

Abstract
Introduction
Neurofibrillary tangles (NFT) composed of Tau are hallmarks of neurodegeneration in Alzheimer disease. Transgenic mice expressing full-length pro-aggregant human Tau (2N4R Tau-ΔK280, termed TauΔK) or its repeat domain (TauRD-ΔK280, TauRDΔK) develop a progressive Tau pathology with missorting, phosphorylation, aggregation of Tau, loss of synapses and functional deficits. Whereas TauRDΔK assembles into NFT concomitant with neuronal death, TauΔK accumulates into Tau pretangles without overt neuronal loss. Both forms cause a comparable cognitive decline (with onset at 10mo and 12mo, respectively), which is rescued upon switch-off of transgene expression. Since methylene blue (MB) is able to inhibit Tau aggregation in vitro, we investigated whether MB can prevent or rescue Tau-induced cognitive impairments in our mouse models. Both types of mice received MB orally using different preventive and therapeutic treatment protocols, initiated either before or after disease onset. The cognitive status of the mice was assessed by behavior tasks (open field, Morris water maze) to determine the most successful conditions for therapeutic intervention.
Results
Preventive and therapeutic MB application failed to avert or recover learning and memory deficits of TauRDΔK mice. Similarly, therapeutic MB treatment initiated after onset of cognitive impairments was ineffective in TauΔK mice. In contrast, preventive MB application starting before onset of functional deficits preserved cognition of TauΔK mice. Beside improved learning and memory, MB-treated TauΔK mice showed a strong decrease of insoluble Tau, a reduction of conformationally changed (MC1) and phosphorylated Tau species (AT180, PHF1) as well as an upregulation of protein degradation systems (autophagy and proteasome). This argues for additional pleiotropic effects of MB beyond its properties as Tau aggregation inhibitor.
Conclusions
Our data support the use of Tau aggregation inhibitors as potential drugs for the treatment of AD and other tauopathies and highlights the need for preventive treatment before onset of cognitive impairments.
Electronic supplementary material
The online version of this article (doi:10.1186/s40478-015-0204-4) contains supplementary material, which is available to authorized users.
Keywords: Alzheimer disease, Tau mouse model, Behavior, Methylene blue, Therapy
Introduction
Alzheimer disease (AD) is a fatal dementia with progressive accumulation of protein aggregates composed of amyloid-beta (Aβ) and the microtubule-associated Tau protein in brain structures relevant for learning and memory [1]. Despite much progress made in recent years, the details of the neurotoxic cascade are still a matter of debate. However, it is assumed that the accumulation of protein aggregates is closely linked to neurotoxicity and degeneration. Current treatment options for AD are still limited to acetylcholinesterase inhibitors (donepezil, galantamine, rivastigmine), NMDA (N-methyl-D-aspartate) receptor antagonists (memantine) or other supportive strategies ameliorating the symptoms, rather than treating the underlying causes of neurodegeneration [2]. Recently, a number of promising drug candidates targeting different stages of Aβ pathology failed in clinical trials [3,4]. In addition the spreading of Tau pathology (rather than that of Aβ pathology) correlates closely with loss of synapses and neurons, the best correlate of cognitive decline [5-7]. As a result, Tau has become a promising target for therapeutic intervention.
The process of Tau aggregation, from oligomers to neurofibrillary tangles (NFT) is suspected to induce neurodegeneration. Support for this hypothesis came from Tau transgenic mouse models expressing aggregation-prone Tau mutants. Examples are P301S [8-10], P301L [11-13] and ΔK280 [14,15], or, as negative controls, non-aggregating Tau mutants, e.g. ΔK280-PP [16,17]. Aggregation-prone Tau mutants were identified from cases of sporadic and hereditary forms of frontotemporal dementia (FTD). The deletion mutation ΔK280 was originally detected in a sporadic FTD case [18,19] and more recently in an Alzheimer patient [20].
In mice, expression of either full-length pro-aggregant human Tau-ΔK280 (TauΔK) or its repeat domain (TauRDΔK) under control of the CaMKIIα promoter leads to progressive neuropathology. Both models share several common aspects, including the formation of co-aggregates composed of exogenous human and endogenous mouse Tau, loss of synapses accompanied by functional deficits such as cognitive decline and electrophysiological impairments. The most important similarity of both models is that the functional deficits can be recovered after switching-off the expression of pro-aggregant Tau. As a consequence of switch-off, Tau co-aggregates undergo structural reorganization with release and clearance of exogenous Tau, whereas the aggregated endogenous mouse Tau persists for several months. By contrast, the two models clearly differ in the onset and severity of Tau pathology. TauRDΔK is the more toxic Tau species, presumably because of its greater propensity for aggregation based on β-structure. Thus, expression of TauRDΔK leads to early development of silver-positive NFT and neuronal loss inside the hippocampus formation (~5mo), which are nearly absent in TauΔK mice. In addition cognitive decline and loss of synapses occurs earlier in TauRDΔK (~10mo) than in TauΔK mice (~12mo).
Despite the variations between the TauRDΔK and TauΔK mouse models, the studies summarized above share the essential fact that tauopathies as such are largely reversible, provided that the amyloidogenic Tau is removed [14,15]. Thus strategies and substances counteracting Tau aggregation are promising candidates for the treatment of AD and other tauopathies.
Several screens identified low molecular weight compounds, which act as Tau aggregation inhibitors [21-24]. One compound of particular interest is methylene blue (MB). The inhibitory effect of MB applies not only to Tau aggregation [24,25] but also to other proteins involved in neurodegeneration such as huntingtin [26], TDP-43 and alpha-synuclein [27], Aβ [28] and prion protein [29]. MB is an FDA approved drug and has a long history of medical use, mainly as an antiseptic and anti-malaria compound. Biochemically, MB is a bioavailable member of the phenothiazine family with high water solubility. It is a redox-cycling compound, relatively non-toxic and able to pass the blood-brain barrier [30,31]. The first anti-Tau therapy with MB in humans was reported by Wischik et al. in 2008 [32]. In this phase 2 clinical trial, a daily dose of 3x 60 mg Rember™ (a derivative of the oxidized form of MB) over 1 year appeared to show a slow-down of cognitive decline in mild and moderate AD patients. Since none of these studies have been published in a peer-reviewed journal, a skeptical attitude towards the presented results remains in the field. Meanwhile Rember™ was replaced by its more sophisticated successor LMTX™, which is optimized in terms of enhanced tolerability and higher absorption in the intestine as it contains the reduced “leuco” form of MB [33]. Up to now three phase-3 trials have been announced for LMTX™, one for the treatment of bvFTD or Pick disease and two studies for AD. First results are expected for late 2015 or early 2016 (as stated by TauRx Therapeutics [34]).
In parallel a number of preclinical studies in various model organisms were performed to elucidate MB’s neuroprotective mechanism of action. However, the results published remain controversial. While MB ameliorates the neurotoxic phenotype in a C. elegans model of tauopathy with aggregated Tau in neurons [35], it was reported to be ineffective in zebrafish expressing P301L mutant Tau without aggregated Tau [36]. In mice, MB showed beneficial phenotypic effects in models of Huntington disease [26] and in models of AD [37-39], but failed in models of amyotrophic lateral sclerosis [40,41]. In addition MB’s effect on neuronal pathology is still under debate. In case of Tau pathology there is a debate on whether MB acts on soluble or insoluble Tau species [39,42,43] and whether additional mechanisms such as enhancing general protein clearance via autophagy or proteasomes, improving energy metabolism, or other effects on ATP/GTP binding proteins play a role [38,44-46].
The present study aims to contribute to the discussion how MB can ameliorate Tau-induced pathological changes and (more importantly) cognitive impairment in inducible pro-aggregant mice. In addition our purpose is to refine treatment paradigms with respect to treatment initiation, concentration and duration. To this end pro-aggregant TauΔK and TauRDΔK mice were treated orally with MB using different long- and short-term treatment protocols to determine the most successful conditions for therapeutic intervention. The effect of MB on cognition was evaluated in different behavior tasks (open field and Morris water maze) and the brain pathology of MB-treated and untreated mice were compared to gain insights into MB’s mode of action.
Materials and methods
Generation of pro-aggregant Tau transgenic mice
Transgenic mice expressing pro-aggregant human full-length Tau (2N4R, Tau441, with deletion mutant ΔK280, 441-1 = 440 amino acids, here termed TauΔK) or pro-aggregant Tau repeat domain (construct K18, 4R, residues 244-372, termed TauRDΔK) were generated as described [16,17] (Additional file 1: Figure S1). Briefly, responder mice carrying either the Tau-transgene together with a luciferase reporter were crossbred with the CaMKIIα-tTA transactivator mice [47] to obtain double-transgenic mice with constitutive expression of luciferase and pro-aggregant TauΔK or TauRDΔK. All bigenic offspring were heterozygous and had an identical C57BL/6 genetic background. Non-transgenic littermates were used as controls. The transgene expression of bigenic mice started roughly around birth (0mo) concomitant with the onset of CaMKIIα activity. The expression was measured in vivo by bioluminescence imaging of luciferase activity. All animal procedures were approved in accordance with the German Animal Welfare Act.
In vivo bioluminescence imaging of luciferase activity
In vivo bioluminescence imaging to quantify luciferase activity and estimate expression strength of Tau transgenes was performed using an Ivis Lumina II system (Caliper Life Science) as described [15]. Briefly, mice received an intraperitoneal injection of 150 mg/kg D-luciferin/PBS (Caliper Life Science) 10 min prior to imaging and were anesthetized using 2% isoflurane (Abbott). A sequence of images was collected using a highly sensitive CCD camera. The bioluminescence emission was analyzed and quantified by the Living Image 4.0 software (Caliper Life Science).
Oral methylene blue treatment of Tau transgenic mice
Methylene blue (MB, C16H18CIN3S * 3 H2O, Sigma) was administered ad libitum via the drinking water supplemented with saccharin (Huxol, 1 tablet per 200 ml). Mice received a daily MB-dose of 40 or 20 mg/kg based on a daily drinking volume of ~5-6 ml and a body weight of 25-35 g. The concentration of the MB drinking solution was 0.25 mM or 0.5 mM, respectively. TauΔK mice were treated using a daily dose of 20 mg/kg MB. In all cases Tau expression started at birth (~0mo); one group of TauΔK mice received MB for 14.5mo starting at 1.5mo of age. A second group was administered MB for 6mo, starting at 9mo of age and a third group received MB for 3mo, starting at 15mo of age. TauRDΔK mice received a daily dose of 20 mg/kg MB for 3mo and 14.5mo starting at 12mo or 1.5mo of age, respectively. Another group of TauRDΔK mice was treated with 40 mg/kg MB for 3mo, starting at 12mo of age. MB-treated groups were accompanied by groups of vehicle treated (H2O + saccharin) Tau transgenic littermates and by groups of wild-type littermates (MB or vehicle treated). Each group was composed of 6-11 age and gender matched animals.
Behavior tasks
Housing conditions: prior to behavior experiments, mice were single-housed under standard conditions with food, water and MB ad libitum in a room with inverted 12 h light/dark cycle to assure testing in the nocturnal phase of the animals.
Open field test: the open field consists of a 50 x 50 cm arena divided into 20 x 20 cm center, a 5 cm wall zone and a 10 cm border zone. Each mouse was placed into the center of the box and could freely explore the arena for 15 min while being tracked by a video system (Viewer II, Biobserve). The following parameters were analyzed: activity, distance moved, mean velocity, time spent in the center zone and distance to wall. Activity was defined as amount of active time (%) during the duration of stay, in which the mouse’s movement speed exceeded the activity threshold. The activity threshold defines a certain velocity limit to distinguish active from inactive behavior (1 cm/s).
Morris water maze (MWM): before starting the MWM experiment, a 2 days pretraining protocol was conducted to habituate the mice to swimming and climbing onto a hidden platform. To avoid any interference with the MWM learning, the pretraining was performed in a different room and apparatus than used for the MWM. Spatial memory abilities were examined in the standard hidden-platform acquisition and retention version of the Morris water maze [48]. A 180 cm circular pool was filled with water opacified with non-toxic white paint (Biofa Primasol 3011). The tank was divided into four quadrants: target (T), right adjacent (R), opposite (O), and left adjacent (L). A 15 cm round platform was hidden 1 cm beneath the surface of the water at a fixed position in the center of the target quadrant. The pool was surrounded by landmarks attached to the walls to facilitate orientation. Each mouse performed 4 swimming trials per day (maximum duration 90s, 10 min inter-trial interval) for five consecutive days. The time required to locate the hidden platform (escape latency), path length and swimming speed were determined. On acquisition day 3, 4, 5, as well as 2 days after the end of acquisition, a probe trial was conducted without platform. Acquisition and probe trials were recorded and analyzed by the Viewer II video tracking system (Biobserve).
