs of St3gal5−/− mice were fused after P12. Arrows indicate the fused stereocilia. (B) TEM images of the IHC stereocilia in St3gal5+/+ and −/− mice at P17.
We next examined the morphology of IHC hair bundles using both SEM (Fig. 5A) and TEM (Fig. 5B). Until P10, IHC stereocilia of St3gal5−/− mice were structurally normal. However, by P12 some IHC stereocilia were stuck together and bent (Fig. 5A, arrows). Most degenerated by P14. TEM at P17 revealed giant and fused stereocilia with thick, long rootlets that penetrated the cuticular plate (Fig. 5B).
Irregular expression of PTPRQ and myosin VI in stereocilia of St3gal5 null mice
Myosin VI is normally concentrated at the base of stereocilia, where it interacts with protein tyrosine phosphatase receptor Q (PTPRQ) to maintain organization of the cell surface coat and structure of the overall hair bundle (25). We used immunostaining to evaluate involvement of gangliosides in the formation of this basal PTPRQ-myosin VI complex (Fig. 6). As shown in Figure 6B, PTPRQ was exclusively localized to the base of stereocilia in wild-type mice, but in St3gal5−/− mice, PTPRQ was maldistributed along the fused stereocilia.
Figure 6.
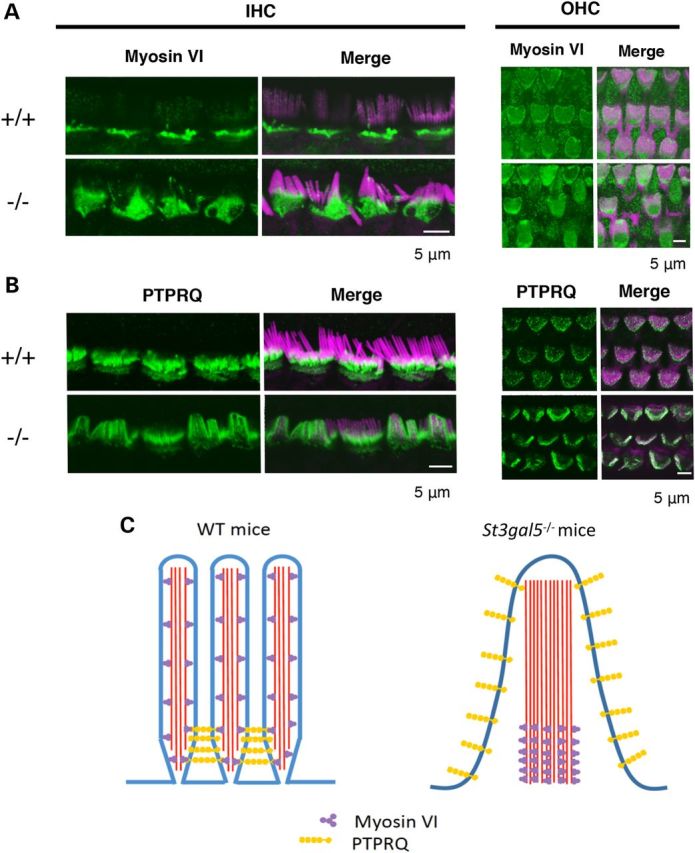
Dislocalization of PTPRQ and myosinVI in the stereocilia of St3gal5 null mice. Confocal images showing stereocilia of IHCs and OHCs of St3gal5+/+ and −/− mice stained for myosinVI (A) PTPRQ (B) (green) and F-actin (phalloidin, magenta). (C) Schematic images for dislocalization of PTPRQ and myosin VI in the IHC stereocilia of St3gal5−/− mouse. The red color of phalloidin staining (A and B) has been converted to magenta using Adobe Photoshop CC2014 software.
In fused IHC stereocilia of St3gal5−/− mice, myosin VI was present from the base to the midshaft but absent from top regions (Fig. 6A), suggesting loss of normal ciliary motor action. In St3gal5−/− OHCs, expression of myosin VI was concentrated on the surface of the cuticular plate, close to vestigial kinocilia (Fig. 6A). Our observations suggest that membrane domain enriched with GM3 are crucial to the formation and proper localization of the stereocilia PTPRQ-myosin VI complex. In the absence of GM3, localization of this complex is disrupted (Fig. 6C), and this may have important structural and functional consequences for both inner and outer hair cells.
