

Abstract
IMPORTANCE
Latino populations have one of the highest prevalences of type 2 diabetes worldwide.
OBJECTIVES
To investigate the association between rare protein-coding genetic variants and prevalence of type 2 diabetes in a large Latino population and to explore potential molecular and physiological mechanisms for the observed relationships.
DESIGN, SETTING, AND PARTICIPANTS
Whole-exome sequencing was performed on DNA samples from 3756 Mexican and US Latino individuals (1794 with type 2 diabetes and 1962 without diabetes) recruited from 1993 to 2013. One variant was further tested for allele frequency and association with type 2 diabetes in large multiethnic data sets of 14 276 participants and characterized in experimental assays.
MAIN OUTCOME AND MEASURES
Prevalence of type 2 diabetes. Secondary outcomes included age of onset, body mass index, and effect on protein function.
RESULTS
A single rare missense variant (c.1522G>A [p.E508K]) was associated with type 2 diabetes prevalence (odds ratio [OR], 5.48; 95% CI, 2.83–10.61; P = 4.4 × 10−7) in hepatocyte nuclear factor 1-α (HNF1A), the gene responsible for maturity onset diabetes of the young type 3 (MODY3). This variant was observed in 0.36% of participants without type 2 diabetes and 2.1% of participants with it. In multiethnic replication data sets, the p.E508K variant was seen only in Latino patients (n = 1443 with type 2 diabetes and 1673 without it) and was associated with type 2 diabetes (OR, 4.16; 95% CI, 1.75–9.92; P = .0013). In experimental assays, HNF-1A protein encoding the p.E508K mutant demonstrated reduced transactivation activity of its target promoter compared with a wild-type protein. In our data, carriers and noncarriers of the p.E508K mutation with type 2 diabetes had no significant differences in compared clinical characteristics, including age at onset. The mean (SD) age for carriers was 45.3 years (11.2) vs 47.5 years (11.5) for noncarriers (P = .49) and the mean (SD) BMI for carriers was 28.2 (5.5) vs 29.3 (5.3) for noncarriers (P = .19).
CONCLUSIONS AND RELEVANCE
Using whole-exome sequencing, we identified a single low-frequency variant in the MODY3-causing gene HNF1A that is associated with type 2 diabetes in Latino populations and may affect protein function. This finding may have implications for screening and therapeutic modification in this population, but additional studies are required.
The estimated prevalence of type 2 diabetes in Mexican adults was 14.4% in 2006,1 making it one of the leading causes of death in Mexico.2 Based on statistics from 1999–2002, the standardized prevalence of diagnosed diabetes was 10% in Mexican Americans and 5.2% in whites.3 Although environmental factors such as lifestyle and diet likely explain the majority of this health disparity, it was recently found that genetic variants in the gene SLC16A11 (NCBI NC_000017.11) were associated with higher rates of type 2 diabetes in Latinos.4 Latinos, defined as persons who trace their origin to Central and South America, and other Spanish cultures, fall on a continuum of Native American and European genetic ancestry.4 Identifying genetic factors associated with type 2 diabetes in Latino populations could increase understanding of its pathophysiology, improve risk prediction, and focus treatment choice based on knowledge of the underlying biology of the disease.
Type 2 diabetes is typically diagnosed after age 40 years, is caused by the combined action of genetic susceptibility and environmental factors, is associated with obesity, and is polygenic. Genome-wide association studies for typical type 2 diabetes forms have identified more than 70 distinct genetic loci carrying common variants that are associated with modest differences in prevalence of the disease.5–7 Because these common variants explain a small fraction of the estimated heritability, it is hypothesized that low-frequency or rare variants of strong effects, not captured by genome-wide association studies but amenable to sequencing approaches, contribute in a meaningful proportion to the genetic architecture of the disease. To date, low-frequency variants with near-complete penetrance have not been found in whole-exome sequencing studies of type 2 diabetes,8,9 although a recent whole-genome sequencing study found rare variants associated with type 2 diabetes prevalence in an Icelandic population.10
To explore the association of rare protein-coding genetic variants with type 2 diabetes in the Latino population, we performed whole-exome sequencing (which captures both common and rare genetic variants in the protein-coding regions of genes) on case-control studies composed of individuals of Mexican or another Latino ancestry, with replication in a separate multiethnic data set.
Methods
Study Design and Patients
This study was performed as part of the Slim Initiative in Genomic Medicine for the Americas (SIGMA) Type 2 Diabetes Consortium, whose goal is to characterize the genetic basis of type 2 diabetes in Mexican and Latin American populations drawn from 4 studies4,11–13 (Table 1, details of these studies are provided in the Supplement). All participant shad either Mexican or other Latino ancestry based on self-report and verification using principal component analysis of genotype data. Replication studies included individuals from a multiethnic study (Type 2 Diabetes Genetic Exploration by Next-Generation Sequencing in Multi-Ethnic Samples [T2D-GENES] and Genetics of T2D [GoT2D]) and an on going collection of Mexican participants from 18 indigenous groups for genetic studies (Diabetes in Mexico Study 2 [DMS2]) (eTable 1, details of these studies are provided in Supplement). Diagnosis of type 2 diabetes followed the American Diabetes Association criteria. Each participant provided written informed consent for genetic investigation. All contributing studies were approved by their respective local ethics committees.
Table 1.
Characteristics of Cohorts Comprising the SIGMA Type 2 Diabetes Whole-Exome Sequence Project
| Source | Sample Location | Study Design | No. of Participants | No (%) of Men | Mean (SD) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Age, y | Age of Onset, y | BMI | Fasting Glucose, mg/dL | Proportion With Native American Ancestry | |||||
| UNAM/INCMNSZ Diabetes Study,4 2014 | Mexico City, Mexico | Prospective cohort | |||||||
| Controls | 539 | 206 (38.2) | 55.0 (9.4) | 28.4 (3.8) | 86.4 (7.2) | 0.75 (0.10) | |||
| Type 2 diabetes | 533 | 216 (40.5) | 55.3 (12.5) | 43.8 (11.2) | 28.5 (4.4) | 0.78 (0.11) | |||
| Diabetes in Mexico Study,4 2014 | Mexico City, Mexico | Prospective cohort | |||||||
| Controls | 459 | 119 (25.9) | 52.4 (7.7) | 28.0 (4.6) | 90.1 (7.2) | 0.67 (0.18) | |||
| Type 2 diabetes | 509 | 168 (33.0) | 55.5 (11.1) | 47.2 (10.6) | 29.0 (5.4) | 0.79 (0.12) | |||
| Mexico City Diabetes Study,11,12 2005 and 2011 | Mexico City, Mexico | Prospective cohort | |||||||
| Controls | 526 | 204 (38.8) | 62.3 (7.5) | 29.4 (4.8) | 90.1 (9.0) | 0.69 (0.14) | |||
| Type 2 diabetes | 270 | 110 (40.7) | 64.0 (7.5) | 55.0 (9.7) | 29.9 (5.5) | 0.67 (0.15) | |||
| Multiethnic Cohort,1 2000 | Los Angeles, California | Prospective cohort | |||||||
| Controls | 438 | 212 (48.5) | 59.3 (7.2) | 26.9 (4.3) | 0.53 (0.09) | ||||
| Type 2 diabetes | 482 | 227 (47.0) | 58.7 (7.2) | NA | 29.8 (5.7) | NA | 0.58 (0.08) | ||
| Overall SIGMA | |||||||||
| Controls | 1962 | 742 (37.8) | 57.3 (8.9) | 28.3 (4.5) | 88.2 (9.0) | 0.67 (0.15) | |||
| Type 2 diabetes | 1794 | 719 (40.1) | 57.6 (10.6) | 47.5 (11.5) | 29.1 (5.2) | 0.71 (0.15) | |||
Abbreviations: BMI, body mass index, calculated as weight in kilograms divided by height in meters squared; NA, not available; UNAM/INCMNSZ, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Universidad Nacional Autónoma de México.