Biochemical analysis of brain tissue
Sarcosyl-extraction, total protein preparation and western blots were performed as described previously [14,15]. Depending on the 1st antibody, 2-20 μg of total protein or 3 μl of sarcosyl extraction lysates from brain tissue were loaded for the detection with pan-Tau antibody K9JA (1:20000, Dako A-0024), the human Tau specific antibody TauY9 (1:2000, Enzo), phospho-Tau antibodies 12E8 (pSer262/pSer356, 1:500, Elan), PHF1 (pSer396/pSer404, 1:500, gift from Dr. P. Davies) and antibodies against synaptophysin (1:20000, Sigma), synapsin1 (1:2000, Novus Biologicals), PSD95 (1:2000, Dianova), Beclin (1:10000, Santa Cruz), HSC70 (1:10000, Abcam), lamp2a (1:2000, Abcam), PSMD13 (1:2000, Abcam). Blots were normalized by the concentration of β-actin (1:20000, Sigma).
Histology
Immunohistochemistry was performed on 5 μm paraffin sections as described [14,15]. The following antibodies were used for light microscopy: MC1 (conformational epitope, aa 5-15 + 312-322, 1:10, gift of Dr. P. Davies, Albert Einstein College, NY) and AT180 (pThr231/pSer235, 1:1000, Pierce). Secondary antibodies as well as the avidin-biotinylated peroxidase complex were provided by the Vectastain Universal Elite ABC kit (Vector laboratories) and DAB (Dako) was used to visualize the antibody labeling. Gallyas silver impregnation was performed as described [14,15]. Immunofluorescence: the primary mouse monoclonal OXPHOS antibody cocktail (1:250, MitoScience) followed by a secondary anti-mouse DyLight 650 antibody (1:500, Thermo Fisher Scientific) was applied. Nuclei counterstain was performed with Syto13 (1:4000, Life Technologies GmbH). Stainings imaged by a LSM510 Meta confocal microscope (Zeiss) using lasers, beam splitters, and filters according to the fluorophores.
Statistics
Open field test: MB-treated, untreated and control groups were compared by one-way ANOVA with post hoc Newman-Keuls multiple comparison test. Morris water maze: MB-treated, untreated and control groups were compared by two-way repeated ANOVA followed by a post hoc Fisher LSD multiple comparison test. Asterisks indicate differences between treated and untreated TauΔK mice (MWM acquisition). For analysis of probe trials a two-tailed t-test against chance level (25%) or a one-way ANOVA with post-hoc Newman-Keuls multiple comparison test was done. Protein amounts were compared by one way ANOVA with post hoc Newman-Keuls multiple comparison test or by an unpaired two-tailed t-test. Numbers of samples are indicated in figure legends. All data are presented as group mean values with standard error of mean (SEM), the accepted level of significance was p < 0.05. Statistical comparisons were performed using STATISTICA 10.0 software (StatSoft), graphs were designed using Prism 5.0 software (GraphPad). *: p < 0.05, **: p < 0.01, ***: p < 0.001.
Results
Characteristics of pro-aggregant Tau transgenic mice
Inducible mice with constant expression of full-length pro-aggregant TauΔK or repeat domain pro-aggregant TauRDΔK develop a progressive neuropathology including prominent cognitive deficits. Importantly, cognitive deficits as well as synaptic impairments recover after switching off the expression of human Tau [14,15], demonstrating that Tau-induced pathology can be reversed in principle. These studies provide the rationale for treatment of pro-aggregant mice using Tau-directed drugs.
From 3 months (mo) of age onwards (or after 3mo of Tau expression), repeat domain TauRDΔK mice show a pronounced neuropathology, especially in terms of Tau aggregation and neuronal death. By comparison, the brain pathology is much less pronounced in full-length TauΔK mice. These mice develop a pre-tangle pathology indicated by the conformation-dependent antibody MC1 [49] but lack a silver-positive NFT pathology and neuronal loss in the hippocampus. The difference is consistent with the fact that TauRDΔK lacks the flanking regions (Additional file 1: Figure S1), which leads to a higher β-propensity, causing efficient aggregation and a stronger neurotoxicity than full-length TauΔK at comparable expression levels. Note that TauRDΔK cannot react with antibody MC1, since it lacks part of the epitope; the same holds for other diagnostic antibodies (e.g. AT8) with epitopes outside the repeat domain. Therefore, the appearance of MC1 or AT8 reactivity in TauRDΔK mice originates from endogenous mouse Tau, which has been transformed to a pathological state, triggered by exogenous TauRDΔK. By contrast, the epitope of antibody 12E8 (pS262 + pS356) lies within the repeats and is present in all Tau variants.
Due to the specific design of the transgenic DNA the expression of human TauΔK and TauRDΔK can be deduced from luciferase activity by bioluminescence imaging (BLI) in living mice. This opens the possibility to preselect animals with comparable Tau expression prior to experiments in order to reduce inter-individual variations, which is important since Tau levels influence the severity of pathology [50].
Pharmacokinetic properties of MB in mice
To check bioavailability and lifetime of MB in mice, MB levels in plasma and brain were measured after intravenous (i.v.) application of 30 mg/kg MB or oral application of 45 mg/kg MB (Additional file 2: Figure S2) in wild-type C57BL/6 mice (n = 3 per time point). MB levels were evaluated by quantification of tetramethylthioninium-ion (TMT-ion) concentrations using liquid chromatography - mass spectrometry (LC-MS/MS). TMT-ion equals MB without chloride and 3*H2O.
After i.v. administration, peak concentrations in plasma and brain were observed at 5 to 15 min post application and pharmacokinetic analyses over 24 h revealed MB half-lives of t1/2 = 4.4 h in plasma and t1/2 = 3.0 h in brain (Additional file 2: Figure S2a). Intravenous application of MB led to a reduced general condition and apathy of the mice, which may point to an acute toxic effect of the bolus. In contrast, oral administration of MB was well-tolerated and led to peak concentrations in plasma and brain within the first 2 hours post application. Pharmacokinetic analyses over 24 h exhibited MB half-lives of t1/2 = 4.7 h in plasma and t1/2 = 30.6 h in brain. The oral bioavailability was determined at ~18% (Additional file 2: Figure S2b). Furthermore, the results show a concentration of MB inside the brain, with ~30-fold and ~60-fold higher MB levels in brain tissue compared to plasma at 24 h post i.v. or oral application, respectively.
MB application strategies
The goal of the study was to optimize MB treatment conditions in order to get the maximum beneficial effect on learning and memory performance of Tau transgenic mice. To this end we followed different treatment strategies (Figure 1). The major difference between the various treatment protocols is the initial time point of intervention, namely before or after onset of cognitive impairments, which start typically at ~12mo of age in TauΔK mice and at ~10mo of age in TauRDΔK mice. The key question was, how much Tau pathology can be allowed while still preventing cognitive decline by MB treatment.
Figure 1.
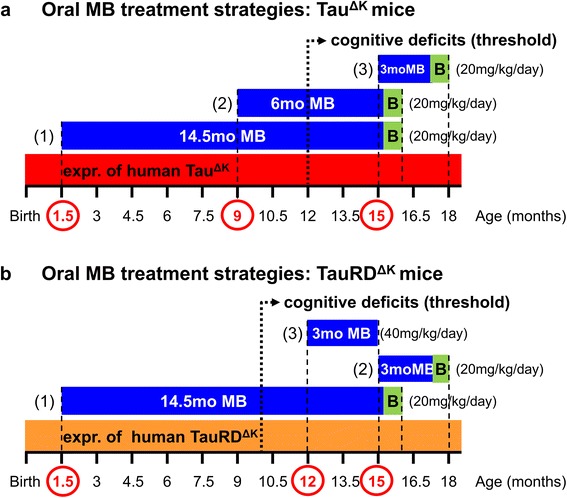
Oral MB treatment strategies of mice. (a) Full-lengthTauΔK mice received MB for 14.5mo starting at 1.5mo of age, before onset of Tau pathological changes and cognitive decline (protocol 1, 1.5 mo Tau pathology, 14.5 mo preventive MB, 20 mg/kg), for 6mo starting at 9mo of age, shortly before the onset of cognitive impairments (protocol 2, 9 mo Tau pathology, 6 mo preventive MB, 20 mg/kg) and for 3mo starting at 15mo of age, at a time point, when learning and memory deficits are already present (protocol 3, 15 mo Tau pathology, 3 mo therapeutic MB, 20 mg/kg). The constant expression of TauΔK throughout the entire life-span in the absence of doxycycline is depicted as red bar. The onset of progressive cognitive failure starting ~12 months of age is represented by the dotted arrow. (b) TauRDΔK received MB for 14.5mo starting at the age of 1.5mo, before onset of Tau pathology and cognitive impairment (protocol 1, 1.5 mo Tau pathology, 14.5 mo preventive MB, 20 mg/kg) and for 3mo starting at 15mo of age, after the onset of cognitive decline (protocol 2, 15mo Tau pathology, 3mo therapeutic MB, 20 mg/kg). An excessive MB dose of 40 mg/kg was applied for 3mo at the age of 12-15mo (protocol 3, excessive MB, 40 mg/kg). The constant expression of TauRDΔK throughout life-span in the absence of doxycycline is depicted as orange bar. The onset of cognitive deficits starting ~10mo of age is represented by the dotted arrow. Periods and initiation of MB treatment are indicated by blue bars and red circles on the time axis, including period of behavioral testing during the final month (green box). The daily MB dose (20 or 40 mg/kg) for each treatment protocol is given in brackets. MB was administered via the drinking water. MB: methylene blue; mo: months; B: behavior tests; expr.: expression.
MB application strategies for pro-aggregant full-length TauΔK mice
In preventive treatment protocol (1), MB application was initiated at 1.5mo of age and continued for 14.5mo beyond the anticipated onset of cognitive deficits typically around ~12mo of age in this mouse strain. The aim was to target Tau-induced changes very early on, before any neuropathological and cognitive changes occur (Figure 1a, preventive MB for 14.5mo).
In preventive treatment protocol (2), MB administration was initiated at 9mo of age, a time point ~3mo before the expected onset of cognitive impairment but with presence of early TauΔK-induced pathological changes such as conformational change, missorting and phosphorylation of Tau (Figure 1a, preventive MB for 6mo).
Finally, therapeutic treatment protocol (3) aimed to test whether therapeutic intervention by MB after onset of cognitive decline has the potential to rescue the existing neuropathology and phenotype (Figure 1a, therapeutic MB for 3mo). Therefore MB application was initiated at 15mo of age (~3mo after onset of cognitive impairments) and continued for 3mo. In this respect, treatment protocol (3) was closely analogous to the aforementioned Tau switch-off experiments, which resulted in a complete rescue of functional deficits [14,15]. All TauΔK mice were treated using an oral daily dose of 20 mg/kg MB, which was administered via the drinking water.
MB application strategies for pro-aggregant repeat domain TauRDΔK mice
Similar to TauΔK animals, TauRDΔK mice received MB (20 mg/kg) for 14.5mo, starting at 1.5mo of age, long before the appearance of Tau pathology and cognitive impairments (Figure 1b, protocol 1, preventive MB for 14.5mo).
In therapeutic treatment protocol (2), MB (20 mg/kg) application started at 15mo of age and continued for 3mo (Figure 1b, therapeutic MB for 3mo). At the age of 15mo, TauRDΔK mice exhibit strong neuropathological changes as described above and show severe learning and memory deficits.
Finally, TauRDΔK mice received an excessive MB dose of 40 mg/kg for 3mo at the age of 12-15mo to further evaluate the effect of MB on TauRDΔK aggregation (Figure 1b, therapeutic protocol (3), excessive MB for 3mo).