Discussion
Gangliosides and mammalian auditory development
Humans lacking all a- and b-series gangliosides have a profound disturbance of auditory function, described here as the neonatal onset hearing impairment, weak or absent MEMRs and DPOAEs, abnormal ABR morphology and thresholds, absent CMs, and delayed CAEPs. Absent DPOAEs and CMs indicate severe OHC impairment. Although the presence of ABRs in seven of eight patients indicates at least partial IHC function, numerous abnormalities revealed by MEMR and ABR testing suggest there is significant dysfunction of IHCs, central auditory pathways or both. As further evidence of this, ABR thresholds are generally higher than expected if OHC dysfunction alone explain hearing loss. Wave reversal observed in many patients indicates that hearing is more impaired at high versus low and middle frequencies (26).
In contrast, humans with autosomal recessive B4GALNT1 deficiency have no hearing loss, visual impairment or epileptic encephalopathy, but suffer from progressive spastic paraplegia (13–17,27). This suggests that (i) each ganglioside has a particular distribution and functional relevance within the nervous system; (ii) there may be limited functional redundancy among various ganglioside species; and (iii) GM3 is indispensable to both the cochlear and neural phases of sound processing in mammals.
In this study, we exploited differential patterns of GSL expression among wild-type, St3gal5−/− and B4galnt1−/− mice to explore cochlear mechanisms of hearing loss that could be specifically attributed to deficiency of GM3 (18). Wild-type mice have a marked increase of gangliosides and other GSLs in the cochlea during early postnatal maturation (Fig. 1D), suggesting these molecules contribute to hearing onset. After auditory maturation, GM3 and GM1 show a distinctive distribution among cellular elements of the organ of Corti (i.e. IHC, OHC, Dieters cell, pillar cell) as summarized in Table 3. Both IHCs and OHCs express GM3, which is preferentially distributed to the apical surface, cuticular plate and stereocilia, whereas only IHCs express GM1, which is localized to their apical surface. In St3gal5−/− mice, degeneration of OHCs precedes that of IHCs, and this may reflect relative enrichment of GM3 in OHCs.
St3gal5−/− and B4galnt1−/− mice have predictable patterns of GSL expression within the organ of Corti. Cochlea of B4galnt1−/− mice have high GM3 content, intact hair bundle structure and support normal hearing. In contrast, cochlea of St3gal5−/− mice are devoid of GM3, develop microstructural and degenerative changes as early as P3, and do not support auditory function. B4galnt1/Siat8 double null mice that express only GM3 have an acoustic startle response (28), and we confirmed that Siat8−/− mice lacking b-series gangliosides (e.g. GD3) hear normally (unpublished data).
All GM3 synthase-deficient patients tested with a newborn hearing screen failed, confirming that auditory deficits in humans with GM3 deficiency are present early in life. However, no structural abnormality of OHC or IHC stereocilia is evident in St3gal5−/− mice at P0. Thus GM3 may play its most critical role in postnatal maturation rather than embryogenesis of the auditory system. Consistent with this, auditory function of St3gal5−/− mice, although impaired at the onset of hearing, is completely abolished by P17 (18), at which time TEM reveals pathological remodeling of stereocilia (Fig. 5B).
Glycocalyx integrity and membrane cycling in GM3-deficient mice
Apical membranes of stereocilia are covered with a glycocalyx composed of acidic glycoproteins and glycolipids (including gangliosides) that contain sialic acid (29). This creates a dense negative charge distribution within the glycocalyx that normally prevents fusion of adjacent stereocilia. In experimental animals, aminoglycoside administration reduces the expression of sialoglycoconjugates in the OHC glycocalyx (30) and is associated with fusion of stereocilia (31). Fused stereocilia in St3gal5−/− mice may similarly reflect loss of the normal repulsive charge barrier between adjacent stereocilia, mediated in part by GM3 and its a- and b-series derivatives.
In the cuticular plate region of St3gal5−/− OHCs, we observed depletion of actin filaments, weak connections between the plasma membrane and actin cortical layer (23) and numerous membrane blebs. Abundant blebs are also seen in aminoglycoside-treated organ of Corti (32,33). In both cases, blebs may arise from imbalance of endocytosis and exocytosis (24,34,35). Since endocytosis in OHCs is clathrin-independent, mediated by caveolae and glycolipid rafts (24,36,37), the depletion of GM3 from ganglioside-dependent membrane organization may disrupt membrane recycling or vesicle transport to cause bleb formation (Fig. 4).
