

Abstract
Background
Mycobacterium tuberculosis continues to kill more people than any other bacterium. Although its archetypal host cell is the macrophage, it also enters, and survives within, dendritic cells (DCs). By modulating the behaviour of the DC, M. tuberculosis is able to manipulate the host’s immune response and establish an infection. To identify the M. tuberculosis genes required for survival within DCs we infected primary human DCs with an M. tuberculosis transposon library and identified mutations with a reduced ability to survive.
Results
Parallel sequencing of the transposon inserts of the surviving mutants identified a large number of genes as being required for optimal intracellular fitness in DCs. Loci whose mutation attenuated intracellular survival included those involved in synthesising cell wall lipids, not only the well-established virulence factors, pDIM and cord factor, but also sulfolipids and PGL, which have not previously been identified as having a direct virulence role in cells. Other attenuated loci included the secretion systems ESX-1, ESX-2 and ESX-4, alongside many PPE genes, implicating a role for ESX-5. In contrast the canonical ESAT-6 family of ESX substrates did not have intra-DC fitness costs suggesting an alternative ESX-1 associated virulence mechanism. With the aid of a gene-nutrient interaction model, metabolic processes such as cholesterol side chain catabolism, nitrate reductase and cysteine-methionine metabolism were also identified as important for survival in DCs.
Conclusion
We conclude that many of the virulence factors required for survival in DC are shared with macrophages, but that survival in DCs also requires several additional functions, such as cysteine-methionine metabolism, PGLs, sulfolipids, ESX systems and PPE genes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1569-2) contains supplementary material, which is available to authorized users.
Keywords: Mycobacterium tuberculosis, dendritic cells, transposon library, cholesterol, phenolic glycolipids, sulfolipid, phthiocerol dimycolates, ESX systems, nitrate reductase, PPE genes
Introduction
Mycobacterium tuberculosis is one of the world’s most successful human pathogens, with almost 10 million new cases every year [1] and more than 2 billion people latently infected. People are usually infected with M. tuberculosis by inhaling respired droplets that pass through the upper respiratory tract and deposit the bacilli in the alveoli. Here they are phagocytosed by a range of cells including neutrophils [2,3], macrophages [3] and dendritic cells (DCs) [3,4], all of which contribute to a co-ordinated immunological response to the infection that is often protective. However in some cases M. tuberculosis is able to manipulate both the innate, and the subsequent adaptive immune response, to establish an infection that may persist for a lifetime.
Although all these cells engulf M. tuberculosis into a membrane bound phagosome, the fundamental functional differences between them result in different phagosomal environments and ultimately different outcomes for each type of phagocyte. For example the neutrophil phagosome remains relatively alkaline and generates a larger oxidative burst [5] than that of macrophages. This is associated with accelerated, necrotic neutrophil death and the signalling of an inflammatory response that is capable of both activating macrophages and causing DCs to mature [6,7].
The primary host of M. tuberculosis is often considered to be the unactivated alveolar macrophage. M. tuberculosis is able to survive and proliferate in these cells by inhibiting the bactericidal processes of phagosomal fusion [8,9] and acidification [10], and by altering its metabolism to utilise intracellular substrates such as amino acids, fatty acids and cholesterol [11-13]. M. tuberculosis also manipulates the cells’ behaviour by modulating immune signalling [14] and antigen presentation [15], and by controlling the timing and mode of cell death [16,17]. Many bacterial genes have been identified as involved in these specific virulence processes [18], both by examining individual mutants, and by using genome-wide genetic screens [19-22].
M. tuberculosis are also phagocytosed by immature DCs [23,24], causing them to mature and migrate to the draining lymph nodes where antigens are presented to T cells. M. tuberculosis is capable of modulating much of this maturation cascade [25], by for instance lowering MHCI and MHCII presentation to CD8+ and CD4+ T cells respectively [26], and by altering cytokine profiles [27]. In contrast to macrophages, DCs are thought to be able to limit intracellular M. tuberculosis proliferation [28-30], despite M. tuberculosis disrupting phagosomal trafficking and preventing phagosomal fusion [31]. Transcriptional studies [29,32] indicate that the DC phagosomal environment has relatively limited connectivity with host cell recycling and biosynthetic pathways and so is nutrient limited, particularly with respect to amino acids and carbon substrates [29]. Other bactericidal stresses such as acidification, and reactive oxygen and nitrogen species that are associated with bactericidal activity in activated macrophages appear to be limited or absent in DCs [29,32,33]. Despite these studies, we still know relatively little about the phagosomal DC environment, and the interactions between it and the bacillus.
In spite of the importance of DCs in the host's response to M. tuberculosis infection, there have been no comprehensive studies of the genetic requirements of M. tuberculosis to survive inside DCs comparable to those performed in macrophages [20,34,35]. In this study we use an M. tuberculosis genome-scale transposon mutant library, generated in the clinical W-Beijing isolate, GC1237 [36], to infect human monocyte-derived DCs and monitored mutant fitness by parallel sequencing. These data comprehensively describe, for the first time, the mycobacterial genes required for survival in the DC and so identify cellular functions and components that are critical to the interplay between the DC host cell and M. tuberculosis pathogen.