After MB treatment, mice were tested for their general motor and exploration behavior using an open field test and for their spatial reference learning and memory performance in a Morris water maze (MWM) test. In the open field test, no differences between MB-treated and untreated pro-aggregant Tau mice or wild-type (WT) mice were observed for average velocity and distance covered throughout all experimental setups. In addition swimming speed did not differ between these groups in the MWM test. Thus, we exclude that motor deficiencies of pro-aggregant Tau mice account for the differences detected in behavioral readouts (see below). This issue is of concern since some published Tau-transgenic mice developed motor deficits caused by transgene expression in the spinal cord due to Thy-1.2 or PrP promoters [11,13], which is avoided here by the use of the CaMKIIα promoter.
MB treatment of pro-aggregant full-length TauΔK mice
Preventive treatment starting 11mo before cognitive decline
In this protocol, MB treatment started shortly after birth and long before the expected onset of cognitive decline (~12mo), and continued beyond this for a total of 14.5mo (including the final month of behavioral testing, Figure 1a).
Regarding open field activity, the 14.5mo MB treatment resulted in significant group differences, such that untreated TauΔK mice were less active than WT animals or MB-treated TauΔK mice (Figure 2a, p = 0.004, Additional file 3: Figure S3). In contrast, no group differences were observed for anxiety-related parameters (time in center, distance to wall), suggesting that preventive MB treatment did not have anxiolytic effects (Additional file 4: Figure S4).
Figure 2.
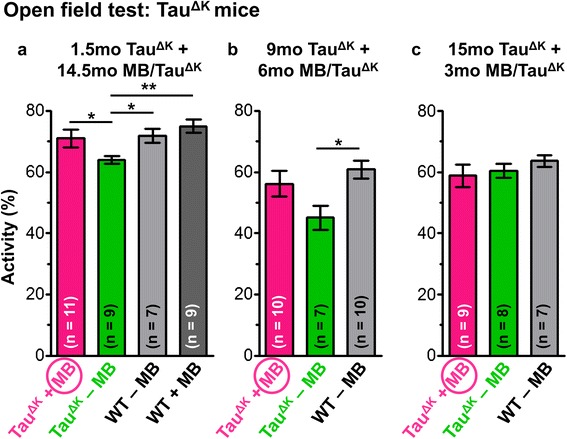
Exploration behavior of MB-treated TauΔK mice. Total activity (%) within 15 min is analyzed by an open field test after various MB treatment periods. (a) Preventive MB treatment for (a) 14.5mo and for (b) 6mo results in preservation of exploration behavior of TauΔK mice similar to wild-type (WT) animals, whereas untreated TauΔK mice show a significantly reduced activity. By contrast no group-differences are observed after therapeutic MB treatment for 3mo (c). Bars represent mean values ± SEM. Statistics: one-way analysis of variances with post-hoc Newman-Keuls multiple comparisons test. Asterisks indicate significant differences between groups; *: p < 0.05; **: p < 0.01; mo: months.
During MWM acquisition, WT mice and MB-treated pro-aggregant full-length TauΔK mice showed a superior learning performance over untreated TauΔK mice (WT vs. TauΔK - MB: p = 0.025). Importantly, MB-treated TauΔK mice demonstrated a similar learning efficiency than WT animals (WT vs. TauΔK + 14.5mo MB: p = 0.532) and performed significantly better than untreated TauΔK at day 4 and day 5 (TauΔK - MB vs. TauΔK + 14.5mo MB; day 4: p = 0.025; day 5: p = 0.018) (Figure 3a). In probe trials, MB-treated TauΔK mice exhibited a higher preference for the target quadrant and a more precise localization of the target platform position in comparison to untreated TauΔK mice (Additional file 5: Figure S5). The results indicate a preservation of cognitive abilities upon preventive MB treatment initiated before the onset of Tau pathological changes and cognitive decline. Interestingly, WT mice receiving MB for 14.5mo, showed a slightly superior learning performance over untreated WT mice (Figure 3a), pointing towards an additional beneficial effect of MB on brain metabolism.
Figure 3.
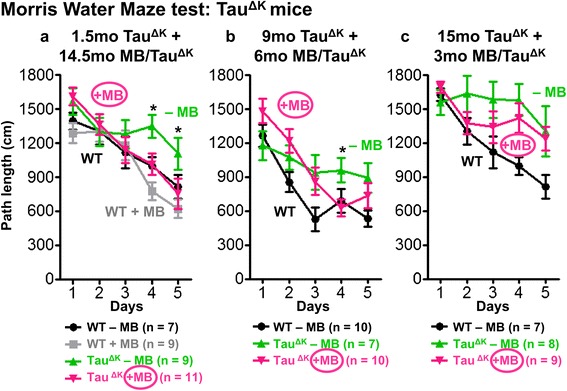
Spatial learning of MB-treated TauΔK mice. Morris water maze (MWM) acquisition shows spatial learning abilities as indicated by path lengths (cm) of mice after MB application using different preventive and therapeutic treatment paradigms. Significant group differences are observed between untreated TauΔK mice and WT animals throughout all experiments, demonstrating impaired learning abilities upon expression of pro-aggregant Tau (a-c). By contrast TauΔK mice treated with MB for 14.5mo (a) and 6mo (b) behave comparable to controls indicating preservation of cognitive functions. Short-term MB treatment for 3mo has no beneficial impact on the learning performance of TauΔK mice (c). Data shows mean path length ± SEM. Statistics: two-way repeated measure analysis of variances with post hoc Fishers LSD multiple comparisons test. Asterisks indicate differences between MB-treated and untreated TauΔK mice; *: p < 0.05; n: number of mice.
MB application for 14.5mo (started at 1.5mo of age) reduced insoluble TauΔK and endogenous mouse Tau, compared with untreated TauΔK mice (Figure 4). Similarly, a decrease of conformationally-changed Tau (epitope MC1, 5-15 + 312-322, Figure 5, Additional file 6: Figure S6) and a reduction of phosphorylated Tau (epitope AT180, pThr231 + pSer235 and epitope PHF1, pSer396 + pSer404) was observed after 14.5mo MB treatment (Figure 5, Figure 6, Additional file 6: Figure S6). By contrast, Tau phosphorylated at the KXGS motifs inside the repeat domain (epitope 12E8, pSer262 + pSer356) was increased in MB-treated animals (Figure 6). Note that Tau phosphorylation at the KXGS motifs causes detachment of Tau from microtubules, but also protects Tau against aggregation [51].
Figure 4.
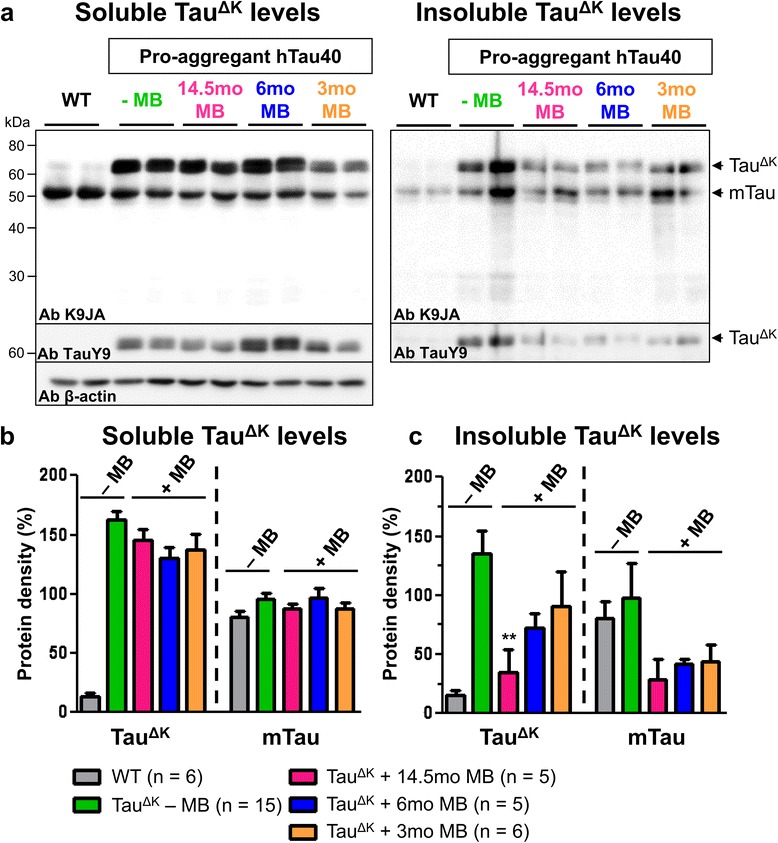
MB treatment reduces detergent insoluble TauΔK and mouse Tau. Sarcosyl-extraction of soluble and insoluble Tau from cortex tissue. (a) Protein levels of soluble and insoluble Tau species in untreated TauΔK mice compared to TauΔK mice receiving MB for 14.5mo, 6mo and 3mo detected by the pan-Tau antibody K9JA and the human Tau specific antibody TauY9. MB administration reduces levels of detergent-insoluble human and mouse Tau. (b) Quantification of soluble Tau levels in cortex lysates shows a minor but non-significant decrease of soluble Tau species upon MB treatment. (c) Quantification of insoluble Tau levels in cortex lysates demonstrates a clear decrease of insoluble human and mouse Tau in MB-treated mice. Bars represent mean protein densities (%) ± SEM; statistics: one-way analysis of variances with post-hoc Newman-Keuls multiple comparisons test. Asterisks indicate significant differences in comparison to untreated TauΔK mice; **: p < 0.01; Ab: antibody; mTau: mouse Tau; n, number of samples.
Figure 5.
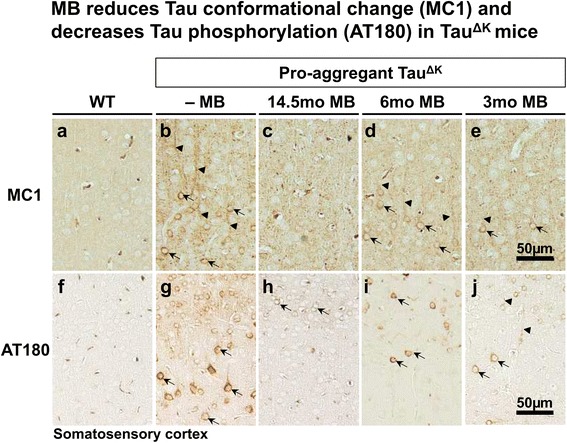
Histological analysis of conformationally changed and phosphorylated Tau in MB-treated mice. (a-e) MC1 immunoreactivity (epitope 5-15 + 312-322) indicates a pathological Tau conformation. MC1 positive neurons are prominent in somatosensory cortex (SSCx) of untreated TauΔK mice with missorting of Tau to cell soma (arrows) and apical dendrites (arrowheads). In contrast MB treatment, especially preventive MB treatment for 14.5mo, clearly reduces MC1 immunoreactivity in TauΔK mice. (f-j) Histological analysis of phosphorylated Tau using the AT180 antibody (dual phosphorylation epitope pThr231 + pSer235). Untreated TauΔK mice display a massive mislocalization of phosphorylated Tau to cell soma (arrows) and apical dendrites (arrowheads) of SSCx neurons, whereas MB treatment diminishes the extent of AT180 phosphorylation. Scale bar: 50 μm.
Figure 6.