GM3-enriched membrane organization, PTPRQ-myosin VI complex localization and hair cell morphology
In the normal organ of Corti, functional PTPRQ-myosin VI complexes at the base of stereocilia appear critical to maintaining hair cell morphology and function (25). PTPRQ is a shaft connector located at the tapered base of stereocilia, with an extracellular domain containing 18 fibronectin III (FNIII) repeats, a membrane spanning domain, and a cytoplasmic domain that has both phosphatidylinositol and tyrosine phosphatase activities (38,39). Myosin VI is an actin-based motor protein that controls interactions between the plasma membrane and actin cytoskeleton (40). Mice lacking PTPRQ are deaf and lack tapering at the base of stereocilia, which become fused (25). In deaf, myosin VI-deficient mice, comparable structural changes of stereocilia are accompanied by maldistribution of PTPRQ along the length of the stereocilia membrane (25,41,42). We observed a similar morphological disruption of stereocilia and maldistribution of PTPRQ in St3gal5−/− mice (Fig. 6B), indicating a common underlying mechanism.
In renal membranes, myosin VI has a regulatory association with Na+/H+ exchanger three within microdomains called lipid rafts (43). These lipid rafts, enriched in cholesterol, gangliosides and other GSLs, are important for the maintenance of brush border microvilli architecture and function, and also influence the activity of many transmembrane proteins (44–50). Furthermore, the existence of ganglioside-rich membrane domains colocalized with PTPRQ has been observed in the basal tapers of bull frog hair cells (51). Based on observations from GM3-deficient humans and mice, we postulate that membrane organization enriched with GM3 are essential to the functional interactions among PTPRQ, myosin VI, actin and the cytoskeleton of inner and outer hair cell stereocilia (Fig. 6C).
Taken together, our results show that normal spatial and temporal expression of GM3 is critical to the structural integrity and function of auditory hair cells. These findings provide a foundation for understanding mechanisms of hearing loss in humans with GM3 synthase deficiency, but are not sufficient to explain the full constellation of neurological findings in these patients. Clearly, systemic GM3 deficiency also interferes with cerebral myelination and electrical activity (Fig. 2). This certainly affects central auditory function and also explains the severe psychomotor delay, epilepsy and visual impairment seen in children homozygous for ST3GAL5 c.694C>T. Future studies should more carefully delineate the role of lipid rafts generally, and GM3 specifically, in the development and physiology of the central nervous system.
Although hearing is already impaired at birth in humans with GM3 synthase deficiency, our observations of St3gal5−/− mice suggest that a critical postnatal window may exist during which hair cell remodeling and degeneration could be rescued by ganglioside repletion therapy. This will prove challenging for at least three reasons: (i) purified GM3 is prohibitively expensive, and not currently available in sufficient quantities to offer human subjects; (ii) bioavailability of intestinal GM3 is unknown but may be quite limited, making enteral GM3 therapy impractical; and (iii) even if GM3 is successfully delivered to the circulation, it may not efficiently cross biological barriers into the organ of Corti or central nervous system. Cochlear and neural tissues are believed to make their own GSLs, and it is not clear that gangliosides from the circulation could be routed in adequate abundance to their proper location to restore function of OHCs, IHCs, neurons and oligodendrocytes. Nevertheless, these are soluble questions and the answers are important; we have experiments underway to answer them.
Materials and Methods
Study subjects
Study of human subjects was approved by the Lancaster General Hospital Institutional Review Board and parents consented in writing on behalf of their children. We studied eight children, ages 4.1 ± 2.3 years (four females), all of whom were members of the Old Order Amish community of Pennsylvania and homozygous for c.694C>T mutations of ST3GAL5. All patients received longitudinal pediatric care at the Clinic for Special Children (Strasburg, PA).
Auditory testing
We performed tympanometry testing with a 226-Hz probe tone, and ipsilateral MEMRs were measured between 80–100 dB HL at 0.5, 1, 2 and 4-kHz (Titan, Interacoustics). Distortion product otoacoustic emissions were obtained using the ILO (Otodynamic) ‘eight points/octave’ function. 2f1–f2 were recorded for f2 varying from 842 Hz to 7996 Hz and intensities of the primaries were kept constant across the frequency range (f1 = 65 dB SPL, f2 = 55 dB SPL). The f1/f2 frequency ratio was 1.22.
ABRs were elicited using 100 µs air-conduction clicks and recorded from a two-channel four-electrode montage (mastoid-high forehead-mastoid) with the Eclipse system (Interacoustics). Responses were first obtained at 80 dB normal hearing level (nHL). The intensity was increased by 10 dB when deemed appropriate to better visualize a response. When responses were present, intensities were then decreased in 10 dB steps to obtain a response threshold.