SI conversion factor: To convert fasting glucose from mg/dL to mmol/L, multiply by 0.0555.
Genetic Studies
Sample Selection and Whole-Exome Sequencing
In total, 3862 samples were selected for whole-exome sequencing from a larger data set of 8214 samples previously genotyped with the OMNI 2.5 array (Illumina).4 To increase representation of genetic variation not queried in studies of European populations, selection criteria for whole-exome sequencing was based on the proportion of Native American ancestry estimated from principal component analysis of genotype data (eMethods section and eFigures 1 and 2 in the Supplement). Whole-exome sequencing was performed on blood DNA from these samples using Sure-Select Human All Exonv2.0(Illumina),44-Mb–baited target. Raw reads were mapped with the Burrows-Wheeler Aligner, reprocessed with Picard to recalibrate base quality scores and perform local realignment around known indels. Genetic variants were called with the Genome Analysis Toolkit Unified Genotyper module14 and were filtered to remove likely artifacts using several quality-control metrics such as mean coverage, concordance of nonreference genotypes with array data, and missing rate as specified in the eMethods section in the Supplement. Independent replication was sought in whole-exome sequence data from the T2D-GENES and GoT2D projects, which together sequenced 13 098 individuals from 5 ethnic groups (Europeans, East Asians, African Americans, South Asians, and Latinos).
Statistical Analyses
We used the liability threshold model, which models participants as having an unobserved continuous phenotype called liability.15 We computed the residual value of the liability after accounting for the part that can be predicted by each participant’s age and body mass index (BMI) using LTSOFT software (http://www.hsph.harvard.edu/alkes-price/software).16 Significance was evaluated with the residual liabilities as outcome using an expedited mixed linear model,17 which adjusts for sex, ancestry (eFigure 3 in the Supplement), and relatedness via a variance-component matrix with 2-sided tests. Odds ratios (ORs) were estimated using logistic regression models on type 2 diabetes status adjusting for age, BMI, and ancestry as specified in the eMethods section in the Supplement. The experiment-wide statistical significance threshold was set to P < 5 × 10−8 to adjust for then umber of variants evaluated. In addition to single-variant testing, the sequence kernel association test18 and collapsing tests19 were used to test the possibility of genes and groups of genes associated to disease susceptibility via aggregation of rare variants.
Results of all functional experiments are expressed as means (SDs), and experiments were performed on at least 3 independent occasions unless otherwise specified. Statistical analyses were performed using the 2-tailed t test, and P <.05 was considered significant for these functional studies.
Functional Studies
Plasmids, Cell Culture, and Transfections
Details of functional studies are specified in the eMethods section in the Supplement. The human liver hepatocyte nuclear factor 1α (HNF1A) complementary DNA in expression vector pcDNA3.1/HisC (NCBI Entrez Gene BC104910.1) was used for all cell studies.20 Firefly luciferase reporter vectors (pGL3) included promoter sequences for the rat albumin (pGL3-RA), human HNF4A (NCBI Entrez Gene 3172) P2 (pGL3-HNF4AP2), and mouse Glut2 (pGL3-GLUT2) genes. Renilla luciferase reporter construct pRL-SV40 (GenBank AF025845.2) was used as an internal control. The HNF-1A mutants were made using the QuikChange Site-Directed XL Mutagenesis Kit (Stratagene). HeLa cells and MIN6 β-cells were grown as previously described,20,21 and transfected according to manufacturers’ recommendations using the Metafectene Pro (Biontex-USA) or Lipofectamine 2000 (Life Technologies), respectively.
Transactivation and Protein Expression Analyses
Transcriptional activity was measured 24 hours after transfection using the Dual-Luciferase Reporter Assay System (Promega Biotech) on a Chameleon luminometer (Hidex). To measure HNF-1A protein levels, transfected HeLa cells were lysed in passive lysis buffer (Promega Biotech) and proteins were analyzed (from 2.5 μg of total protein) by SDS-PAGE and immunoblotting using an HNF-1A-tag (anti-Xpress antibody, Life Technologies).
DNA Binding Studies
The HNF-1A protein was produced in a coupled in vitro transcription/translation System (TnT-T7, Promega Biotech). The level of binding of HNF-1A proteins to a radio labeled rat albumin oligonucleotide was investigated by electrophoretic mobility shift assays as previously described.22
Immunofluorescence
Analysis of nuclear vs cytosol localization of HNF-1A proteins was performed in 500 cells using an HNF-1A-tag (anti-Xpress antibody) and Alexa Fluor 488 (Life Technologies) essentially as reported previously.20
Results
Study Participants
Demographic and clinical characteristics of the 3756 participants in the discovery cohort are shown in Table 1. Only 2% of type 2 diabetes cases had onset before 25 years, and 81% of them were overweight or obese (BMI >25, calculated as weight in kilograms divided by height in meters squared).
Genetic Studies
Exome-wide Search for Low-Frequency Variants Associated With Type 2 Diabetes
Our hybrid selection libraries covered 76% of sequenced targets at 20x depth of coverage with a mean of 67.17x. The concordance of nonreference genotypes between the sequence data and the array data was 0.995. After quality control of sequence data, 1 190 196 variants were observed in the whole-exome sequencing data of 3756 samples (1794 type 2 diabetes cases and 1962 controls; eTable 2 in the Supplement). Of these, 264 995 variants were observed in at least 2 of our samples but absent in the 1000 Genomes Project23 and the Exome Sequencing Project24 (eTable 3 in the Supplement).
In our single-variant association analyses, a cluster of linked common missense variants in SLC16A11 were consistently associated with type 2 diabetes prevalence (P = 2.08 × 10−10) as had been previously reported in genome-wide association studies by the SIGMA T2D Consortium and others (eFigure 4A and eTable 4 in the Supplement).4,25
Among variants with minor allele frequency of less than 5%, a single missense variant departed from the null distribution (eFigure 4B in the Supplement). This variant encoded an NCBI NP_000536.5:p.E508K (p.E508K) substitution (NCBI NC_000012.12:c.1522G>A; chr12:121437091_G>A) in exon 8 of HNF1A, the gene responsible for the maturity onset diabetes of the young type 3 (MODY3) subtype of MODY3 (Mendelian Inheritance in Man No. 142410). The p.E508K variant was observed in 37 type 2 diabetes cases (1 in homozygous form) and in 7 participants without diabetes (OR, 5.48; 95% CI, 2.83–10.61; P = 4.4 × 10−7; Figure 1 and Figure 2 and eFigure 5 in the Supplement).
Figure 1. Discovery and Replication of the HNF1A p.E508K Variant.

Forest plot showing odds ratio estimates and 95% confidence intervals at p.E508K (squared boxes) from the 4 SIGMA studies, the SIGMA pooled mega-analysis, the replication studies, and the overall meta-analysis. Odds ratios for the meta-analyses are represented with a diamond. SIGMA mega-analysis represents the combined results from the 4 SIGMA studies. DMS indicates Diabetes in Mexico Study; MCDS, Mexico City Diabetes Study; MEC, Multiethnic Cohort; UIDS, Universidad Nacional Autónoma de México/Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán Diabetes Study; T2D-GENES, Type 2 Diabetes Genetic Exploration by Next-Generation Sequencing in Multi-Ethnic Samples.
aRepresents data from the current article.