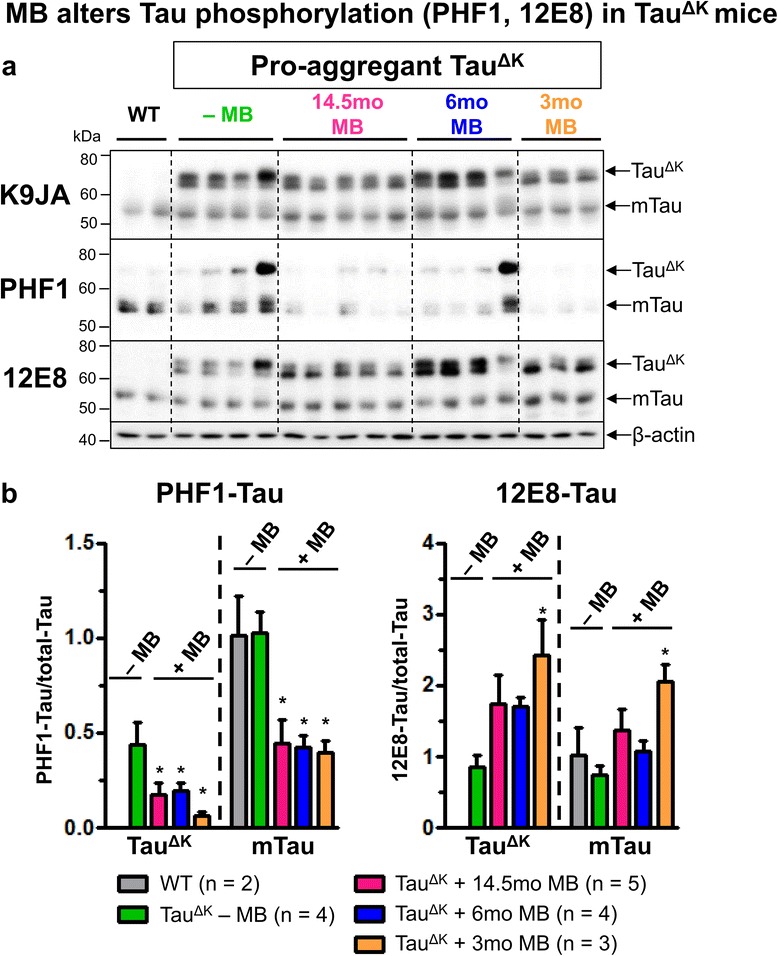
MB application alters levels of phosphorylated Tau. (a) Levels of total Tau (pan-Tau antibody K9JA), PHF1 phosphorylated (epitope pSer396/pSer404) and 12E8 (epitope pSer262/pSer356) phosphorylated Tau in cortex homogenates of MB-treated and untreated TauΔK mice. MB administration for 14.5mo, 6mo and 3mo results in a decrease of PHF1 phospho-Tau as compared to untreated TauΔK mice. By contrast an increase of 12E8 phospho-Tau inside the repeat domain of Tau is observed after MB treatment, indicating detachment from microtubules. (b) Determination of relative protein densities by quantification of (a). Levels of phosphorylated Tau were normalized to total Tau levels. Bars represent mean values ± SEM. Statistics: one-way analysis of variances with post-hoc Newman-Keuls multiple comparisons test. Asterisks indicate significant differences in comparison to untreated TauΔK mice; *: p < 0.05; n, number of samples.
Previously, we described a close relationship between expression of pro-aggregant Tau, synaptic failure inside the hippocampus formation and cognitive decline [14,15]. While untreated pro-aggregent TauΔK mice showed a consistent reduction in pre- and postsynaptic proteins (i.e. synapsin 1, synaptophysin, PSD95), MB treatment for 14.5mo preserved pre- and postsynaptic protein levels (Figure 7), suggesting a protective effect of MB on the synaptic integrity which likely contributes to the preservation of cognition.
Figure 7.
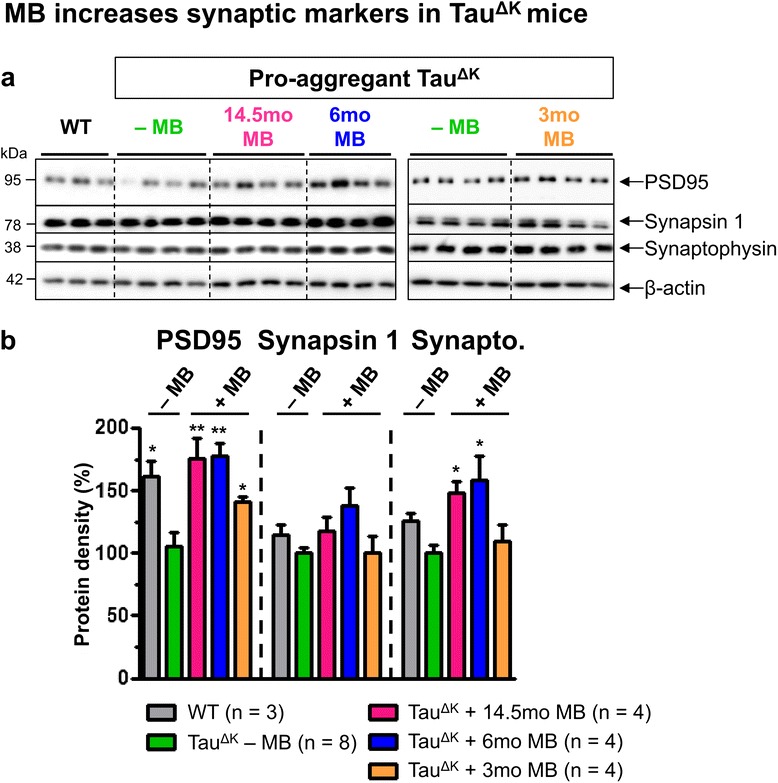
Recovery of synaptic marker proteins in hippocampus of MB-treated mice. (a) Levels of pre- and post-synaptic proteins synaptophysin, synapsin 1 and postsynaptic density 95 (PSD95) in hippocampus lysates of age-matched untreated and MB-treated TauΔK mice. β-actin serves as loading control. (b) Determination of relative protein densities by quantification of (a). Untreated TauΔK mice show a constant decrease of synapsin 1, synaptophysin and PSD95 levels in comparison to WT, due to constant expression of pro-aggregant TauΔK. In contrast a recovery of pre- and postsynaptic markers is observed after preventive MB application for 14.5mo and 6mo, whereas therapeutic MB treatment for 3mo causes a partial rescue of post- but not of presynaptic markers. Protein levels were normalized to β-actin. Bars represent mean values ± SEM. Statistics: one-way analysis of variances with post-hoc Newman-Keuls multiple comparisons test. Asterisks indicate significant differences in comparison to untreated TauΔK mice; *: p < 0.05; **: p < 0.01; n, number of samples.
Clearance of proteins via autophagy is a major pathway to maintain neuronal homeostasis and health. Preventive MB treatment of TauΔK mice for 14.5mo increased levels of beclin, heat shock cognate protein 70 (HSC70) and lysosome-associated membrane protein 2a (Lamp2a) (Figure 8), indicating an upregulation of autophagy. While beclin is involved in the initiation of the autophagosome formation during macroautophagy, HSC70 (a constitutively expressed molecular chaperone) and Lamp2a (lysosomal receptor) play a role in chaperone-mediated autophagy (CMA) of Tau (Wang et al., 2009). Another prominent pathway for protein degradation is the ubiquitin-proteasome system. As judged by an increased level of PSMD13 (regulatory subunit of the 26S proteasome), 14.5mo MB treatment enhanced proteasome function in comparison to WT and untreated TauΔK mice (Figure 8). Taken together, the tendency of MB to preserve or enhance protein degradation may counteract the continuous accumulation of toxic Tau species and contribute to maintenance of protein homeostasis.
Figure 8.
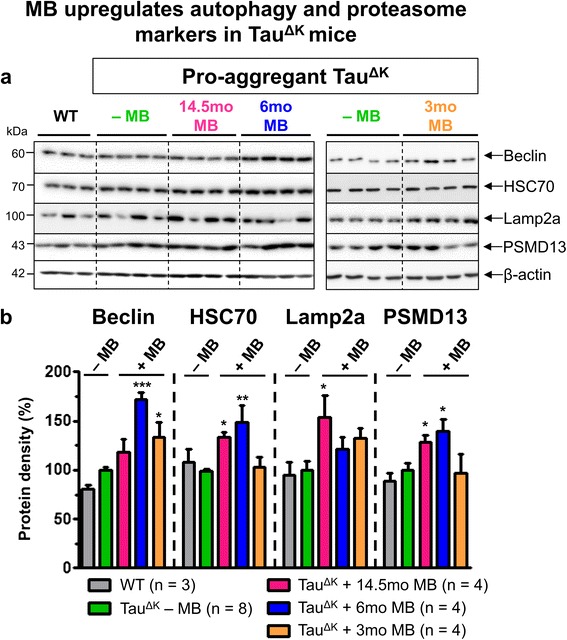
Preventive MB administration increases protein degradation. (a) Levels of autophagy- (beclin, HSC70), lysosome- (lamp2a), and proteasome-related (PSMD13) proteins in hippocampus lysates of age-matched untreated TauΔK mice compared to MB-treated TauΔK mice for 14.5mo, 6mo and 3mo. β-actin serves as loading control. (b) Quantification of (a). While both preventive MB treatment strategies (14.5mo and 6mo MB) increase levels of beclin, HSC70, Lamp2a and PSMD13, therapeutic MB treatment for 3mo leads to a partial increase of beclin and Lamp2a whereas levels of HSC70 and PSMD13 remain unaffected in comparison to untreated TauΔK mice. Protein levels were normalized to β-actin. Bars indicate mean values ± SEM. Statistics: one-way analysis of variances with post-hoc Newman-Keuls multiple comparisons test. Asterisks indicate significant differences in comparison to untreated TauΔK mice; *: p < 0.05; **: p < 0.01; ***: p < 0.001; n, number of samples.
MB’s role as electron carrier and thus redox-cycling compound is widely discussed in the literature [52]. However, we did not detect obvious differences between MB-treated and untreated TauΔK mice regarding mitochondria distribution and proteins of the electron transport chain (Additional file 7: Figure S7), indicating that MB did not alter this route of neuronal energy production.
In summary, if preventive treatment was started at an early age the cognitive abilities of TauΔK mice were essentially preserved, well beyond the expected onset of decline and in spite of the continued expression of the toxic mutant full-length TauΔK.
Preventive treatment starting 3mo before cognitive decline
Preventive MB administration (protocol 2, started ~3mo before onset of cognitive decline and continued for 6mo) showed an increase of open field activity of TauΔK mice in comparison to the reduced activity of untreated TauΔK mice (Figure 2b, Additional file 3: Figure S3). No group differences concerning anxiety related parameters were observed (Additional file 4: Figure S4). In MWM acquisition, both untreated TauΔK and 6mo MB-treated TauΔK mice performed less well than WT mice (WT vs. TauΔK - MB: p = 0.017; WT vs. TauΔK + 6mo MB: p = 0.036, Figure 3b). However, TauΔK mice with 6mo MB application showed a better learning performance compared to untreated TauΔK on day 4 (TauΔK - MB vs. TauΔK + 6mo MB: p = 0.042, Figure 3b). In addition they were able to localize the target quadrant and platform position more precisely than untreated TauΔK animals (Additional file 5: Figure S5), implying a minor beneficial effect of MB on cognition.
Neuropathological analysis revealed a reduction of insoluble TauΔK and mouse Tau (Figure 4), a decrease of conformationally-changed Tau (MC1) and phosphorylated Tau (AT180, PHF1) after 6mo of MB administration (Figure 5, Figure 6, Additional file 6: Figure S6). In addition, synaptic and autophagy marker proteins as well as the proteasome marker PSMD13 were increased after 6mo MB (Figure 7, Figure 8) but mitochondria remained unaffected (Additional file 7: Figure S7).
Thus, if preventive treatment was started at adult age with progressive tau pathology but still before the anticipated onset of cognitive decline, cognitive abilities were protected to a certain extent, in spite of the continued expression of toxic TauΔK.
Therapeutic treatment starting 3mo after cognitive decline
Finally, we tested the therapeutic potential of MB to reverse cognitive deficits ~3mo after onset (Figure 1a, protocol 3). MB treatment for 3mo did not enhance open field activity of TauΔK mice. Thus, treated TauΔK mice behaved similarly to untreated TauΔK animals (Figure 2c). WT mice showed a slight but non-significant increase in open field activity compared to transgenic animals, which was most pronounced within the first 5 minutes of the experiment (Additional file 3: Figure S3) and probably due to an advanced age of the mice during testing. Anxiety-related parameters were unaffected (Additional file 4: Figure S4).
An MWM test indicated a severe learning impairment of 3mo MB-treated TauΔK mice similar to untreated TauΔK mice and in contrast to WT animals, which improved consistently on each acquisition day (WT vs. TauΔK - MB: p = 0.001; WT vs. TauΔK + 3mo MB: p = 0.011, Figure 3c). In addition, MB-treated mice did not show any preference for the target quadrant (Additional file 5: Figure S5), underlining the persistence of cognitive deficits after 3mo MB.
Nevertheless, in spite of the failure in improving cognitive defects, MB treatment was still able to reduce sarcosyl-insoluble Tau, although to a minor extent in case of TauΔK compared to preventive MB treatment paradigms (Figure 4). In addition, conformationally changed Tau (MC1) and phosphorylated Tau (AT180, PHF1) were clearly reduced after 3mo MB treatment (Figure 5, Figure 6, Additional file 6: Figure S6).