Condensation and rarefaction clicks were used to distinguish the CM from the compound action potential. The rate of stimulation was 27.7/s, low pass filter was 1500 Hz and high pass filter was 100 Hz, with gain set at 20 K. The presence of a wave was only established when at least two different recordings (for each polarity of the click) were available at the same or different intensity to verify reproducibility.
CAEPs were recorded with the Intelligent Hearing System device following the delivery of the stimulus/da/. Stimuli were delivered at 80 dB SPL to each ear separately through insert earphones. Responses were recorded using a two-channel, four-electrode montage (mastoid-high forehead-mastoid). The recording window was 400 ms. For each ear, 60 responses were averaged three times to calculate a grand average.
Mice
All animal studies were approved by Institutional Review Boards of Tohoku Pharmaceutical University. St3gal5−/− mice were generated as previously described (18,52). Mutant mice were maintained on a C57BL6 background by heterozygous mating to generate littermate controls and PCR genotyping of St3gal5 alleles was performed as previously described (18). Briefly, St3gal5 genotypes were determined by Southern blot and PCR analysis of genomic DNA isolated from embryonic stem cells and tail biopsies. Primer pairs used for genotyping included 5′-GGAATCCATCCCTTTTCTCACAGAG-3′ and 5′-TGAACTCACTTGGCATTGCTGG-3′ for detection of the wild-type allele (exon 2) and 5′-ACTGGGCACAACAGACAATCGG-3′ and 5′-TGGATACTTTCTCGGCAGGAGC-3′ for the knockout allele (neomycin resistance gene). B4galnt1−/− mice were established from B4galnt1 and B4galnt2 double null mice supplied by the Consortium for Functional Glycomics (www.functionalglycomics.org). Genotypes were using PCR from tail-tissue DNA (53). Both types of null mice were backcrossed with C57BL/6 mice over >11 generations.
Immunohistochemistry
Animals were kept under deep anesthesia and briefly perfused with physiological saline and 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4). Each cochlea was rapidly removed from the temporal bones and fixed in 4% paraformaldehyde in 0.1 M PB for 1 h at room temperature or overnight at 4°C. For whole mounts, the organ of Corti was dissected out of the cochlea and the Reissner's membrane and tectorial membrane removed to expose the sensory epithelium. For sectioning, cochlea were decalcified in 10% ethylenediamine tetraacetic acid in Tris buffer (pH 7.4) and washed in PB, equilibrated overnight in 40% sucrose and then embedded and rapidly frozen in a mixture of equal parts 40% sucrose in PB and Tissue-Tek OCT compound (Sakura Finetechnical Co.). Cochlea were cryosectioned at 5 μm thickness. Sections and dissected cochlea were immunostained with anti-ganglioside GM3 (GMR6, Seikagaku Co. Ltd.), FITC-conjugated cholera toxin B-subunit (SIGMA), anti-myosin VI (Proteus BioSciences), anti-PTPRQ (gifted by Drs Sakaguchi and Kachar). Alexa 488-conjugated secondary antibodies, Alexa 594-conjugated phalloidin and propidium iodide were from Invitrogen. FITC-conjugated secondary anti-IgM antibody was from Vector Laboratories. Images were collected using Olympus FLUOVIEW confocal microscope.
TUNEL staining
Apoptotic cells in the cochlea were detected using In Situ Cell Death Detection Kit, POD (Roche). Apoptotic cleavage of genomic was identified by TUNEL, following the manufacturer's instructions. Positive controls for apoptosis were obtained by treating cochlear sections with DNase I.
Scanning and TEM
For SEM, cochlea were dissected to expose the organ of Corti and were fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in 30 mm HEPES buffer (pH 7.3) for 2 h at room temperature. Postfixation was performed by rotating tissues in 1% osmium tetroxide in 0.1 M sodium cacodylate buffer for 1 h at 4°C. Samples were dehydrated through a graded series of ethanol, substituted with t-butyl alcohol, freeze-dried, coated with platinum, and viewed in a JEOL JSM-T330A scanning microscope at 20 kV. For TEM, cochlea were fixed in 2.5% glutaraldehyde in 0.1 M PB (pH 7.4) for 2 h at room temperature. Postfixation was performed by rotating tissues in 1% osmium tetroxide in 0.1 M PB for 1 h at 4°C. Samples were dehydrated through a graded series of ethanol, equilibrated with propylene oxide and imbedded in Quetol-812 resin. Blocks were cured for 2 d at 60°C and sectioned with glass and diamond knives at 90–200 nm thickness. Sections were mounted on copper grids, double stained with uranyl acetate followed by lead citrate, and viewed in a JEOL transmission electron microscope at 70 kV.