Figure 2. The HNF-1A Protein With a Heat Map of Diabetes-Associated Mutations.
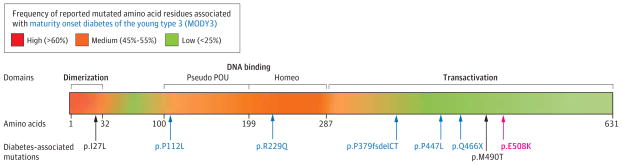
The dimerization, DNA binding, and transactivation domains of the HNF-1A protein49–51 are highlighted. The position of the p.E508K mutation is shown as well as a common variant (p.I27L), MODY3 mutations studied (p.P112L, p.R229Q, p.P379fsdelCT, p.P447L, p.Q466X), and a rare variant associated with type 2 diabetes (p.M490T). The overlaid heat map illustrates how many of the amino acid residues of each HNF-1A domain have been reported to be mutated and hence due to the monogenic diabetes form MODY3. Domain areas in red have a higher concentration of reported mutations than areas in orange and green. Pseudo POU indicates protein domain that includes short sequence motifs similar to regions in the POU family of transcriptional activators; Homeo, protein homeodomain that binds DNA in a sequence-specific manner.
In our replication effort, the p.E508K variant was found in the T2D-GENES Latino group26,27 but entirely absent in all other populations, showing a nominally significant association with increased prevalence for type 2 diabetes (7 affected carriers and 1 nonaffected carrier; OR, 5.61; 95% CI, 1.34–23.49; P = .0013). After de novo genotyping 1178 additional Mexican self-identified indigenous individuals (DMS2, further details are provided in the Supplement), we observed 9 affected carriers and 4 nonaffected carriers (OR, 3.50; 95% CI, 1.17–10.44; P = .0183). Combined, the 2 replication studies identified 15 affected carriers and 5 nonaffected controls (OR, 4.16; 95% CI, 1.75–9.92; P = .0013). Combining all available data yielded 52 affected carriers and 12 nonaffected controls (OR, 4.96; 95% CI, 2.93–8.38; an experiment-wide P = 2.39 × 10−9 ; Figure 1).
We found no evidence for p.E508K in the 1092 samples of the 1000 Genomes Project,23 the 6503 samples in the Exome Sequencing Project24 or in 11 160 non-Latino samples in the T2D-GENES and GoT2D data sets. Analysis of local ancestry in our data indicates that all p.E508K carriers in our studies carry at least 1 segment of inferred Native American ancestry (eTable 5 in the Supplement).
In group tests that included combinations of rare (MAF <1%) nonsynonymous, loss-of-function variants, or both in up to 15 469 genes (eTables 6 and 7 in the Supplement), we found no significant associations after removing the effect of the HNF1A p.E508K variant. The aggregated effect of these potentially functional variants in 2 gene-sets of 13 MODY genes and 70 previously implicated type 2 diabetes genes were similarly negative after removing the effect of the HNF1A p.E508K variant (eTables 8 and 9 in the Supplement).
Functional Studies
Mutations in HNF1A that cause MODY diabetes alter protein function through reduced transactivation, decreased binding to DNA, or disrupted nuclear localization.20 Because p.E508K is located in the HNF-1A transactivation domain, we investigated its effect on transactivation using a reporter construct assay in HeLa cells. Protein carrying p.E508K was compared with a wild-type HNF-1A variant as well as 4 other HNF-1A variants in the DNA-binding or transactivation domains: p.M490T, which has been observed in 1 patient with type 2 diabetes,28 and 3 mutations (p.P447L, p.P379fsdelCT, and p.R229Q) previously identified in patients with MODY3.29 The p.E508K mutant demonstrated lower transcriptional activity on the HNF-1A-responsive rat albumin promoter than wild-type HNF-1A (P < .0001) or p.M490T. However, the 3 MODY3 mutants showed greater reductions in transactivation (Figure 3). Similar reductions in p.E508K transcriptional activation were found in MIN6 cells (eFigure 6A in the Supplement), and using 2 different reporter constructs (GLUT2 and HNF4A promoters; eFigure 6B in the Supplement). The p.E508K mutant protein bound to an HNF-1A binding site-containing oligonucleotide with equal affinity to the wild-type protein (Figure 4 and eFigure 6C in the Supplement), whereas 2 MODY3-associated mutants with mutations in the DNA-binding domain, p.P112L and p.R229Q, demonstrated impaired DNA binding (Figure 4).20
Figure 3. Transcriptional Activation of HNF-1A p.E508K as Measured by the Expression of the Firefly Luciferase Reporter Gene.

HeLa cells were transient transfected with nonmutant or mutant HNF1A plasmids and reporter plasmids pGL3-RA and pRL-SV40. Measurements are given in fold activity relative to wild-type. Each point represents the mean (error bars indicate 95% CIs) of 9 readings. TA indicates variants that affect the transactivation domain; DNA bind, the DNA binding domain; and pcDNA3.1, the empty pcDNA3.1 vector. All values were P < .05 compared with wild-type activity.
Figure 4. DNA Binding of HNF-1A p.E508K to the Rat Albumin Promoter as Studied by Electrophoretic Mobility Shift Assay.
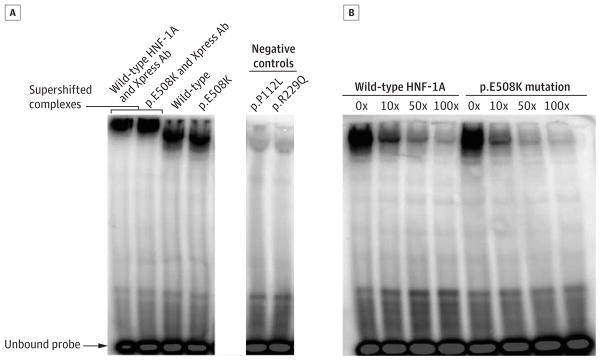
Xpress-epitope-tagged wild-type and p.E508K mutant proteins incubated with a radiolabeled DNA fragment containing the HNF-1A-binding site in the rat albumin promoter. A, Two HNF-1A mutants (p.P112L and p.R229Q) with impaired DNA-binding were included as negative controls. Addition of the anti-Xpress antibody induced a supershift (a reduction in mobility of protein-DNA complex due to antibody binding, relative to protein-DNA complex alone) for the DNA-protein complexes, confirming the identity of HNF-1A within the complexes. B, A competition assay was performed by adding increasing amounts (0x, 10x, 50x, or 100x) of radiolabeled DNA fragment, confirming the identity of the radiolabeled probe.
Compared with wild-type HNF-1A, the p.E508K mutant demonstrated slightly impaired nuclear targeting, with an increased proportion of cells displaying both cytosolic and nuclear staining. The shift in nuclear localization was less than that observed using the cytosol-retained HNF-1A mutant p.Q466X (Figure 5 and eFigure 6D in the Supplement). Expression of the p.E508K protein was 47.5% lower than that of wild-type HNF-1A (P = 1.03×10−5; eFigure 6E in the Supplement).
Figure 5. Intracellular Localization of HNF-1A p.E508K in Transiently Transfected HeLa cells and MIN6 β cells.