By contrast, the 3mo MB application was not able to reverse the decline of presynaptic markers synaptophysin and synapsin 1 and the protective effect on postsynaptic PSD95 was less pronounced than in preventive MB treatment protocols (Figure 7). These results point towards sustained synaptic malfunction, which may underlie cognitive impairment independently of the reversal of biochemical Tau parameters. In addition, 3mo MB treatment only partly affected protein degradation systems. Whereas an increase in beclin and Lamp2a levels was detected, levels of HSC70 and PSMD13 were comparable to untreated age-matched TauΔK mice (Figure 8). As before, no effect on mitochondria was observed (Additional file 7: Figure S7).
Preventive and therapeutic MB treatments of TauRDΔK mice
In contrast to full-length TauΔK mice, animals expressing the repeat domain TauRDΔK develop a pronounced brain pathology in terms of Tau aggregation (neurofibrillary tangles) and neuronal loss and exhibit an earlier onset of cognitive decline ~10mo of age. This is consistent with the fast aggregation of the protein in vitro caused by the higher propensity for β-structure, and the absence of the N-and C-terminal flanking domains (Additional file 1: Figure S1). As in the case of full-length TauΔK, we tested different treatment protocols for TauRDΔK mice (Figure 1b).
We performed preventive MB treatment for 14.5 mo, starting at 1.5mo of age, long before onset of cognitive decline (Figure 1b, protocol 1), as well as therapeutic treatment for 3 mo, starting 5mo after onset of cognitive decline (at age 15 mo) (Figure 1b, protocol 2). Both treatments failed to decrease the level of insoluble Tau (exogenous and endogenous, Additional file 8: Figure S8) and did not protect synapses or induce protein degradation via autophagy (data not shown).
Surprisingly, even long-term preventive treatment of TauRDΔK mice (14.5mo) failed to reverse Tau-induced cognitive deficits (Figure 9). MB-treated TauRDΔK mice did not learn the location of the hidden platform, similar to untreated TauRDΔK animals (Figure 9a), and did not show a significant preference for the target quadrant in subsequent probe trials during MWM (Figure 9b). Thus, 20 mg/kg/day MB was not sufficient to suppress Tau aggregation and subsequent neurotoxic processes initiated by TauRDΔK.
Figure 9.
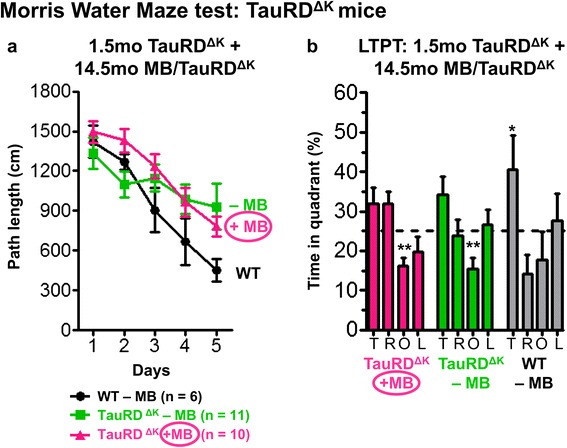
Preventive MB treatment does not rescue cognitive decline of TauRDΔK mice. MWM test shows cognitive abilities of TauRDΔK mice after preventive MB application for 14.5mo. (a) MB-treated as well as untreated TauRDΔK animals show impaired learning abilities similar to untreated TauRDΔK littermates, as indicated by increased path lengths to reach the hidden platform in MWM acquisition in comparison to WT. Data shows mean path length ± SEM. (b) Preventive MB treatment for 14.5mo does not result in a higher preference of the target quadrant as compared to untreated TauRDΔK mice and controls. Bars represent mean values ± SEM. Statistics: two-tailed one sample t-test against chance level of 25%; *: p < 0.05; **: p < 0.01; n = number of animals; T: target quadrant; R: right quadrant; O: opposite quadrant; L: left quadrant; LTPT: long-term probe trial.
To investigate whether higher doses of MB would influence TauRDΔK pathology, the mice were treated with an excessive dose of MB (40 mg/kg/day MB) for 3mo (Figure 1b, protocol 3, ~12-15mo of age, after cognitive decline). This actually resulted in an increase of aggregated Tau species in the hippocampus of TauRDΔK mice as detected by sarcosyl extraction and Gallyas silver staining (Figure 10). However, phosphorylated Tau was unaltered and autophagy partly reduced (data not shown). This detrimental effect of high doses MB was further confirmed by cell culture experiments. Inducible N2a cells expressing pro-aggregant TauRDΔK [53] accumulated aggregated Tau species in response to high concentrations of MB (25-100nM) in a dose-dependent manner (Additional file 9: Figure S9). Especially MB concentrations >100nM caused a massive loss of N2a cells, indicating a toxic gain of function in cells.
Figure 10.
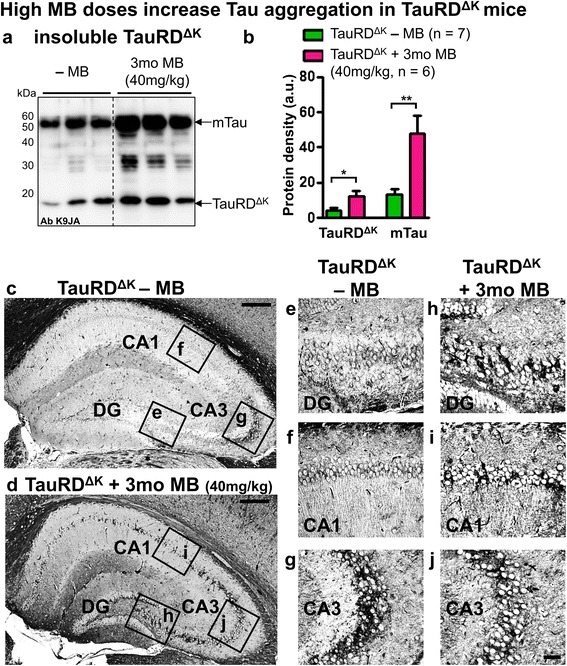
High MB-doses increase Tau aggregation. (a) Sarcosyl-extraction of insoluble Tau from cortex tissue of TauRDΔK mice. MB application using a daily dose of 40 mg/kg for 3mo results in an increase of sarcosyl-insoluble human TauRDΔK and endogenous mouse Tau (antibody K9JA). (b) Quantification of (a) shows a ~2-3 fold increase of insoluble Tau after high-dose MB treatment for 3mo. Bars represent mean values ± SEM. Statistics: two-tailed t-test; *: p < 0.05; **: p < 0.01; n, number of samples. (c, d) Gallyas silver staining of Tau aggregates shows an increase of tangle-positive cells inside the hippocampus formation of MB-treated TauRDΔK mice (40 mg/kg for 3mo). Scale bar: 200 μm. Boxed areas indicate close-ups presented in (e-j). Higher magnification of dentate gyrus (DG), CA3 and CA1 hippocampal areas of untreated TauRDΔK mice (e-g) in comparison to corresponding areas of MB-treated TauRDΔK mice (h-j). CA3: region III cornus ammonis; CA1: region I cornus ammonis. Scale bar: 40 μm.
Discussion
In this study we analyzed different protocols to determine optimal conditions for the preservation of cognitive abilities in pro-aggregant Tau transgenic mice treated with the Tau-aggregation inhibitor Methylene Blue (MB). Pro-aggregant full-length human TauΔK or repeat domain TauRDΔK was expressed in inducible transgenic mice from birth onwards, leading to onset of cognitive decline at ~12mo or 10mo, respectively. The questions to be tested were: can treatment before the expected onset prevent the cognitive decline? Can treatment after the onset rescue the decline?
The results show that (1) MB preserves cognition in full-length pro-aggregant TauΔK mice upon preventive treatment, provided that the intervention starts before the onset of behavioral deficits, whereas treatment initiated after onset of cognitive decline does not rescue. In addition, preventive MB application starting at young age (~1.5mo) has a greater impact on the cognitive performance than preventive MB application initiated at advanced age (~9mo) but shortly before onset of cognitive impairments. (2) MB reduces Tau aggregation, Tau phosphorylation and pathological Tau conformation in TauΔK mice. (3) MB protects synapses and enhances protein degradation by autophagy but has no obvious effect on mitochondria in TauΔK mice. (4) MB fails to prevent cognitive deficits in pro-aggregant repeat domain TauRDΔK mice, even when applied shortly after birth, presumably because the high aggregation propensity of TauRDΔK overwhelms the protective actions of the drug. (5) MB administered at high doses increases Tau aggregation rather than counteracting it.
Absorption and distribution of MB in mice
Methylene blue was well tolerated in wild-type mice after oral application and reached plasma and brain concentrations beyond the detection level over a time course of 24 h. At 24 h post application, MB brain levels were ~30-60-fold higher as compared to MB plasma levels after i.v. and oral administration, respectively. The data demonstrates that MB is well absorbed, readily crosses the blood-brain barrier and concentrates in the nervous tissue, confirming earlier reports of MB pharmacokinetics in mice [39]. In good agreement to previous studies analyzing pharmacokinetic profiles of MB in humans [54,55], MB peak-levels in plasma and brain were observed 1-2 h after oral intake. In this study, MB plasma half-life times of 4-5 h and brain half-life times ~3 h or ~30 h were calculated after i.v. or oral MB application. In comparison, human MB plasma half-life times in the range of ~5-6 h [2,54] to ~18 h [55] were determined, whereas half-life times in human brains are not available. In addition the total bioavailability of MB seems to be lower in mice (~20%) in comparison to humans (~70%, [55]). These variations may arise from different absorption and distribution properties of MB in rodents, which further complicate a direct comparison of human and rodent MB dosing despite FDA surface area conversion tables [56]. Furthermore, pharmacokinetic analyses presented in this study reflect absorption and distribution of MB in wild-type mice, which may slightly differ in aged TauΔK and TauRDΔK animals. Since Tau transgenic mice develop an age-dependent Tau neuropathology, alterations of the blood-brain barrier permeability cannot be excluded.
MB’s therapeutic potential depends on the time point of intervention and Tau aggregation
Tau aggregation requires detachment of Tau from microtubules, which is increased by Tau phosphorylation inside the repeat domain [57]. Especially disease- and stress-related conditions induce mislocalization of unbound Tau into postsynaptic sites, which is connected to increased phosphorylation and aggregation of Tau and impaired microtubule interactions [58,59]. In further consequence, accumulation of phosphorylated Tau in dendritic spines causes functional impairments, which finally lead to the loss of synapses [60].
The process of Tau aggregation includes several stages: the repeat-domain of Tau contains motifs with increased tendency to form β-structures, which function as aggregation-prone templates and are enhanced by mutations such as ΔK280 [61]. In seeded assembly, Tau nuclei trigger further aggregation by interaction with other Tau repeat domains from both exogenous human and endogenous mouse Tau, resulting in the co-assembly into neurofibrillary tangles.
After switch-off, the synthesis of new pro-aggregant β-structure seeds is blocked, while existing aggregates (containing human and mouse Tau) are gradually cleared by the intracellular machinery. However, endogenous mouse Tau is still continuously expressed and can be incorporated into existing seeds and polymers. Thus the remaining aggregates mainly consist of mouse Tau, which is less toxic to synapses [14].
According to this scheme, removing or blocking amyloidogenic Tau counteracts Tau pathology. As a consequence of this “genetic treatment”, cognitive impairment can be rescued by an overall elimination of toxic Tau, even after cognitive decline has set in. This holds for both models, pro-aggregant full length Tau (TauΔK) and repeat domain Tau (TauRDΔK) [14,15]. By contrast, aggregation inhibitors such as MB interfere with the aggregation process but do not affect de novo synthesis of pro-aggregant toxic Tau. This allows the formation of new β-structured seeds, which continue to aggregate. This would explain why MB can prevent pathology if administered at an early time point (because this keeps the seeds for aggregation below a critical threshold) but is inefficient if administered too late.