Auditory brainstem responses
Mice (4 weeks of the age) were anesthetized with ketamine hydrochloride and xylazine hydrochloride. ABRs were recorded by using stainless steel electrodes placed at the vertex (positive), behind the bilateral ears (negative), and at the lower back (ground). Tone burst stimuli (rise/fall time: 0.2 ms; plateau time: 1 ms; repetitive rate: 11 Hz) were applied from the speaker (PT-R100; Pioneer) by using a function generator (DPS-725; Dia Medical) for which the system could provide 100 dB SPL at each frequency. Evoked responses were filtered with a band pass of 200 to 3 kHz, and the average 200 sweeps was recorded by using a single processor (Neuropack Micro; Nihon Kohden). ABR waveforms were recorded in decreasing 5 dB SPL intervals from a maximum amplitude until no waveforms could be visualized.
GSL (ganglioside) analysis
Cochlea were dissected from wild-type, B4galnt1−/−, and St3gal5−/− mice after perfusion with PBS, then lyophilized. Dried tissues were crushed, and lipids were extracted from the tissues with the following solvent mixtures: chloroform/methanol, 1:1, 1:2 (v/v). After evaporation, the residue was dissolved in chloroform/methanol/water (30:60:8, v/v). The sample was allowed to absorb to the column packing with DEAE-Sephadex A25 (GE Healthcare). Chloroform/methanol/water (30:60:8, v/v) was applied to a column of same material and then chloroform/methanol/1 N sodium acetate aqueous (30:60:8, v/v). Neutral and acidic lipid fractions were eluted and spotted 2 mg/protein on the high performance-thin-layer chromatogram (HPTLC) plates. The plate of acidic fraction was developed in chloroform/methanol/0.2%CaCl2 (55:45:10, v/v) and the plate of neutral fraction was developed in chloroform/methanol/water (65:25:4, v/v). Glycolipids were detected by spraying the plate with orcinol-sulfuric acid reagent.
LC-MS analysis
The acidic fraction was analyzed by liquid chromatography-ion trap-mass spectrometry with a NH2 column (Inertsil NH2, 3 mm, 1 × 50 mm, GL Science, Japan), flow rate (50 µl/min), a linear gradient made with solvent A: 1 mm ammonium formate in acetonitrile–water (95:5, v/v), solvent B: 50 mm ammonium formate in acetonitrile–water (50:50, v/v), and a time program: at 0 and 5 min (5% of B), 20 min (75% of B), 25 min (90% of B) and 30 min (90% of B) (54). The MS1 chromatogram was monitored with theoretical values of [M – H]−1 or [M − 2H]−2 for each ganglioside (54). The structures indicated in the figure is tentative, and based on information with only molecular related ions indicated with black and, in addition, carbohydrate-related fragment ions obtained by MS2 analysis indicated with red, and requires the consideration of possibility of d20:1 sphingosine instead of d18:1 in the ceramides.
Statistical analysis
Values in the text are the means ± SD. Data were compared using Student's t-test for two-group comparison or ANOVA for multigroup comparison. Significant differences were post hoc analyzed using Scheffé's test. Differences were considered significant at P < 0.05.
Conflict of Interest statement. None declared.
Funding
This work was supported by research grants for a Grant-in-Aid for Scientific Research (B) (to J.-i.I.) and for Scientific Research on Innovative Areas (no. 23110002, Deciphering sugar chain-based signals regulating integrative neuronal functions) from MEXT, Japan, the Mizutani Research Foundation for Glycoscience (J.-i.I.), The Naito Foundation (J.-i.I.), the ONO Medical Research Foundation (J.-i.I.), the Uehara Memorial Foundation (J.-i.I.), and by Grant-in-Aid for Japan Society for the Promotion of Science Fellows (M.Y.). This work was also supported by MEXT-Supported Program for the Strategic Research Foundation at Private Universities. The Clinic for Special Children is supported by generous charitable contributions from the Amish and Mennonite communities it serves.