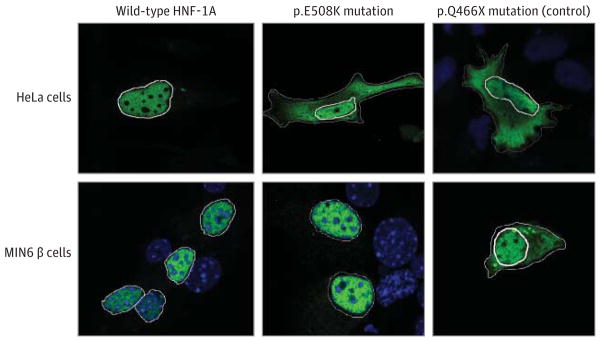
Cells were transfected for 48 hours and Xpress-epitope-tagged HNF-1A proteins detected with anti-Xpress antibody and Alexa488 (green). DNA staining (DAPI) is shown in blue. A previously reported HNF-1A mutant, p.Q466X, with impaired nuclear localization was included as a control. For the purpose of clarity, the nuclei have been marked with a solid white line. To illustrate cytosolic accumulation, the cell membrane has been marked with a dotted white line for mutants p.E508K and p.Q466X.
Clinical Characteristics of p.E508K Carriers
When comparing p.E508K carriers with noncarriers among the 3756 participants in our study, we did not observe statistically significant differences in the mean (SD) age of diabetes onset: 45.3 (11.2) years vs 47.5 (11.5) years, P = .49; BMI, 28.2; (5.5) vs 29.3 (5.3), P = .19; waist circumference in men, 92.9 (7.0) cm vs 99.3 (11.0) cm, P = .14 or women, 98.0 (13.9) cm vs 99.7 (13.9) cm, P = .64; or in fasting glucose levels, 176.5 (84.6) mg/dL vs 165.7 (75.6) mg/dL, P = .43 (To convert fasting glucose from mg/dL to mmol/L, multiply by 0.0555; Table 2 and Figure 6).
Table 2.
Phenotypic Characteristics of 3756 Participants From the SIGMA Studies According to Type 2 Diabetes Status and p.E508K Carrier Status
| Mean (SD)
|
P Value Carriers vs Noncarriers
|
|||||
|---|---|---|---|---|---|---|
| Type 2 Diabetes
|
Controls
|
|||||
| p.E508K (n = 37) | p.E508 (n = 1757) | p.E508K (n = 7) | p.E508 (n = 1955) | Type 2 Diabetes | Controls | |
| Age, y | 55.9 (9.6) | 57.6 (10.7) | 54.3 (9.2) | 57.3 (8.9) | .34 | .34 |
|
| ||||||
| Age at onset, y | 45.3 (11.2) | 47.5 (11.5) | .49 | |||
|
| ||||||
| Men | 11 | 707 | 3 | 739 | ||
|
| ||||||
| Women | 26 | 1050 | 4 | 1216 | ||
|
| ||||||
| Fasting glucose, mg/dL | 176.5 (84.6) | 165.7 (75.6) | 86.4 (9.0) | 88.2 (9.0) | .43 | .37 |
|
| ||||||
| BMI | 28.2 (5.5) | 29.3 (5.3) | 27.1 (3.5) | 28.3 (4.5) | .19 | .55 |
|
| ||||||
| Waist, cm | ||||||
|
| ||||||
| Men | 92.9 (7.0) | 99.3 (11.1) | 90.5 (19.8) | 97.6 (9.7) | .14 | .64 |
|
| ||||||
| Women | 98.0 (13.9) | 99.7 (13.9) | 95.5 (7.8) | 94.9 (13.3) | .64 | .88 |
|
| ||||||
| Waist to hip ratio, cm | ||||||
|
| ||||||
| Men | 0.96 (0.05) | 0.97 (0.07) | 0.96 (NA)a | 0.97 (0.10) | .54 | .88 |
|
| ||||||
| Women | 0.93 (0.07) | 0.92 (0.08) | 0.91 (0.05) | 0.90 (0.09) | .90 | .85 |
Abbreviations: BMI, body mass index, calculated as weight in kilograms divided by height in meters squared; NA, not applicable.
SI conversion factor: To convert fasting glucose from mg/dL to mmol/L, multiply by 0.0555.
Only 1 participant with this measurement.
Figure 6. Phenotypic Distribution of p.E508K Carriers.

The scatterplot shows the age of onset and the body mass index (BMI) for each p.E508K carrier (filled circle) with type 2 diabetes in the discovery studies with data on age of onset and BMI available (n = 29). The vertical and horizontal lines represent classical thresholds for the clinical diagnosis of MODY3 (age of onset <25 years and BMI<25). Histograms showing distributions of BMI and age of diabetes onset 1274 SIGMA discovery cohort participants (p.E508K carriers and noncarriers with Type 2 diabetes) are shown on the left and below the scatterplot. In the box-and-whisker plots, the central horizontal line indicates median, with box extremes indicating the first and third quartiles. The whiskers indicate maximum and minimum values after removal of outliers (unfilled circles).
Discussion
We performed whole-exome sequencing in 3756 individuals of Mexican and Mexican American ancestry and performed an exome-wide search for low-frequency and rare variants associated with type 2 diabetes. The only rare variant with a significant association with type 2 diabetes prevalence was the p.E508K variant in HNF1A, the gene responsible for MODY3. The effect size of the variant (OR, 4.96; 95% CI, 2.93–8.38) was the largest observed to date for any diabetes variant with a frequency more than 1 in 1000. This association was replicated in 2 independent cohorts of Latinos and Mexicans with an OR of similar magnitude. We also demonstrated, using transiently transfected cell models, reduced levels of transactivation activity for p.E508K compared with wild-type HNF-1A. As shown in binding assays, this reduction in activity was not driven by differences in DNA-binding affinity but may be attributable to reduced protein expression and altered nuclear localization of the mutant protein.
MODY is a monogenic cause of diabetes, which usually manifests at earlier ages (<25 years) and presents in nonobese patients.30 Each MODY family carries a rare coding mutation in 1 of 13 genes that has an autosomal dominant pattern of transmission.30 Mutations in the known MODY genes are thought to explain between 0.18% and 1.8% of all type 2 diabetes cases.31–34
The p.E508K variant has been reported in 2 published articles,35,36 both reporting on individuals with MODY. In 1 case, a family member had early onset diabetes (age 17 years), and carried both HNF1A p.E508K and a mutation in HNF4A, p.R80Q. The father from whom p.E508K was inherited was diagnosed with type 2 diabetes at age 57 years.35,36 The finding of these variants in patients with MODY suggested that they might be high-penetrance alleles. Our study in large populations without ascertainment bias for early-onset showed that p.E508K was associated with a 5-fold increase in prevalence, but incomplete penetrance. Moreover, in our study, carriers of p.E508K did not show early-onset of type 2 diabetes, were indistinguishable from the wider type 2 diabetes population in adiposity or glycemia, and thus did not fulfill classical MODY3 diagnostic criteria (Table 2, Figure 6). These data are consistent with the possibility that p.E508K is a weaker allele than some other MODY3 mutations and that ascertainment bias may have led to overestimation of the effects of this and other MODY mutations, as suggested previously.28
A private mutation (G319S) in HNF1A has been found in Oji-Cree populations associated with early-onset type 2 diabetes.37 Also, a very rare frameshift deletion in HNF1A, 290fsdelC, was recently associated with MODY and type 2 diabetes in the Icelandic population.10,38
Our study surveyed variants across the majority of protein-coding exons in a sizable population, providing the highest-resolution scan to date of the contribution of protein-coding genetic variation to type 2 diabetes. Our study had 80% power to detect variants with the OR and carrier frequency of p.E508K (5-fold and 1% in the population). For variants of higher frequency, our power was sufficient to detect a smaller effect (80% power for variants with frequency >2% and OR>3.3). We performed both single-variant analysis and burden tests that combined rare variants in each gene. Only 1 rare coding variant and 1 gene showed significant association with type 2 diabetes prevalence. These data suggest that low-frequency variants in coding regions explain only a small fraction of the heritability of type 2 diabetes.