Several studies have confirmed the efficacy of MB in models of tauopathy when administered at early time points. A recovery of Tau-induced pathology and related locomotor phenotype was reported for a C. elegans model of tauopathy [35]. In Tau transgenic mice improvements of learning and memory behavior as a consequence of MB application was reported for Tau-P301S mice [62] and rTg4510 (Tau-P301L) mice [39]. In the successful treatment protocol used by Stack et al. (2014), MB (4 mg/kg/day) was administered for 10mo, starting at 1mo of age. Typically, Tau-P301S mice (line PS19) develop cognitive deficits ~6mo of age [8,63]. Thus, MB treatment was initiated ~5mo before the onset of cognitive impairment, and therefore this can be considered as a preventive strategy. O’Leary et al. (2010) reported an improved cognitive performance of rTg4510 mice after 3mo of MB treatment (10 mg/kg/day) starting at 3mo of age. Both studies are consistent with our results, which demonstrate a beneficial effect of MB in TauΔK mice, when administered using preventive application strategies.
The data underscores the need of early biomarkers (e.g. from cerebrospinal fluid, brain imaging, etc.), which would identify asymptomatic patients at a point where toxic Tau aggregates have not yet reached a critical level, so that aggregation inhibitors have a chance to keep them from becoming super-critical [64,65].
MB interference in TauΔK and TauRDΔK mice
Why is MB able to counteract the pathology of full-length TauΔK but not that of repeat domain TauRDΔK? Removal of the flanking domains outside the repeats of Tau is known to enhance Tau aggregation [53,66,67] because this exposes the pro-aggregant hexapeptide motifs of Tau which are normally shielded by the N- and C-terminal domains [68]. Tau aggregates of repeat domain TauRDΔK mice are positive for the amyloid marker thioflavin S, and fibers can be visualized by electron microscopy. By contrast, mice expressing full-length TauΔK lack these characteristics because this protein aggregates more slowly, has a low expression level, and does not reach the stage of mature Gallyas-positive tangles inside the hippocampus. Nevertheless, the “pre-tangles” contain smaller aggregates and oligomers, which are sufficient to cause synaptic decay and cognitive decline, with later onset than mice expressing the repeat domain.
Figure 11 illustrates a hypothetical progression model for both full-length TauΔK and repeat domain TauRDΔK mice. In both models, progressive Tau aggregation (red line) due to continuous Tau expression proceeds to a threshold indicating the onset of cognitive impairment (“point of no return”, marked by X, Figure 11a,b). In the case of pro-aggregant full-length TauΔK mice, the “point of no return” is reached at the age of ~12mo (Figure 11a). The slope of the aggregation can be reduced by MB (black lines corresponding to treatment protocols 1, 2, 3). If this takes place before the “point of no return”, cognitive impairment is delayed to a much later age (protocols 1, 2). If treatment starts after the “point of no return”, cognitive decline cannot be rescued, in spite of the slower aggregation rate (protocol 3).
Figure 11.
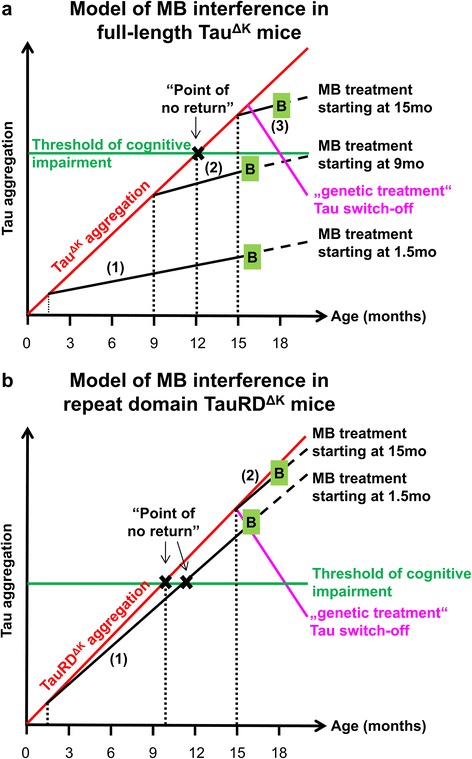
Models of MB interference during disease progression in full-length TauΔK and repeat domain TauRDΔK mice. (a) In TauΔK mice unaltered Tau aggregation due to continuous expression and accumulation of toxic Tau reaches the threshold of cognitive impairment at the age of ~12mo. Once the “point of no return” is passed, TauΔK mice exhibit cognitive impairments and synaptic decay [15]. Preventive application of MB, starting before the critical threshold is reached, efficiently interferes with TauΔK aggregation. Thus, the disease progression is slowed down and the onset of cognitive decline postponed. However, therapeutic MB application initiated at 15mo of age, beyond the “point of no return” and after the onset of cognitive impairment, is not able to rescue the cognitive phenotype. By contrast, “genetic treatment” by TauΔK switch-off can reduce aggregation and reverse the cognitive decline. (b) TauRDΔK exhibits stronger aggregation properties than TauΔK, due to a higher β-propensity. Thus, TauRDΔK mice show a faster disease progression with onset of cognitive decline ~10mo of age [14]. MB’s anti-aggregation properties are not strong enough to efficiently retard the aggregation process, even if applied at early age (1.5mo). Consequently, the “point of no return” is barely delayed in TauRDΔK mice, and therapeutic treatment is ineffective. Similar to TauΔK, genetic treatment by TauRDΔK switch off is still able to reverse the cognitive phenotype even after onset of cognitive impairment. Red line: unaltered progression of Tau aggregation; green line: threshold of cognitive impairment; black X: “point of no return”; solid black lines: MB treatment periods of mice; magenta lines: “genetic treatment” by switch off of Tau expression; B, green boxes: behavior tests and termination of experiments; dotted black lines: assumed further progression of Tau aggregation; (1, 2, 3): MB treatment protocols according to Figure 1; mo: months.
The principle in case of TauRDΔK is similar, with the major difference that Tau aggregation is stronger and leads to a faster disease progression with earlier onset of cognitive deficits ~10mo of age (Figure 11b). Here the anti-aggregation potential of MB is not strong enough to retard Tau aggregation significantly and postpone the onset of cognitive impairment, even if treatment is started at early age (protocol 1). In contrast to treatment with an aggregation inhibitor, “genetic treatment” by switching-off TauΔK or TauRDΔK reverses the slope (due to ongoing protein degradation) so that cognitive decline can be rescued (Figure 11a + b, magenta line, [14,15,69]).
Direct interference of MB and Tau
MB is a redox-recycling agent and interacts with exposed cysteines, nucleotide-binding proteins and others; this may explain its many uses in medicine [70]. With regard to Tau, MB acts as an aggregation inhibitor [24,25,30] by a mechanism based on the oxidation of cysteine residues C291 and C322. They lie within the Tau repeat domain and affect the conformations and interactions leading to aggregation [71,72]. In addition, MB has been reported to lower the level of Tau by interacting with the chaperone hsp70 [73].
Beside MB (IC50: 1.9 μM, [24]), other potent Tau aggregation inhibitors have been identified (reviewed in [21,74-76]). Especially rhodanines (e.g. bb14, IC50 = 0.67 μM) and phenylthiazolyl-hydrazides (PTHs, i.e. BSc3094, IC50 = 1.6 μM) efficiently inhibit Tau aggregation and show a low cytotoxicity in cell-based assays [77,78], in C. elegans [35] or in hippocampal slice culture models of tauopathy [79].
General effects of MB on cell metabolism
The fact that MB-treated WT mice show a somewhat better learning performance compared to untreated WT mice argues for a general increase in cell metabolism or clearance of toxic proteins upon MB treatment. Especially modulation of autophagy and subsequent protein degradation via lysosomes is a promising therapeutic target in neurodegenerative diseases since autophagy is a major pathway for the clearance of long-lived and aggregated proteins [80,81]. Indeed, aggregated forms of Tau are cleared preferentially by autophagy [82]; however, the situation is less well defined for toxic oligomeric forms of Tau where both degradation pathways may contribute [83]. Several studies suggest an effect of MB on different protein degradation pathways. While Congdon and colleagues show an mTOR-dependent induction of macroautophagy mediated by MB [44], Medina et al. (2011) report an increase of chymotrypsin- and trypsin-like activities of the proteasome after MB treatment. In addition, inhibition of the Hsp70 ATPase activity by MB facilitated chaperone-mediated clearance of Tau [73].
The upregulation of different degradation mechanisms by MB was also observed in our experiments, as judged by different marker proteins. While upregulation of beclin is related to increased macroautophagy, higher levels of HSC70 and Lamp2a argue for an induction of CMA, and the rise in PSMD13 points to an increase of proteasome activity. In this respect, MB provides additional beneficial effects which may enhance MB’s neuroprotective properties. In contrast we were not able to confirm a protective effect of MB in terms of oxidative stress as described by others [62]. These discrepancies may be due to the use of another transgenic mouse model, which differs by cognitive phenotype as well as in brain pathology.
Importance of the dose
We and others [62] observed that high doses of MB (40 mg/kg/day) are detrimental and rather increase pathology. A hormetic dose-response to MB is discussed in the literature, suggesting that MB’s beneficial effect may be limited to a certain range of concentration, whereas concentrations below or beyond the optimal range are ineffective or even lead to detrimental effects. In rats, an optimum concentration of 4 mg/kg improves cognitive function, whereas lower (1 mg/kg) and higher (10 mg/kg) doses were ineffective [84]. In addition, an excessive dose of 50 mg/kg resulted in a decrease of cognitive functions below control levels, indicating a detrimental effect of MB [84]. This hormetic dose-response was attributed to MB’s auto-oxidizing properties. In vivo at low concentrations (<5 μM), reduced MBH2 and oxidized MB are in equilibrium and form a reversible reduction-oxidation system, which functions as an electron cycler and scavenger of free radicals. By this mechanism, MB has the potential to donate electrons to the mitochondrial respiratory chain, which finally increases energy production. On the contrary, higher MB concentrations (>5-10 μM) eliminate the equilibrium and electrons are taken away from the respiratory chain. This results in inhibition of cytochrome c oxidation, either directly via reduction of oxygen [85] or indirectly via inhibition of nitric oxide synthase (NO) [86].
Currently attempts are underway to further improve MB in terms of bioavailability and tolerance. A new formulation of Rember™ named LMTX™ provides MB in a stabilized, reduced “leuco-form”, which shows a higher absorbance from the intestine and therefore increases the effective dose of MB in the brain [4,33,34]. Another strategy is the development of MB-loaded hydrophobic nanoparticles suitable for blood-brain barrier permeation, which may provide a steady-state drug release instead of variable drug concentrations after intake of a single daily dose [87].
Conclusion
In summary, we observed several effects of MB, which may together contribute to the improvement of cognition in treated Tau-transgenic mice. The most crucial parameters in our experiments are (i) the time point of intervention and (ii) the aggregation propensity of Tau. A preventive strategy (initiated before onset of cognitive decline) can be successful, provided that the aggregation propensity is not too strong, which is the case for full-length TauΔK but not for TauRDΔK. However, in neither case can the pathology be reversed by MB if therapeutic treatment is started after the onset of cognitive decline.
In other words, MB treatment is no longer effective when the destruction of synaptic functions has already progressed beyond the “point of no return”. The direct effect of MB on Tau aggregation is one of the key events in the defense against Tau-induced functional changes since MB’s potency decreases with increasing β-propensity of Tau. Additionally, beneficial side effects of MB that enhance neuronal cell metabolism reinforce the neuroprotective impact of MB.
Thus, our study supports the use of MB as Tau aggregation inhibitor for treatment of AD and other tauopathies but clearly emphasizes the need for early preventive intervention prior to irreversible synaptic damage.
Acknowledgements
We thank Dr. A. Haemisch and his team at the animal facility at the University of Hamburg Medical School for their continuous help in mouse breeding. We gratefully acknowledge reagents from Prof. Dr. E. Kandel (Columbia University, New York, NY; CaMKIIα-tTA transgenic mice), Dr. P. Seubert (Elan Pharma, South San Francisco, CA; 12E8 antibody) and Dr. P. Davies (Albert Einstein College, Bronx, NY; MC1 and PHF1 antibodies). This research was supported by MPG, DZNE, EU-FP7/Memosad, Tau Consortium and Katharina-Hardt-Stiftung.