References
- 1.Roberts W.M., Howard J., Hudspeth A.J. (1988) Hair cells: transduction, tuning, and transmission in the inner ear. Annu. Rev. Cell Biol., 4, 63–92. [DOI] [PubMed] [Google Scholar]
- 2.Hudspeth A.J. (1997) How hearing happens. Neuron, 19, 947–950. [DOI] [PubMed] [Google Scholar]
- 3.Hakomori S. (1981) Glycosphingolipids in cellular interaction, differentiation, and oncogenesis. Annu. Rev. Biochem., 50, 733–764. [DOI] [PubMed] [Google Scholar]
- 4.Schengrund C.L. (1990) The role(s) of gangliosides in neural differentiation and repair: a perspective. Brain. Res. Bull., 24, 131–141. [DOI] [PubMed] [Google Scholar]
- 5.Kawai H., Allende M.L., Wada R., Kono M., Sango K., Deng C., Miyakawa T., Crawley J.N., Werth N., Bierfreund U., Sandhoff K., Proia R.L. (2001) Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J. Biol. Chem., 276, 6885–6888. [DOI] [PubMed] [Google Scholar]
- 6.Hakomori S.I. (2002) The glycosynapse. Proc. Natl. Acad. Sci. U.S.A., 99, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Proia R.L. (2004) Gangliosides help stabilize the brain. Nat. Genet., 36, 1147–1148. [DOI] [PubMed] [Google Scholar]
- 8.Handa Y., Ozaki N., Honda T., Furukawa K., Tomita Y., Inoue M., Furukawa K., Okada M., Sugiura Y. (2005) GD3 synthase gene knockout mice exhibit thermal hyperalgesia and mechanical allodynia but decreased response to formalin-induced prolonged noxious stimulation. Pain, 117, 271–279. [DOI] [PubMed] [Google Scholar]
- 9.Takamiya K., Yamamoto A., Furukawa K., Yamashiro S., Shin M., Okada M., Fukumoto S., Haraguchi M., Takeda N., Fujimura K., et al. (1996) Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc. Natl. Acad. Sci. U.S.A., 93, 10662–10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiavegatto S., Sun J., Nelson R.J., Schnaar R.L. (2000) A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Exp. Neurol., 166, 227–234. [DOI] [PubMed] [Google Scholar]
- 11.Inoue M., Fujii Y., Furukawa K., Okada M., Okumura K., Hayakawa T., Furukawa K., Sugiura Y. (2002) Refractory skin injury in complex knock-out mice expressing only the GM3 ganglioside. J. Biol.Chem., 277, 29881–29888. [DOI] [PubMed] [Google Scholar]
- 12.Yamashita T., Wu Y.P., Sandhoff R., Werth N., Mizukami H., Ellis J.M., Dupree J.L., Geyer R., Sandhoff K., Proia R.L. (2005) Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc. Natl. Acad. Sci. U.S.A., 102, 2725–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson M.A., Cross H., Proukakis C., Priestman D.A., Neville D.C., Reinkensmeier G., Wang H., Wiznitzer M., Gurtz K., Verganelaki A., et al. (2004) Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet., 36, 1225–1229. [DOI] [PubMed] [Google Scholar]
- 14.Farukhi F., Dakkouri C., Wang H., Wiztnitzer M., Traboulsi E.I. (2006) Etiology of vision loss in ganglioside GM3 synthase deficiency. Ophthalmic Genet., 27, 89–91. [DOI] [PubMed] [Google Scholar]
- 15.Boccuto L., Aoki K., Flanagan-Steet H., Chen C.F., Fan X., Bartel F., Petukh M., Pittman A., Saul R., Chaubey A., et al. (2013) A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum. Mol. Genet., 23, 418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H., Bright A., Xin B., Bockoven J.R., Paller A.S. (2013) Cutaneous dyspigmentation in patients with ganglioside GM3 synthase deficiency. Am. J. Med. Genet. Part A, 161A, 875–879. [DOI] [PubMed] [Google Scholar]
- 17.Fragaki K., Ait-El-Mkadem S., Chaussenot A., Gire C., Mengual R., Bonesso L., Bénéteau M., Ricci J.E., Desquiret-Dumas V., Procaccio V., et al. (2013) Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency. Eur. J. Hum. Genet., 21, 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshikawa M., Go S., Takasaki K., Kakazu Y., Ohashi M., Nagafuku M., Kabayama K., Sekimoto J., Suzuki S., Takaiwa K., et al. (2009) Mice lacking ganglioside GM3 synthase exhibit complete hearing loss due to selective degeneration of the organ of Corti. Proc. Natl. Acad. Sci .U. S. A., 106, 9483–9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santi P.A., Mancini P., Barnes C. (1994) Identification and localization of the GM1 ganglioside in the cochlea using thin-layer chromatography and cholera toxin. J. Histochem. Cytochem., 42, 705–716. [DOI] [PubMed] [Google Scholar]