Our study has limitations. Current exome-capture methods are imperfect. Additional low-frequency variants associated with type 2 diabetes might have been missed due to incomplete coverage of all human exons, and, by design, this technology does not detect variants in the noncoding majority of the genome. Although a 2% frequency of p.E508K among type 2 diabetes cases could translate into more than 100 000 carriers in Mexico alone, this number is still far from explaining the expected overall genetic contribution to type 2 diabetes. Although our study represents the largest published exome-based survey of type 2 diabetes to date, larger sample sizes will be needed to perform an adequately powered survey of variants at frequencies lower than 1%.39,40
The current study and a recent publication reporting an association of common variants in SLC16A11 with type 2 diabetes in Latinos4 demonstrate the value of studying diverse populations. The HNF1A p.E508K variant has not been reported in other whole-exome sequencing or candidate gene association studies for type 2 diabetes of European9,10,41 and Asian42–45 ancestry. We surveyed a total of 25 663 exomes in this study, both from our own study and collaborating consortia. The p.E508K variant was identified only in individuals from Mexico or in Latinos from the southern United States, indicating that this variant is only found at appreciable frequency in a tightly restricted subset of human populations. Further studies will be required to characterize the fine-scale geographic distribution of p.E508K and its association with type 2 diabetes prevalence in other Latino populations. Our results emphasize that systematic discovery of the genetic determinants of complex disease, especially for rare variants, will require surveys across a wide range of human populations.
The association of the p.E508K variant with type 2 diabetes prevalence in the Latino population has potential clinical implications. Approximately 4 in a thousand people in Latino populations carry p.E508K, and these individuals have a 5-fold increase in prevalence for type 2 diabetes (2.1% in cases, 0.35% in controls). Second, it is known that patients with MODY3 are sensitive to sulfonylureas,46 experiencing improved metabolic control on sulfonylurea therapy compared with insulin,47 in addition to improved quality of life due to reduced injections and capillary glucose measurements. Also, these patients have a 5-fold higher response to the sulfonylurea gliclazide than to metformin, which is the first-line drug of choice for the treatment of type 2 diabetes.48 If this was shown to be the case for carriers of p.E508K, it could motivate choice of sulfonylurea therapy for the estimated 2% of all Latino patients with type 2 diabetes who carry this variant. Because this response may be dependent on additional genetic or environmental factors, further studies are needed to determine whether metformin or a sulfonylurea should be the first line of treatment in these patients.
Conclusions
Using whole-exome sequencing, we identified a single low-frequency missense variant (p.E508K) in HNF1A, the gene responsible for a monogenic, early-onset form of diabetes (MODY3), that was associated with type 2 diabetes prevalence in general populations of Latinos. This rare variant was associated with a 5-fold increase in the prevalence of type 2 diabetes, but it was not associated with an early-onset form of diabetes, and, in our data, affected carriers were clinically indistinguishable from the wider type 2 diabetes population. In vitro, p.E508K negatively affected transcriptional activation, protein expression, and nuclear localization. Further research is warranted to evaluate the clinical relevance of these findings, including the benefits of selective population screening and the choice of genotype-guided therapeutic regimens.
Supplementary Material
Acknowledgments
Funding/Support: The work was conducted as part of the Slim Initiative for Genomic Medicine, a project funded by the Carlos Slim Health Institute in Mexico. The UNAM/INCMNSZ Diabetes Study was supported by Consejo Nacional de Ciencia y Tecnologiía grants 138826, 128877, CONACT-SALUD 2009-01-115250, and a grant from Dirección General de Asuntos del Personal Académico, UNAM, IT 214711. The Diabetes in Mexico Study was supported by Consejo Nacional de Ciencia y Tecnología grant 86867 and by Instituto Carlos Slim de la Salud, A.C. The Mexico City Diabetes Study was supported by National Institutes of Health (NIH) grant R01HL24799 and by the Consejo Nacional de Ciencia y Tenologia grants 2092, M9303, F677-M9407, 251M, and 2005-C01-14502, SALUD 2010-2-151165. The Multiethnic Cohort was supported by NIH grants CA164973, CA054281, and CA063464. The Singapore Chinese Health Study was funded by the National Medical Research Council of Singapore under its individual research grant scheme and by NIH grants R01 CA55069, R35 CA53890, R01 CA80205, and R01 CA144034. The Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) project was supported by NIH grants U01DK085526 and U01DK085501. The San Antonio Mexican American Family Studies (SAMAFS) were supported by R01 DK042273, R01 DK047482, R01DK053889, R01 DK057295, P01 HL045522, and a Veterans Administration Epidemiologic grant (R.A.D). The University of Bergen, Research Council of Norway, KG Jebsen Foundation, Helse Vest, and European Research Council funded the Norwegian team. Dr Mercader was supported by Sara Borrell Fellowship from the Instituto Carlos III, Spain. Dr Estrada was supported by The Netherlands Organization for Scientific Research under the Rubicon fellowship 825.12.023.
Authors: The following investigators of the SIGMA Type 2 Diabetes Consortium take authorship responsibility for the study results: Karol Estrada, PhD; Ingvild Aukrust, PhD; Lise Bjørkhaug, PhD; Noël P. Burtt, PhD; Josep M. Mercader, PhD; Humberto García-Ortiz, PhD; Alicia Huerta-Chagoya, MSc; Hortensia Moreno-Macías, PhD; Geoffrey Walford, MD; Jason Flannick, PhD; Amy L. Williams, PhD; María J. Gómez-Vázquez, BSc; Juan C. Fernandez-Lopez, MSc; Angélica Martínez-Hernández, PhD; Federico Centeno-Cruz, PhD; Elvia Mendoza-Caamal, MD; Cristina Revilla-Monsalve, PhD; Sergio Islas-Andrade, MD, PhD; Emilio J. Córdova, PhD; Xavier Soberón, PhD; María E. González-Villalpando, MD; E. Henderson, MD; Lynne R. Wilkens, DrPH; Loic Le Marchand, MD, PhD; Olimpia Arellano-Campos, MD, PhD; Maria L. Ordóñez-Sánchez, BSc; Maribel Rodríguez-Torres, BSc; Rosario Rodríguez-Guillén, MSc; Laura Riba, MSc; Laeya A. Najmi, MSc; Suzanne B.R. Jacobs, PhD; Timothy Fennell, BSc; Stacey Gabriel, PhD; Pierre Fontanillas, PhD; Craig L. Hanis, PhD; Donna M. Lehman, PhD; Christopher P. Jenkinson, PhD; Hanna E. Abboud, MD; Graeme I. Bell, PhD; Maria L. Cortes, PhD; Michael Boehnke, PhD; Clicerio González-Villalpando, MD; Lorena Orozco, MD, PhD; Christopher A. Haiman, ScD; Teresa Tusié-Luna, MD, PhD; Carlos A. Aguilar-Salinas, MD, PhD; David Altshuler, MD, PhD; Pål R. Njølstad, MD, PhD; Jose C. Florez, MD, PhD; Daniel G. MacArthur, PhD.