Results and Discussion
Generation of a transposon mutant library in M. tuberculosis GC1237
A clinical W-Beijing strain of M. tuberculosis, GC1237 [36], was transduced with the Mycomar phage and plated onto 7H11 to generate a library of approximately 2 × 105 independent insertional mutants. This was a similar size to equivalent libraries generated previously in the laboratory strain, M. tuberculosis H37Rv [37]. Subsequent parallel sequencing of the transposon insertion sites and analysis with accumulation curves predicted that the library contained transposon insertion into 55,516 of the 62,488 predicted TA dinucleotides of the M. tuberculosis GC1237 genome (Additional file 1: Figure S1).
The dynamics of M. tuberculosis infection of DCs
The ability of intracellular M. tuberculosis to proliferate in DCs remains poorly characterised. Some studies have described intracellular growth [38-40] in DCs, while others have reported only static bacterial loads [29,30]. To clarify these contradictory observations, and to place the results of the library selection in context, we examined the dynamics of the M. tuberculosis library during the DC infection.
Dendritic cells (differentiated from donor monocytes, and with surface markers characteristic of immature DCs (Additional file 2: Figure S2)) were infected with the transposon library for 4 h at a multiplicity of infection (MOI) of 5, resulting in uptake of approximately 8 × 105 cfu per well (a number sufficient to minimise stochastic sampling errors), and equivalent to 23% of DCs containing a bacillus (Inset of Figure 1a). During the first 72 hours the number of DCs decreased to 27% of their initial number (44% of those in the uninfected control) (Figure 1b), behaviour similar to that observed by both Ryan et al. [41] and Abdalla et al. [42]. Total intracellular bacterial numbers in the DC cultures suggested that there was only limited bacterial growth during the infection (Inset of Figure 1a). However when intracellular M. tuberculosis counts were normalised to the number of live DC (Figure 1b) the number of M. tuberculosis per cell did increase during the first 72 h from an average of 0.24 cfu per DC to an average of 1.5 cfu per DC (Figure 1a). These bacterial loads are very similar to those that have been observed in DCs isolated from M. tuberculosis infected mice [4]. This rapid increase in the intracellular bacterial load was likely due, at least in part, to the re-uptake of bacilli released from lysed DCs and observed in the extracellular fraction (Figure 1c). Between day 3 and day 7 the DC population stabilised and the average bacillary load per DC did not rise significantly (although donor-to-donor variability was considerable) indicating only minimal intracellular bacterial growth (Figure 1a and Figure 1b).
Figure 1.
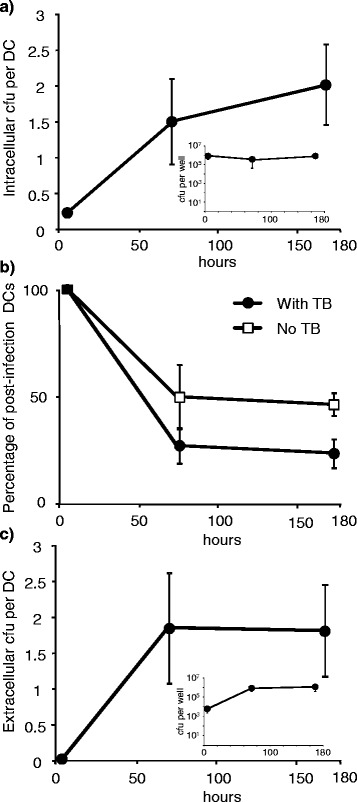
Dynamics of intracellular and extracellular M. tuberculosis and their dendritic cell host. The number of (a) intracellular M. tuberculosis per DC (b) the percentage of viable post-infection DCs and (c) the number of extracellular M. tuberculosis per DC. Total numbers of bacteria per well are shown in the plot insets. Error bars are standard errors of the mean from 5 separate experiments each with different PBMC donors.
The genetic requirements of M. tuberculosis for DC infection
To assess, in parallel, the relative fitness of each mutant within DCs, DNA from the intracellular M. tuberculosis mutants was extracted 4 hours, 3 days and 7 days after infection, and the position and frequency of transposon inserts determined.
Our data identifies a large number of mutants as having fitness costs during survival in DCs (Additional file 4). Of the genes identified as non-essential in vitro [43], mutations in 982 genes had fitness costs after 3 days and 916 after 7 days (Figure 2a); these gene sets correlated strongly with each other (Spearman’s rank-order correlation = 0.856, p < 1 × 10−6, Figure 2b), indicating that much of the selection took place during the first 3 days, with only a few extra genes having fitness effects between days 3 and 7. This perhaps reflects the stability of the DC and M. tuberculosis populations during this time (Figure 1).
Figure 2.