- 20.Casciola-Rosen L., Rosen A., Petri M., Schlissel M. (1996) Surface protrusions on apoptotic cells are sites of enhanced procoagulant activity: implications for coagulation events and antigenic spread in systemic lupus erythematosus. Proc. Natl. Acad. Sci. U. S. A., 93, 1624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barros L.F., Kanaseki T., Sabirov R., Morishima S., Castro J., Bittner C.X., Maeno E., Ando-Akatsuka Y., Okada Y. (2003) Apoptotic and necrotic protrusions in epithelial cells display similar neck diameters but different kinase dependency. Cell Death Differ., 10, 687–697. [DOI] [PubMed] [Google Scholar]
- 22.Morelli A., Chiozzi P., Chiesa A., Ferrari D., Sanz J.M., Falzoni S., Pinton P., Rizzuto R., Olson M.F., Di Virgilio F. (2003) Extracellular ATP causes ROCK I-dependent protrusion formation in P2X7-transfected HEK293 cells. Mol. Biol. Cell, 14, 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi X., Gillespie P.G., Nuttall A.L. (2005) Na+ influx triggers protrusion formation on inner hair cells. Am. J. Physiol. Cell Physiol., 288, C1332–C1341. [DOI] [PubMed] [Google Scholar]
- 24.Griesinger C.B., Richards C.D., Ashmore J.F. (2004) Apical endocytosis in outer hair cells of the mammalian cochlea. Eur. J. Neurosci., 20, 41–50. [DOI] [PubMed] [Google Scholar]
- 25.Sakaguchi H., Tokita J., Naoz M., Bowen-Pope D., Gov N.S., Kachar B. (2008) Dynamic compartmentalization of protein tyrosine phosphatase receptor Q at the proximal end of stereocilia: implication of myosin VI-based transport. Cell. Motil. Cytoskeleton, 65, 528–538. [DOI] [PubMed] [Google Scholar]
- 26.Coats A.C., Martin J.L. (1977) Human auditory nerve action potentials and brain stem evoked responses: effects of audiogram shape and lesion location. Arch. Otolaryngol., 103, 605–622. [DOI] [PubMed] [Google Scholar]
- 27.Harlalka G.V., Lehman A., Chioza B., Baple E.L., Maroofian R., Cross H., Sreekantan-Nair A., Priestman D.A., Al-Turki S., McEntagart M.E., et al. (2013) Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain, 136, 3618–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tajima O., Egashira N., Ohmi Y., Fukue Y., Mishima K., Iwasaki K., Fujiwara M., Inokuchi J., Sugiura Y., Furukawa K., et al. (2009) Reduced motor and sensory functions and emotional response in GM3-only mice: emergence from early stage of life and exacerbation with aging. Behav. Brain. Res., 198, 74–82. [DOI] [PubMed] [Google Scholar]
- 29.Luft J.H. (1971) Ruthenium red and violet. II. Fine structural localization in animal tissues. Anat. Rec., 171, 369–415. [DOI] [PubMed] [Google Scholar]
- 30.de Groot J.C., Hendriksen E.G., Smoorenburg G.F. (2005) Reduced expression of sialoglycoconjugates in the outer hair cell glycocalyx after systemic aminoglycoside administration. Hear. Res., 205, 68–82. [DOI] [PubMed] [Google Scholar]
- 31.Takumida M., Wersäll J., Bagger-Sjöbäck D., Harada Y. (1989) Observation of the glycocalyx of the organ of Corti: an investigation by electron microscopy in the normal and gentamicin-treated guinea pig. J. Laryngol. Otol., 103, 133–136. [PubMed] [Google Scholar]
- 32.Goodyear R.J., Gale J.E., Ranatunga K.M., Kros C.J., Richardson G.P. (2008) Aminoglycoside-induced phosphatidylserine externalization in sensory hair cells is regionally restricted, rapid, and reversible. J. Neurosci., 28, 9939–9952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gale J.E., Marcotti W., Kennedy H.J., Kros C.J., Richardson G.P. (2001) FM1–43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J. Neurosci., 21, 7013–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richardson G.P., Russell I.J. (1991) Cochlear cultures as a model system for studying aminoglycoside induced ototoxicity. Hear. Res., 53, 293–311. [DOI] [PubMed] [Google Scholar]
- 35.Forge A., Richardson G. (1993) Freeze fracture analysis of apical membranes in cochlear cultures: differences between basal and apical-coil outer hair cells and effects of neomycin. J. Neurocytol., 22, 854–867. [DOI] [PubMed] [Google Scholar]