Affiliations of Authors: Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, Massachusetts (Estrada, Burtt, Mercader, Flannick, Williams, Jacobs, Fontanillas, Altshuler, Florez, MacArthur); Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston (Estrada); Department of Medicine, Harvard Medical School, Boston, Massachusetts (Estrada, Walford, Altshuler, Florez, MacArthur); KG Jebsen Center for Diabetes Research, Department of Clinical Science, University of Bergen, Bergen, Norway (Aukrust, Bjørkhaug, Najmi, Njølstad); Department of Pediatrics, Haukeland University Hospital, Bergen, Norway (Bjørkhaug, Njølstad); Department of Biomedicine, University of Bergen, Bergen, Norway (Aukrust); Center for Human Genetic Research and Diabetes Research Center (Diabetes Unit), Massachusetts General Hospital, Boston (Mercader, Walford, Altshuler, Florez); Joint BSC-CRG-IRB Research Program in Computational Biology, Barcelona Supercomputing Center, Barcelona, Spain (Mercader); Instituto Nacional de Medicina Genómica, Tlalpan, Mexico City, Mexico (García-Ortiz, Fernandez-Lopez, Martínez-Hernández, Centeno-Cruz, Mendoza-Caamal, Córdova, Soberón, Orozco); Instituto de Investigaciones Biomédicas, UNAM Unidad de Biología Molecular y Medicina Genómica, UNAM/INCMNSZ, Coyoacán, Mexico City, Mexico (Huerta-Chagoya, Riba, Tusié-Luna); Universidad Autónoma Metropolitana, Tlalpan, Mexico City, Mexico (Moreno-Macías); Centro de Estudios en Diabetes, Unidad de Investigacion en Diabetes y Riesgo Cardiovascular, Centro de Investigacion en Salud Poblacional, Instituto Nacional de Salud Publica, Mexico City, Mexico (M. E. González-Villalpando, C. González-Villalpando); Department of Molecular Biology, Harvard Medical School, Boston, Massachusetts (Flannick, Altshuler); Department of Biological Sciences, Columbia University, New York, New York (Williams); Department of Biostatistics, Center for Statistical Genetics, University of Michigan, Ann Arbor (Boehnke); Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles (Henderson, Haiman); Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Sección XVI, Tlalpan, Mexico City, Mexico (Gómez-Vázquez, Arellano-Campos, Ordóñez-Sánchez, Rodríguez-Torres, Rodríguez-Guillén, Tusié-Luna, Aguilar-Salinas); Department of Genetics, Harvard Medical School, Boston, Massachusetts (Altshuler); Center for Human Genetic Research, Massachusetts General Hospital, Boston (Altshuler); Department of Biology, Massachusetts Institute of Technology, Cambridge (Altshuler); Unidad de Investigación Médica en Enfermedades Metabólicas, CMN SXXI, Instituto Mexicano del Seguro Social, Mexico City (Revilla-Monsalve, Islas-Andrade); Epidemiology Program, University of Hawaii Cancer Center, Honolulu (Wilkens, Le Marchand); Center for Medical Genetics and Molecular Medicine, Haukeland University Hospital, Bergen, Norway (Najmi); The Genomics Platform, The Broad Institute of Harvard and MIT, Cambridge, Massachusetts (Fennell, Gabriel); Human Genetics Center, University of Texas Health Science Center at Houston (Hanis); Department of Medicine, University of Texas Health Science Center at San Antonio (Lehman, Jenkinson, Abboud); Department of Human Genetics, University of Chicago, Chicago, Illinois (Bell); Department of Medicine, University of Chicago, Chicago, Illinois (Bell); Broad Institute of Harvard and MIT, Cambridge, Massachusetts (Cortes).
Author Contributions: Dr Estrada had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Estrada, Aukrust, Bjørkhaug, Burtt, Orozco, Haiman, Tusié-Luna, Altshuler, Njølstad, MacArthur, Williams, Islas-Andrade, M. González-Villalpando, Hanis, Florez, Boehnke.
Acquisition, analysis, or interpretation of data: Estrada, Aukrust, Bjørkhaug, Burtt, Mercader, Garcia-Ortiz, Huerta-Chagoya, Moreno-Macías, C. González-Villalpando, Orozco, Salinas, Altshuler, Njølstad, MacArthur, Flannick, Cortes, Williams, Gómez-Vázquez, Fernandez-Lopez, Martínez-Hernández, Centeno-Cruz, Mendoza-Caamal, Revilla-Monsalve, Córdova, Soberón, Henderson, Wilkens, Marchand, Arellano-Campos, Ordóñez-Sánchez, Torres, Rodríguez-Guillén, Riba, Walford, Najmi, Jacobs, Fennell, Gabriel, Fontanillas, Jiménez-Morales, Hanis, Florez, Lehman, Jenkinson, Abboud, Bell, Boehnke.
Drafting of the manuscript: Estrada, Mercader, Garcia-Ortiz, Huerta-Chagoya, Moreno-Macías, Orozco, Altshuler, Njølstad, MacArthur, Cortes, Martínez-Hernández, Centeno-Cruz, Islas-Andrade, Córdova, Henderson, Arellano-Campos, Najmi, Gabriel, Jiménez-Morales.
Critical revision of the manuscript for important intellectual content: Estrada, Aukrust, Bjørkhaug, Burtt, Mercader, C. González-Villalpando, Orozco, Haiman, Tusié-Luna, Salinas, Altshuler, Njølstad, MacArthur, Flannick, Williams, Gómez-Vázquez, Fernandez-Lopez, Mendoza-Caamal, Revilla-Monsalve, Soberón, M. González-Villalpando, Wilkens, Marchand, Torres, Rodríguez-Guillén, Riba, Walford, Jacobs, Fennell, Gabriel, Fontanillas, Hanis, Florez, Lehman, Jenkinson, Abboud, Bell, Boehnke.
Statistical analysis: Estrada, Mercader, Garcia-Ortiz, Huerta-Chagoya, Moreno-Macías, Orozco, Haiman, Altshuler, MacArthur, Flannick, Williams, Gómez-Vázquez, Fernandez-Lopez, Walford, Najmi, Fennell, Fontanillas, Boehnke.
Obtained funding: Orozco, Tusié-Luna, Altshuler, Njølstad, Cortes, Soberón, Wilkens, Hanis, Florez, Lehman, Boehnke.
Administrative, technical, or material support: Aukrust, Bjørkhaug, Burtt, Orozco, Tusié-Luna, Salinas, Altshuler, Njølstad, MacArthur, Flannick, Cortes, Fernandez-Lopez, Martínez-Hernández, Centeno-Cruz, Mendoza-Caamal, Revilla-Monsalve, Islas-Andrade, Córdova, Ordóñez-Sánchez, Torres, Rodríguez-Guillén, Riba, Jiménez-Morales, Florez, Lehman, Jenkinson, Abboud, Bell.
Study supervision: Aukrust, Bjørkhaug, Burtt, C. González-Villalpando, Orozco, Tusié-Luna, Altshuler, Njølstad, MacArthur, M. González-Villalpando, Riba, Gabriel, Florez.
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest and none were reported.
Role of the Sponsors: The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
The SIGMA Type 2 Diabetes Consortium: Writing Team: Karol Estrada, PhD, Ingvild Aukrust, PhD, Lise Bjørkhaug, PhD, Noël P. Burtt, PhD, Josep M. Mercader, PhD, Humberto García-Ortiz, PhD, Alicia Huerta-Chagoya, MSc, Hortensia Moreno-Macías, PhD, Geoffrey Walford, MD, Jason Flannick, PhD, Amy L. Williams, PhD, Michael Boehnke, PhD, Clicerio González-Villalpando, MD, Lorena Orozco, MD, PhD, Christopher A. Haiman, ScD, Teresa Tusié-Luna, MD, PhD, Carlos A. Aguilar-Salinas, MD, PhD, David Altshuler, MD, PhD, Pål R. Njølstad, MD, PhD, Jose C. Florez, MD, PhD, Daniel G. MacArthur, PhD.