Abbreviations
- Aβ
Amyloid-beta
- AD
Alzheimer disease
- ALS
Amyotrophic lateral sclerosis
- ATP
Adenosine triphosphate
- BLI
Bioluminescence imaging
- bvFTD
Behavioral variant of frontotemporal dementia
- CaMKIIα
Calcium/calmodulin-dependent protein kinase IIα
- CMA
Chaperone-mediated autophagy
- FDA
Food and Drug Administration
- FTD
Frontotemporal dementia
- GFAP
Glial fibrillary acidic protein
- HSC70
Heat shock cognate protein 70
- ΔK280
Tau mutant with deletion of lysine 280
- Lamp2a
Lysosome-associated membrane protein 2a
- MB
Methylene blue
- mo
Months
- mTOR
mammalian target of rapamycin (protein kinase)
- MWM
Morris water maze
- NFT
Neurofibrillary tangle
- NMDA
N-methyl D-aspartate
- NMR
Nuclear magnetic resonance
- PHF
Paired helical filament
- PSD95
Postsynaptic density 95
- PSMD13
a regulatory subunit of the 26S proteasome
- SSCx
Somatosensory cortex
- TauΔK
full-length human Tau (2N4R, 441 residues) with ΔK280 deletion mutation
- TauRDΔK
Tau 4-repeat domain (construct K18, residues 244-372) with ΔK280 mutation
- tTA
tetracycline transactivator
Additional files
Domains of human full-length TauΔK and repeat domain TauRDΔK. Diagram of the domains of human full-length TauΔK (2N4R, amino acids 1-441) and repeat domain TauRDΔK (amino acid 244-372). A pro-aggregant variant of TauΔK or TauRDΔK with deletion of lysine 280 (ΔK280) is expressed in inducible Tau transgenic mice. The hexapeptide motifs (PHF6*, PHF6) are located in the repeat domain (R1-R4) and show a high tendency to form β-structure. Adapted from [27].
Pharmacokinetic analysis of MB after intravenous and oral application. (a) Profiles of TMT-ion concentrations after intravenous (i.v.) application of 30 mg/kg MB in plasma (ng/ml, red circles) and brain tissue (ng/g, blue squares) measured by LC-MS/MS over 24 h. For i.v. application, TMT-ion half-life times of t½ = 4.4 h in plasma and t½ = 3.0 h in brain are determined. (b) Profiles of TMT-ion concentrations after oral administration of 45 mg/kg MB in plasma (ng/ml, red circles) and brain tissue (ng/g, blue squares) measured by LC-MS/MS over 24 h. For oral administration, TMT-ion half-life times of t½ = 30.6 h in brain and t½ = 4.7 h in plasma are calculated. Final TMT-ion concentrations at 24 h post application are given in nM for each curve. Data represents mean values ± SEM, n = 3 per time point. MB: methylene blue, TMT-ion: tetramethylionium-ion (= MB without chloride and 3*H2O), LC-MS/MS: liquid chromatography – mass spectrometry.
Open field activity of MB-treated mice within intervals. (a) Open field activity within 3 intervals à 5 min. Untreated TauΔK mice show a decreased exploration behavior in comparison to WT. Mice treated with MB for (a) 14.5mo and (b) 6mo show a considerably higher activity within interval 2 and 3 as compared to untreated TauΔK mice, whereas no difference between MB-treated and untreated mice is observed for 3mo of MB application (c). Bars represent mean values ± SEM.
MB application does not influence anxiety-related parameters. No differences are detected between untreated and MB-treated TauΔK mice for anxiety-related parameters such as average distance to wall (a) and total time in center (b) throughout all experimental conditions, suggesting that MB does not exert anxiolytic properties. Bars represent mean values ± SEM. Statistics: one-way analysis of variances with post-hoc Newman-Keuls multiple comparisons test. n.s.: not significant.
MWM long-time probe trial - localization of the target platform. Preventive treatment with MB (14.5mo or 6mo) results in a higher preference of the target quadrant in a long term probe trial (a) and in a more precise localization of the target platform during probe trials as indicated by time in target platform (b). In contrast therapeutic MB application for 3mo does not increase the overall preference of MB-treated TauΔK mice for the target quadrant, albeit it seems to improve the ability to localize the platform position to some extent, suggesting a minor effect of MB. Bars represent mean values ± SEM. Statistics: (a) two-tailed one sample t-test against chance level of 25%; *: p < 0.05; **: p < 0.01; ***: p < 0.001; (b) two-way repeated measure analysis of variances with post hoc Fishers LSD multiple comparisons test. T: target quadrant; R: right quadrant; O: opposite quadrant; L: left quadrant; PT: probe trial; LTPT: long-term probe trial.
Histological analysis of conformationally changed and phosphorylated Tau in MB-treated mice. (a-e) MC1 staining (epitope 5-15 + 312-322) indicates a pathological Tau conformation in somatosensory cortex neurons of untreated TauΔK mice with missorting of Tau to cell soma and apical dendrites (b1-b3). In contrast MB treatment for 14.5mo (c1-c3), 6mo (d1-d3) and 3mo (e1-e2) reduces MC1 immunoreactivity in TauΔK mice. Wild-type (WT) animals are MC1-negative (a1). Scale bar: 50 μm. n = 3 animals per condition. (f-j) Staining of phosphorylated Tau (antibody AT180, dual phosphorylation epitope pThr231 + pSer235) in somatosensory cortex of MB-treated and untreated TauΔK mice. Untreated TauΔK mice exhibit numerous AT180-positive neurons with massive mislocalization of phosphorylated Tau to cell soma and apical dendrites (g1-g3), whereas MB treatment for 14.5mo (h1-h3), 6mo (i1-i3) and 3mo (j1-j3) diminishes the extent of AT180 phosphorylation. Wild-type (WT) animals are AT180-negative (f1). Scale bar: 50 μm. n = 3 animals per condition.
MB shows no influence on mitochondria in CA3 region of the hippocampus. The intracellular distribution of mitochondria in pyramidal neurons of the hippocampal CA3 region is visualized by fluorescence confocal microscopy. Paraffin sections of wild-type animals (a), untreated TauΔK mice (b) and MB-treated TauΔK mice (c-e) were fixed and stained with OXPHOS antibody cocktail (DyLight 650, red), which was used as a mitochondrial marker. Nuclei were counterstained with Syto13 (green). No difference of mitochondrial immunoreactivity was observed among WT animals and TauΔK mice with or without MB treatment for the time period indicated. Scale bar: 10 μm.
Administration of 20 mg/kg MB for 3mo and 14.5mo does not influence insoluble Tau levels in TauRDΔK mice. Sarcosyl-extraction of insoluble Tau from cortex tissue of TauRDΔK mice. MB administration using a daily dose of 20 mg/kg MB for (a) 3mo and (b) 14.5mo does not alter levels of detergent-insoluble Tau in comparison to untreated TauRDΔK mice as shown by the pan-Tau antibody K9JA. Ab: antibody; mTau: mouse Tau.
MB affects the amount of Tau4RDΔK280 protein in inducible N2a cells. (a) Inhibition of recombinant Tau3RD aggregation by MB monitored in vitro by filter assay and Western blot (adapted from [71]). (b) Examples of confocal micrographs of Tau4RDΔK280 expressing N2a cells after 96 h incubation-/expression time. (c) Western blot of the Tau load in Tau4RDΔK280 expressing N2a cells treated with increasing methylene blue concentrations detected by pan-Tau antibody K9JA (c1) and phosphorylation-depended antibody 12E8 (epitopes pSer262/pSer356) (c2). Expression of Tau4RDΔK280 in N2a cells is induced by addition of doxycycline (1 μg/ml) (Tet-on system) as shown by the pan-Tau antibody K9JA. Monomeric and oligomeric Tau is phosphorylated at KXGS motifs inside the repeat domain as shown by Ab 12E8 (epitope pSer262, pSer356). Incubation of N2a cells with increasing concentrations of MB (25nM, 50nM or 100nM) for 4 days results in a dose-dependent increase of oligomeric Tau species and a subsequent decrease of monomeric Tau4RDΔK280. Monomeric Tau includes the entire Tau repeat domain as well as truncated fragments (F2, F3). (d) Relative quantification of (c1) shows the change in Tau protein densities (monomeric vs. oligomeric Tau) upon treatment of cells with increasing concentrations of MB.
Footnotes
Katja Hochgräfe and Astrid Sydow contributed equally to this work.
Competing interest
The authors declare that they have no competing interest.
Authors’ contributions
KH and AS carried out behavior and molecular studies. SK and PG participated in behavior experiments. DC, OP and DM carried out molecular experiments. FM advised behavioral studies and statistical analyses. MP carried out N2a and pharmacokinetic experiments. EMM designed the study. KH, EM and EMM drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Katja Hochgräfe, Email: hochgraefe@mpasmb.desy.de.
Astrid Sydow, Email: sydow@mpasmb.desy.de.
Dorthe Matenia, Email: matenia@mpasmb.desy.de.
Daniela Cadinu, Email: danielacadinu@gmail.com.
Stefanie Könen, Email: stefanie.koenen@dzne.de.
Olga Petrova, Email: petrova@mpasmb.desy.de.
Marcus Pickhardt, Email: marcus.pickhardt@dzne.de.
Petra Goll, Email: petra.goll@t-online.de.
Fabio Morellini, Email: morellini.fabio@zmnh.uni-hamburg.de.
Eckhard Mandelkow, Email: mand@mpasmb.desy.de.
Eva-Maria Mandelkow, Email: mandelkow@dzne.de.