- 36.Grati M., Schneider M.E., Lipkow K., Strehler E.E., Wenthold R.J., Kachar B. (2006) Rapid turnover of stereocilia membrane proteins: evidence from the trafficking and mobility of plasma membrane Ca(2+)-ATPase 2. J. Neurosci., 26, 6386–6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaneko T., Harasztosi C., Mack A.F., Gummer A.W. (2006) Membrane traffic in outer hair cells of the adult mammalian cochlea. Eur. J .Neurosci., 23, 2712–2722. [DOI] [PubMed] [Google Scholar]
- 38.Wright M.B., Hugo C., Seifert R., Disteche C.M., Bowen-Pope D.F. (1998) Proliferating and migrating mesangial cells responding to injury express a novel receptor protein-tyrosine phosphatase in experimental mesangial proliferative glomerulonephritis. J. Biol. Chem., 273, 23929–23937. [DOI] [PubMed] [Google Scholar]
- 39.Oganesian A., Poot M., Daum G., Coats S.A., Wright M.B., Seifert R.A., Bowen-Pope D.F. (2003) Protein tyrosine phosphatase RQ is a phosphatidylinositol phosphatase that can regulate cell survival and proliferation. Proc. Natl. Acad. Sci. U. S. A., 100, 7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nambiar R., McConnell R.E., Tyska M.J. (2010) Myosin motor function: the ins and outs of actin-based membrane protrusions. Cell. Mol. Life Sci., 67, 1239–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Self T., Sobe T., Copeland N.G., Jenkins N.A., Avraham K.B., Steel K.P. (1999) Role of myosin VI in the differentiation of cochlear hair cells. Dev. Biol., 214, 331–341. [DOI] [PubMed] [Google Scholar]
- 42.Hertzano R., Shalit E., Rzadzinska A.K., Dror A.A., Song L., Ron U., Tan J.T., Shitrit A.S., Fuchs H., Hasson T., et al. (2008) A Myo6 mutation destroys coordination between the myosin heads, revealing new functions of myosin VI in the stereocilia of mammalian inner ear hair cells. PLoS Genet., 4, e1000207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riquier A.D., Lee D.H., McDonough A.A. (2009) Renal NHE3 and NaPi2 partition into distinct membrane domains. Am. J. Physiol. Cell Physiol., 296, C900–C910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hauser H., Howell K., Dawson R.M., Bowyer D.E. (1980) Rabbit small intestinal brush border membrane preparation and lipid composition. Biochim. Biophys. Acta, 602, 567–577. [DOI] [PubMed] [Google Scholar]
- 45.Christiansen K., Carlsen J. (1981) Microvillus membrane vesicles from pig small intestine. Purity and lipid composition. Biochim. Biophys. Acta, 647, 188–195. [DOI] [PubMed] [Google Scholar]
- 46.Simons K., van Meer G. (1988) Lipid sorting in epithelial cells. Biochemistry, 27, 6197–6202. [DOI] [PubMed] [Google Scholar]
- 47.Simons K., Ikonen E. (1997) Functional rafts in cell membranes. Nature, 387, 569–572. [DOI] [PubMed] [Google Scholar]
- 48.Simons K., Toomre D. (2000) Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol., 1, 31–39. [DOI] [PubMed] [Google Scholar]
- 49.Zonta B., Minichiello L. (2013) Synaptic membrane rafts: traffic lights for local neurotrophin signaling? Front . Synaptic. Neurosci., 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Head B.P., Patel H.H., Insel P.A. (2014) Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta, 1838, 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao H., Williams D.E., Shin J.B., Brügger B., Gillespie P.G. (2012) Large membrane domains in hair bundles specify spatially constricted radixin activation. J. Neurosci., 32, 4600–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsukamoto K., Kohda T., Mukamoto M., Takeuchi K., Ihara H., Saito M., Kozaki S. (2005) Binding of Clostridium botulinum type C and D neurotoxins to ganglioside and phospholipid. Novel insights into the receptor for clostridial neurotoxins. J. Biol. Chem., 280, 35164–35171. [DOI] [PubMed] [Google Scholar]
- 53.Sun J., Shaper N.L., Itonori S., Heffer-Lauc M., Sheikh K.A., Schnaar R.L. (2004) Myelin-associated glycoprotein (Siglec-4) expression is progressively and selectively decreased in the brains of mice lacking complex gangliosides. Glycobiology, 14, 851–857. [DOI] [PubMed] [Google Scholar]
- 54.Ikeda K., Taguchi R. (2010) Highly sensitive localization analysis of gangliosides and sulfatides including structural isomers in mouse cerebellum sections by combination of laser microdissection and hydrophilic interaction liquid chromatography/electrospray ionization mass spectrometry with theoretically expanded multiple reaction monitoring. Rapid Commun. Mass Spectrom., 24, 2957–2965. [DOI] [PubMed] [Google Scholar]