Analysis Team: Karol Estrada, PhD, Alicia Huerta-Chagoya, MSc, Humberto García-Ortiz, PhD, Hortensia Moreno-Macías, PhD, Josep M. Mercader, PhD, Jason Flannick, PhD, Amy L. Williams, PhD, María J. Gómez-Vázquez, BSc, Juan C. Fernandez-Lopez, MSc, Noël P. Burtt, PhD, Carlos A. Aguilar-Salinas, MD, PhD, Lorena Orozco, MD, PhD, Teresa Tusié-Luna, MD, PhD, David Altshuler, MD, PhD, Jose C. Florez, MD, PhD, Daniel G. MacArthur, PhD; Whole-Exome Sequenced cohorts: Diabetes in Mexico Study: Humberto García-Ortiz, PhD, Angélica Martínez-Hernández, PhD, Federico Centeno-Cruz, PhD, Elvia Mendoza-Caamal, MD, Cristina Revilla-Monsalve, PhD, Sergio Islas-Andrade, MD, PhD, Emilio J. Córdova, PhD, Xavier Soberón, PhD, Lorena Orozco, MD, PhD. Mexico City diabetes study: Clicerio González-Villalpando, MD, María E. González-Villalpando, MD. Multiethnic cohort study: Christopher A. Haiman, ScD, Brian E. Henderson, MD, Lynne R. Wilkens, DrPH, Loic Le Marchand, MD, PhD. UNAM/INCMNSZ diabetes study: Olimpia Arellano-Campos, MD, PhD, Alicia Huerta-Chagoya, MSc, Maria L. Ordóñez-Sánchez, BSc, Maribel Rodríguez-Torres, BSc, Rosario Rodríguez-Guillén, MSc, Laura Riba, MSc, Teresa Tusié-Luna, MD, PhD, Carlos A. Aguilar-Salinas, MD, PhD.
Functional Studies: Laeya A. Najmi, MSc, Ingvild Aukrust, PhD, Lise Bjørkhaug, PhD, Suzanne B. R. Jacobs, PhD, Pål R. Njølstad, MD, PhD.
Whole-Exome Sequencing: Noël P. Burtt, PhD, Timothy Fennell, BSc, Broad Genomics Platform, Stacey Gabriel, PhD.
Replication Studies: T2D-GENES Consortium: Jason Flannick, PhD, Pierre Fontanillas, PhD, Craig L. Hanis, PhD, Donna M. Lehman, PhD, Christopher P. Jenkinson, PhD, Hanna E. Abboud, MD, Graeme I. Bell, PhD, Jose C. Florez, MD, PhD, David Altshuler, MD, PhD, Michael Boehnke, PhD. Diabetes in Mexico study 2: Humberto García-Ortiz, PhD, Angélica Martínez-Hernández, PhD, Emilio J. Córdova, PhD, Silvia Jiménez-Morales, PhD, Federico Centeno-Cruz, PhD, Elvia Mendoza-Caamal, MD, Cristina Revilla-Monsalve, PhD, Sergio Islas-Andrade, MD, PhD, Xavier Soberón, PhD, Lorena Orozco, MD, PhD.
Scientific and Project Management: Noël P. Burtt, PhD, Maria L. Cortes, PhD.
Steering Committee: David Altshuler, MD, PhD, Jose C. Florez, MD, PhD, Christopher A. Haiman, ScD, Carlos A. Aguilar-Salinas, MD, PhD, Clicerio González-Villalpando, MD, Lorena Orozco, MD, PhD, Teresa Tusié-Luna, MD, PhD.
Additional Contribution: Researchers of the DMS2 study thank Olaf Iván Corro Labra and José Luis de Jesus García Ruíz from the “Comisión Nacional para el Desarrollo de los Pueblos Indígenas” for their support in sample collection, for which they were not compensated.
References
- 1.Villalpando S, de la Cruz V, Rojas R, et al. Prevalence and distribution of type 2 diabetes mellitus in Mexican adult population. Salud Publica Mex. 2010;52(suppl 1):S19–S26. doi: 10.1590/s0036-36342010000700005. [DOI] [PubMed] [Google Scholar]
- 2.Barquera S, Tovar-Guzmán V, Campos-Nonato I, González-Villalpando C, Rivera-Dommarco J. Geography of diabetes mellitus mortality in Mexico. Arch Med Res. 2003;34(5):407–414. doi: 10.1016/S0188-4409(03)00075-4. [DOI] [PubMed] [Google Scholar]
- 3.Cowie CC, Rust KF, Byrd-Holt DD, et al. Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National Health And Nutrition Examination Survey 1999–2002. Diabetes Care. 2006;29(6):1263–1268. doi: 10.2337/dc06-0062. [DOI] [PubMed] [Google Scholar]
- 4.Williams AL, Jacobs SB, Moreno-Macías H, et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506(7486):97–101. doi: 10.1038/nature12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42(7):579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diabetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46(3):234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albrechtsen A, Grarup N, Li Y, et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia. 2013;56(2):298–310. doi: 10.1007/s00125-012-2756-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lohmueller KE, Sparsø T, Li Q, et al. Whole-exome sequencing of 2000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet. 2013;93(6):1072–1086. doi: 10.1016/j.ajhg.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steinthorsdottir V, Thorleifsson G, Sulem P, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet. 2014;46(3):294–298. doi: 10.1038/ng.2882. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzo C, Williams K, Gonzalez-Villalpando C, Haffner SM. The prevalence of the metabolic syndrome did not increase in Mexico City between 1990–1992 and 1997–1999 despite more central obesity. Diabetes Care. 2005;28(10):2480–2485. doi: 10.2337/diacare.28.10.2480. [DOI] [PubMed] [Google Scholar]
- 12.Hunt KJ, Gonzalez ME, Lopez R, Haffner SM, Stern MP, Gonzalez-Villalpando C. Diabetes is more lethal in Mexicans and Mexican-Americans compared to Non-Hispanic whites. Ann Epidemiol. 2011;21(12):899–906. doi: 10.1016/j.annepidem.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolonel LN, Henderson BE, Hankin JH, et al. A multiethnic cohort in Hawaii and Los Angeles. Am J Epidemiol. 2000;151(4):346–357. doi: 10.1093/oxfordjournals.aje.a010213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falconer DS. The inheritance of liability to diseases with variable age of onset, with particular reference to diabetes mellitus. Ann Hum Genet. 1967;31(1):1–20. doi: 10.1111/j.1469-1809.1967.tb01249.x. [DOI] [PubMed] [Google Scholar]
- 16.Zaitlen N, Pasaniuc B, Patterson N, et al. Analysis of case-control association studies with known risk variants. Bioinformatics. 2012;28(13):1729–1737. doi: 10.1093/bioinformatics/bts259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang HM, Sul JH, Service SK, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13(4):762–775. doi: 10.1093/biostatistics/kxs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases. Am J Hum Genet. 2008;83(3):311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bjørkhaug L, Sagen JV, Thorsby P, Søvik O, Molven A, Njølstad PR. Hepatocyte nuclear factor-1 alpha gene mutations and diabetes in Norway. J Clin Endocrinol Metab. 2003;88(2):920–931. doi: 10.1210/jc.2002-020945. [DOI] [PubMed] [Google Scholar]