References
- 1.Selkoe D, Mandelkow E, Holtzman D. Deciphering Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a011460. doi: 10.1101/cshperspect.a011460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Santo SG, Prinelli F, Adorni F, Caltagirone C, Musicco M. A meta-analysis of the efficacy of donepezil, rivastigmine, galantamine, and memantine in relation to severity of Alzheimer’s disease. J Alzheimer’s Dis. 2013;35:349–361. doi: 10.3233/JAD-122140. [DOI] [PubMed] [Google Scholar]
- 3.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 4.Wischik CM, Harrington CR, Storey JM. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol. 2014;88:529–539. doi: 10.1016/j.bcp.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 6.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.WNL.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 7.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 8.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buee L. Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 12.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2 N transgenic mice. J Biol Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- 14.Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D’Hooge R, Alzheimer C, Mandelkow EM. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011;31:2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van der Jeugd A, Hochgrafe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D’Hooge R, Mandelkow EM. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012;123:787–805. doi: 10.1007/s00401-012-0987-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, Schonig K, Bujard H, Haemisch A, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem. 2007;282:31755–31765. doi: 10.1074/jbc.M705282200. [DOI] [PubMed] [Google Scholar]
- 17.Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schonig K, Bujard H, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64:414–421. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Swieten JC, Bronner IF, Azmani A, Severijnen LA, Kamphorst W, Ravid R, Rizzu P, Willemsen R, Heutink P. The DeltaK280 mutation in MAP tau favors exon 10 skipping in vivo. J Neuropathol Exp Neurol. 2007;66:17–25. doi: 10.1097/nen.0b013e31802c39a4. [DOI] [PubMed] [Google Scholar]
- 20.Momeni P, Pittman A, Lashley T, Vandrovcova J, Malzer E, Luk C, Hulette C, Lees A, Revesz T, Hardy J, de Silva R. Clinical and pathological features of an Alzheimer’s disease patient with the MAPT Delta K280 mutation. Neurobiol Aging. 2009;30:388–393. doi: 10.1016/j.neurobiolaging.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulic B, Pickhardt M, Mandelkow E. Progress and developments in tau aggregation inhibitors for Alzheimer disease. J Med Chem. 2013;56:4135–4155. doi: 10.1021/jm3017317. [DOI] [PubMed] [Google Scholar]
- 22.Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VM. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem Biophys Res Commun. 2007;358:1–6. doi: 10.1016/j.bbrc.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pickhardt M, von Bergen M, Gazova Z, Hascher A, Biernat J, Mandelkow EM, Mandelkow E. Screening for inhibitors of tau polymerization. Curr Alzheimer Res. 2005;2:219–226. doi: 10.2174/1567205053585891. [DOI] [PubMed] [Google Scholar]
- 24.Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 25.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sontag EM, Lotz GP, Agrawal N, Tran A, Aron R, Yang G, Necula M, Lau A, Finkbeiner S, Glabe C, Marsh JL, Muchowski PJ, Thompson LM. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington’s disease models. J Neurosci. 2012;32:11109–11119. doi: 10.1523/JNEUROSCI.0895-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K, Yoshida M, Onaya M, Fujishiro H, Akiyama H. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology. 2010;30:170–181. doi: 10.1111/j.1440-1789.2009.01089.x. [DOI] [PubMed] [Google Scholar]
- 28.Necula M, Breydo L, Milton S, Kayed R, van der Veer WE, Tone P, Glabe CG. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry. 2007;46:8850–8860. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- 29.Cavaliere P, Torrent J, Prigent S, Granata V, Pauwels K, Pastore A, Rezaei H, Zagari A. Binding of methylene blue to a surface cleft inhibits the oligomerization and fibrillization of prion protein. Biochim Biophys Acta. 2013;1832:20–28. doi: 10.1016/j.bbadis.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 30.Schirmer RH, Adler H, Pickhardt M, Mandelkow E (2011) “Lest we forget you--methylene blue…”. Neurobiol Aging 32:2325 e2327-2316. doi:10.1016/j.neurobiolaging.2010.12.012S0197-4580(10)00529-4 [DOI] [PubMed]
- 31.Oz M, Lorke DE, Petroianu GA. Methylene blue and Alzheimer’s disease. Biochem Pharmacol. 2009;78:927–932. doi: 10.1016/j.bcp.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 32.Wischik CM, Bentham P, Wischik DJ, Seng KM. O3-04-07: Tau aggregation inhibitor (TAI) therapy with rember™ arrests disease progression in mild and moderate Alzheimer's disease over 50 weeks. Paper presented at the Alzheimer’s Association International Conference. Chicago IL, United States: Alzheimer’s Association International Conference; 2008. [Google Scholar]
- 33.Baddeley TC, McCaffrey J, MDS J, Cheung JK, Melis V, Horsley D, Harrington CR, Wischik CM. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer‘s disease. J Pharmacol Exp Ther. 2015;352:110–118. doi: 10.1124/jpet.114.219352. [DOI] [PubMed] [Google Scholar]
- 34.Wischik C, Siew Choon T, Say Way S, Logan S. TauRx.com About LMTX™ for Alzheimer’s. TauRx Therapeutics Ltd. 3, Shenton Way, #21-04 Shenton House Singapore 068805 Republic of Singapore, and TauRx Research Facility Liberty Building Foresterhill Road Aberdeen AB25 2ZP United Kingdom: TauRx Therapeutics Ltd; 2014. [Google Scholar]
- 35.Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow EM, Schmidt E, Baumeister R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum Mol Genet. 2012;21:3587–3603. doi: 10.1093/hmg/dds190. [DOI] [PubMed] [Google Scholar]
- 36.Van Bebber F, Paquet D, Hruscha A, Schmid B, Haass C. Methylene blue fails to inhibit Tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiol Dis. 2010;39:265–271. doi: 10.1016/j.nbd.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 37.Deiana S, Harrington CR, Wischik CM, Riedel G. Methylthioninium chloride reverses cognitive deficits induced by scopolamine: comparison with rivastigmine. Psychopharmacology (Berl) 2009;202:53–65. doi: 10.1007/s00213-008-1394-2. [DOI] [PubMed] [Google Scholar]
- 38.Medina DX, Caccamo A, Oddo S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21:140–149. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Leary JC, 3rd, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, Jinwal UK, Koren J, Jones JR, 3rd, Kraft C, Peters M, Abisambra JF, Duff KE, Weeber EJ, Gestwicki JE, Dickey CA. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Audet JN, Soucy G, Julien JP. Methylene blue administration fails to confer neuroprotection in two amyotrophic lateral sclerosis mouse models. Neuroscience. 2012;209:136–143. doi: 10.1016/j.neuroscience.2011.12.047. [DOI] [PubMed] [Google Scholar]
- 41.Lougheed R, Turnbull J. Lack of effect of methylene blue in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. PLoS One. 2011;6:e23141. doi: 10.1371/journal.pone.0023141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosokawa M, Arai T, Masuda-Suzukake M, Nonaka T, Yamashita M, Akiyama H, Hasegawa M. Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS One. 2012;7:e52389. doi: 10.1371/journal.pone.0052389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spires-Jones TL, Friedman T, Pitstick R, Polydoro M, Roe A, Carlson GA, Hyman BT. Methylene blue does not reverse existing neurofibrillary tangle pathology in the rTg4510 mouse model of tauopathy. Neurosci Lett. 2014;562C:63–68. doi: 10.1016/j.neulet.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff KE (2012) Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 8:609-622. doi:10.4161/auto.1904819048 [DOI] [PMC free article] [PubMed]
- 45.Miyata Y, Rauch JN, Jinwal UK, Thompson AD, Srinivasan S, Dickey CA, Gestwicki JE. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem Biol. 2012;19:1391–1399. doi: 10.1016/j.chembiol.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, Gromer S. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob Agents Chemother. 2008;52:183–191. doi: 10.1128/AAC.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 48.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 49.Jicha GA, Berenfeld B, Davies P. Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer’s disease. J Neurosci Res. 1999;55:713–723. doi: 10.1002/(SICI)1097-4547(19990315)55:6<713::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 50.Hochgräfe K, Mandelkow EM. Making the brain glow: in vivo bioluminescence imaging to study neurodegeneration. Mol Neurobiol. 2013;47:868–882. doi: 10.1007/s12035-012-8379-1. [DOI] [PubMed] [Google Scholar]
- 51.Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- 52.Oz M, Lorke DE, Hasan M, Petroianu GA. Cellular and molecular actions of Methylene Blue in the nervous system. Med Res Rev. 2011;31:93–117. doi: 10.1002/med.20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- 54.Peter C, Hongwan D, Kupfer A, Lauterburg BH. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur J Clin Pharmacol. 2000;56:247–250. doi: 10.1007/s002280000124. [DOI] [PubMed] [Google Scholar]
- 55.Walter-Sack I, Rengelshausen J, Oberwittler H, Burhenne J, Mueller O, Meissner P, Mikus G. High absolute bioavailability of methylene blue given as an aqueous oral formulation. Eur J Clin Pharmacol. 2009;65:179–189. doi: 10.1007/s00228-008-0563-x. [DOI] [PubMed] [Google Scholar]
- 56.U.S. Food and Drug Administration (2005) Guidance for Industry, Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm078932.pdf.
- 57.Mandelkow EM, Mandelkow E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998;8:425–427. doi: 10.1016/S0962-8924(98)01368-3. [DOI] [PubMed] [Google Scholar]
- 58.Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zempel H, Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014;37:721–732. doi: 10.1016/j.tins.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 60.Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A. 2000;97:5129–5134. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stack C, Jainuddin S, Elipenahli C, Gerges M, Starkova N, Starkov AA, Jove M, Portero-Otin M, Launay N, Pujol A, Kaidery NA, Thomas B, Tampellini D, Beal MF, Dumont M. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum Mol Genet. 2014;23:3716–3732. doi: 10.1093/hmg/ddu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takeuchi H, Iba M, Inoue H, Higuchi M, Takao K, Tsukita K, Karatsu Y, Iwamoto Y, Miyakawa T, Suhara T, Trojanowski JQ, Lee VM, Takahashi R. P301S mutant human tau transgenic mice manifest early symptoms of human tauopathies with dementia and altered sensorimotor gating. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blennow K, Zetterberg H, Fagan AM. Fluid biomarkers in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006221. doi: 10.1101/cshperspect.a006221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jack CR, Jr, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80:1347–1358. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wille H, Drewes G, Biernat J, Mandelkow EM, Mandelkow E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol. 1992;118:573–584. doi: 10.1083/jcb.118.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang BJ, Pickhardt M, Mandelkow E, Mandelkow EM. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci U S A. 2007;104:10252–10257. doi: 10.1073/pnas.0703676104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- 69.Hochgräfe K, Sydow A, Mandelkow EM. Regulatable transgenic mouse models of Alzheimer disease: onset, reversibility and spreading of Tau pathology. Febs J. 2013;280:4371–4381. doi: 10.1111/febs.12250. [DOI] [PubMed] [Google Scholar]
- 70.Ginimuge PR, Jyothi SD. Methylene blue: revisited. J Anaesthesiol, Clin Pharmacol. 2010;26:517–520. [PMC free article] [PubMed] [Google Scholar]
- 71.Akoury E, Pickhardt M, Gajda M, Biernat J, Mandelkow E, Zweckstetter M. Mechanistic basis of phenothiazine-driven inhibition of Tau aggregation. Angew Chem Int Ed Engl. 2013;52:3511–3515. doi: 10.1002/anie.201208290. [DOI] [PubMed] [Google Scholar]
- 72.Crowe A, James MJ, Lee VM, Smith AB, 3rd, Trojanowski JQ, Ballatore C, Brunden KR. Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation. J Biol Chem. 2013;288:11024–11037. doi: 10.1074/jbc.M112.436006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jinwal UK, Miyata Y, Koren J, 3rd, Jones JR, Trotter JH, Chang L, O’Leary J, Morgan D, Lee DC, Shults CL, Rousaki A, Weeber EJ, Zuiderweg ER, Gestwicki JE, Dickey CA. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci. 2009;29:12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bulic B, Pickhardt M, Mandelkow EM, Mandelkow E. Tau protein and tau aggregation inhibitors. Neuropharmacology. 2010;59:276–289. doi: 10.1016/j.neuropharm.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 75.Chang E, Honson NS, Bandyopadhyay B, Funk KE, Jensen JR, Kim S, Naphade S, Kuret J. Modulation and detection of tau aggregation with small-molecule ligands. Curr Alzheimer Res. 2009;6:409–414. doi: 10.2174/156720509789207976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brunden KR, Ballatore C, Crowe A, Smith AB, 3rd, Lee VM, Trojanowski JQ. Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: a focus on tau assembly inhibitors. Exp Neurol. 2010;223:304–310. doi: 10.1016/j.expneurol.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bulic B, Pickhardt M, Khlistunova I, Biernat J, Mandelkow EM, Mandelkow E, Waldmann H. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew Chem Int Ed Engl. 2007;46:9215–9219. doi: 10.1002/anie.200704051. [DOI] [PubMed] [Google Scholar]
- 78.Pickhardt M, Larbig G, Khlistunova I, Coksezen A, Meyer B, Mandelkow EM, Schmidt B, Mandelkow E. Phenylthiazolyl-hydrazide and its derivatives are potent inhibitors of tau aggregation and toxicity in vitro and in cells. Biochemistry. 2007;46:10016–10023. doi: 10.1021/bi700878g. [DOI] [PubMed] [Google Scholar]
- 79.Messing L, Decker JM, Joseph M, Mandelkow E, Mandelkow EM. Cascade of tau toxicity in inducible hippocampal brain slices and prevention by aggregation inhibitors. Neurobiol Aging. 2013;34:1343–1354. doi: 10.1016/j.neurobiolaging.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92–104. doi: 10.1038/cr.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Metcalf DJ, Garcia-Arencibia M, Hochfeld WE, Rubinsztein DC. Autophagy and misfolded proteins in neurodegeneration. Exp Neurol. 2012;238:22–28. doi: 10.1016/j.expneurol.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang M-VM, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cardenas-Aguayo MD, Gomez-Virgilio L, DeRosa S, Meraz-Rios MA (2014) The Role of Tau Oligomers in the Onset of Alzheimer’s Disease Neuropathology. ACS Chem Neurosci [Epub ahead of print]. doi:10.1021/cn500148z [DOI] [PubMed]
- 84.Riha PD, Bruchey AK, Echevarria DJ, Gonzalez-Lima F. Memory facilitation by methylene blue: dose-dependent effect on behavior and brain oxygen consumption. Eur J Pharmacol. 2005;511:151–158. doi: 10.1016/j.ejphar.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 85.Bruchey AK, Gonzalez-Lima F. Behavioral, Physiological and Biochemical Hormetic Responses to the Autoxidizable Dye Methylene Blue. Am J Pharmacol Toxicol. 2008;3:72–79. doi: 10.3844/ajptsp.2008.72.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mayer B, Brunner F, Schmidt K. Inhibition of nitric oxide synthesis by methylene blue. Biochem Pharmacol. 1993;45:367–374. doi: 10.1016/0006-2952(93)90072-5. [DOI] [PubMed] [Google Scholar]
- 87.Jinwal UK, Groshev A, Zhang J, Grover A, Sutariya VB. Preparation and characterization of methylene blue nanoparticles for Alzheimer’s disease and other tauopathies. Curr Drug Deliv. 2014;11:541–550. doi: 10.2174/1567201810666131113102037. [DOI] [PubMed] [Google Scholar]