- 21.Aukrust I, Bjørkhaug L, Negahdar M, et al. SUMOylation of pancreatic glucokinase regulates its cellular stability and activity. J Biol Chem. 2013;288(8):5951–5962. doi: 10.1074/jbc.M112.393769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bjørkhaug L, Ye H, Horikawa Y, Søvik O, Molven A, Njølstad PR. MODY associated with two novel hepatocyte nuclear factor-1alpha loss-of-function mutations (P112L and Q466X) Biochem Biophys Res Commun. 2000;279(3):792–798. doi: 10.1006/bbrc.2000.4024. [DOI] [PubMed] [Google Scholar]
- 23.Abecasis GR, Auton A, Brooks LD, et al. 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara K, Fujita H, Johnson TA, et al. DIAGRAM consortium. Genome-wide association study identifies three novel loci for type 2 diabetes. Hum Mol Genet. 2014;23(1):239–246. doi: 10.1093/hmg/ddt399. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell BD, Kammerer CM, Blangero J, et al. Genetic and environmental contributions to cardiovascular risk factors in Mexican Americans. Circulation. 1996;94(9):2159–2170. doi: 10.1161/01.cir.94.9.2159. [DOI] [PubMed] [Google Scholar]
- 27.Hanis CL, Ferrell RE, Barton SA, et al. Diabetes among Mexican Americans in Starr County, Texas. Am J Epidemiol. 1983;118(5):659–672. doi: 10.1093/oxfordjournals.aje.a113677. [DOI] [PubMed] [Google Scholar]
- 28.Flannick J, Beer NL, Bick AG, et al. Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nat Genet. 2013;45(11):1380–1385. doi: 10.1038/ng.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellard S. Hepatocyte nuclear factor 1 alpha (HNF-1 alpha) mutations in maturity-onset diabetes of the young. Hum Mutat. 2000;16(5):377–385. doi: 10.1002/1098-1004(200011)16:5<377::AID-HUMU1>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 30.Molven A, Njølstad PR. Role of molecular genetics in transforming diagnosis of diabetes mellitus. Expert Rev Mol Diagn. 2011;11(3):313–320. doi: 10.1586/erm.10.123. [DOI] [PubMed] [Google Scholar]
- 31.Eide SA, Raeder H, Johansson S, et al. Prevalence of HNF1A (MODY3) mutations in a Norwegian population. Diabetic Med. 2008;25(7):775–781. doi: 10.1111/j.1464-5491.2008.02459.x. [DOI] [PubMed] [Google Scholar]
- 32.Kropff J, Selwood MP, McCarthy MI, Farmer AJ, Owen KR. Prevalence of monogenic diabetes in young adults. Diabetologia. 2011;54(5):1261–1263. doi: 10.1007/s00125-011-2090-z. [DOI] [PubMed] [Google Scholar]
- 33.Ledermann HM. Maturity-onset diabetes of the young (MODY) at least ten times more common in Europe than previously assumed? Diabetologia. 1995;38(12):1482. doi: 10.1007/BF00400611. [DOI] [PubMed] [Google Scholar]
- 34.Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY)? Diabetologia. 2010;53(12):2504–2508. doi: 10.1007/s00125-010-1799-4. [DOI] [PubMed] [Google Scholar]
- 35.Bellanné-Chantelot C, Carette C, Riveline JP, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes. 2008;57(2):503–508. doi: 10.2337/db07-0859. [DOI] [PubMed] [Google Scholar]
- 36.Forlani G, Zucchini S, Di Rocco A, et al. Double heterozygous mutations involving both HNF1A/MODY3 and HNF4A/MODY1 genes. Diabetes Care. 2010;33(11):2336–2338. doi: 10.2337/dc10-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hegele RA, Cao H, Harris SB, Hanley AJ, Zinman B. The hepatic nuclear factor-1alpha G319S variant is associated with early-onset type 2 diabetes in Canadian Oji-Cree. J Clin Endocrinol Metab. 1999;84(3):1077–1082. doi: 10.1210/jcem.84.3.5528. [DOI] [PubMed] [Google Scholar]
- 38.Kristinsson SY, Thorolfsdottir ET, Talseth B, et al. MODY in Iceland is associated with mutations in HNF-1alpha and a novel mutation in NeuroD1. Diabetologia. 2001;44(11):2098–2103. doi: 10.1007/s001250100016. [DOI] [PubMed] [Google Scholar]
- 39.Zuk O, Schaffner SF, Samocha K, et al. Searching for missing heritability. Proc Natl Acad Sci U S A. 2014;111(4):455–464. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322(5903):881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winckler W, Weedon MN, Graham RR, et al. Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007;56(3):685–693. doi: 10.2337/db06-0202. [DOI] [PubMed] [Google Scholar]
- 42.Fang QC, Zhang R, Wang CR, Lin X, Xiang KS. Scanning HNF-1 alpha gene mutation in Chinese early-onset and/or multiplex diabetes pedigrees [In Chinese] Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2004;21(4):329–334. [PubMed] [Google Scholar]
- 43.Nishigori H, Yamada S, Kohama T, et al. Mutations in the hepatocyte nuclear factor-1 alpha gene (MODY3) are not a major cause of early-onset non-insulin-dependent (type 2) diabetes mellitus in Japanese. J Hum Genet. 1998;43(2):107–110. doi: 10.1007/s100380050049. [DOI] [PubMed] [Google Scholar]
- 44.Tonooka N, Tomura H, Takahashi Y, et al. High frequency of mutations in the HNF-1alpha gene in non-obese patients with diabetes of youth in Japanese and identification of a case of digenic inheritance. Diabetologia. 2002;45(12):1709–1712. doi: 10.1007/s00125-002-0978-3. [DOI] [PubMed] [Google Scholar]
- 45.Yorifuji T, Fujimaru R, Hosokawa Y, et al. Comprehensive molecular analysis of Japanese patients with pediatric-onset MODY-type diabetes mellitus. Pediatr Diabetes. 2012;13(1):26–32. doi: 10.1111/j.1399-5448.2011.00827.x. [DOI] [PubMed] [Google Scholar]
- 46.Søvik O, Njølstad P, Følling I, Sagen J, Cockburn BN, Bell GI. Hyperexcitability to sulphonylurea in MODY3. Diabetologia. 1998;41(5):607–608. doi: 10.1007/s001250050956. [DOI] [PubMed] [Google Scholar]
- 47.Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med. 2009;26(4):437–441. doi: 10.1111/j.1464-5491.2009.02690.x. [DOI] [PubMed] [Google Scholar]
- 48.Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362(9392):1275–1281. doi: 10.1016/S0140-6736(03)14571-0. [DOI] [PubMed] [Google Scholar]
- 49.Baumhueter S, Mendel DB, Conley PB, et al. HNF-1 shares three sequence motifs with the POU domain proteins and is identical to LF-B1 and APF. Genes Dev. 1990;4(3):372–379. doi: 10.1101/gad.4.3.372. [DOI] [PubMed] [Google Scholar]
- 50.Chouard T, Blumenfeld M, Bach I, Vandekerckhove J, Cereghini S, Yaniv M. A distal dimerization domain is essential for DNA-binding by the atypical HNF1 homeodomain. Nucleic Acids Res. 1990;18(19):5853–5863. doi: 10.1093/nar/18.19.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tronche F, Yaniv M. HNF1, a homeoprotein member of the hepatic transcription regulatory network. BioEssays. 1992;14(9):579–587. doi: 10.1002/bies.950140902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.