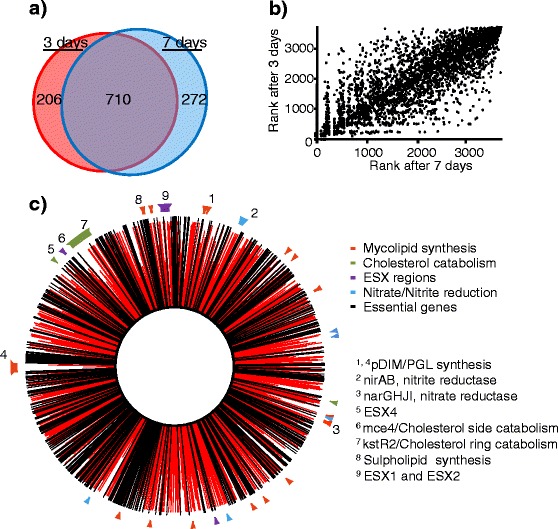
Overview of mutant ‘fitness’ during dendritic cell infection. (a) A Euler diagram (Venn diagram with areas proportional to the values) showing the numbers of mutants with significant fitness costs (p < 0.05) at different times during M. tuberculosis infection of DCs (Additional file 1), (b) a plot of the ‘fitness’ rank of mutations in individual genes 3 days and 7 days after infection, Spearman’s rank-order correlation ρ = 0.856, p < 1 × 10−6. (c) a genome plot of the ‘fitness’ of intracellular M. tuberculosis mutants 7 days after infection. Red lines indicate fitness of the gene knockouts 7 days after DC infection. Black lines are genes assigned as essential in vitro [43] and so excluded from the analysis. Relevant regions of co-functionality are indicated around the perimeter.
These ‘fitness’ associated genes implicate a diverse range of functions and structures as being important for M. tuberculosis survival in DCs, many of which are known virulence factors (p = 4 × 10−8 as reviewed by Forrellad [18]) or have been shown to be important for M. tuberculosis survival in macrophages [21]. Mutants with fitness costs included multi-gene loci associated with specific functional groups such as: the synthesis of lipids e.g. phthiocerol dimycolates (pDIMs) [44], phenolic glycolipids (PGLs) [45] and sulfolipids; cholesterol metabolism [46-48] and ESX systems [49] (Figure 2c and Figure 3). Other functions associated with fitness are more scattered across the genome, but included PPE genes, the genes required for nitrate reduction and genes involved in the metabolism of methionine/cysteine.
Figure 3.
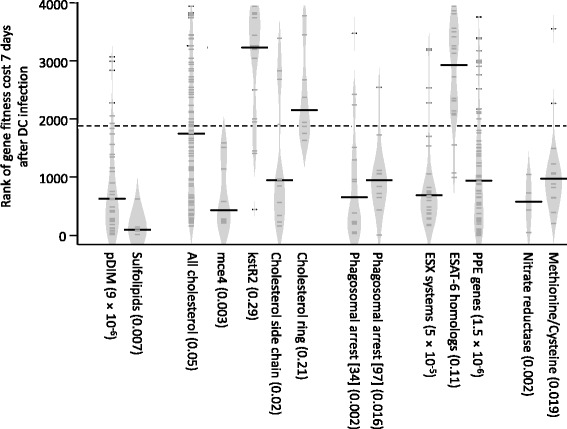
Violin plots of the ‘fitness’ of mutants ordered by functional groups 7 days after dendritic cell infection. Violin plots describing the distribution of the genes within functional gene groupings ranked by their ‘fitness’ associated p values 7 days after DC infection. Violin plots [139] are similar to box plots, but use a kernel density estimation of the probability density, similar to a smoothed distribution histogram (light grey zone), to better illustrate the overall distribution of the data. The probabilities that mutations of the genes of the functional groups had a decreased intracellular fitness, calculated using the Mann–Whitney U test, are given in brackets. Median values are indicated by the bold black lines, each gene by smaller dark grey lines, the dashed line represents the median rank for all non-essential genes [43] in the data-set.
Gene-Nutrient Interactions
Gene-nutrient interaction analysis was used to interpret our genome-scale dataset [50] and to inform further ex silico analysis. This powerful method combines the mutant’s intra-cellular ‘fitness’ data with a metabolic model (GSMN-TB [51]) to make unbiased predictions about the importance of different metabolic pathways. This analysis predicted several bacterial nutrients/pathways to be important during DC infection (Additional file 5) such as cholesterol catabolism, propionyl and valerate degradation (which is likely to be associated with the cholesterol degradation), nitrate reduction, and methionine/cysteine metabolism, as well as several that are well established as being required for intracellular survival, e.g. Fe, O2 and CO2. Although powerful, this approach was somewhat constrained by the redundancy inherent in M. tuberculosis metabolism and by the wide variety of theoretical substrates available to M. tuberculosisin silico and so our interpretation of these outputs remains cautious. It is perhaps most useful when used to independently direct further ex silico analysis.
Cell wall lipids are important for M. tuberculosis survival in DCs
The lipids of the mycobacterial cell wall are well established as important virulence factors both in vivo and in macrophages [18]. The present study confirms that many of these cell wall lipids are also of primary importance for fitness in DCs. Loci involved in their synthesis and modification were amongst the genomic regions most strongly associated with fitness costs.
Mycoserate containing lipids: pDIMs and PGLs. Mycoserates and the related lipids, phthiocerol dimycolates (pDIMs) and phenolic glycolipids (PGLs) are recognised as some of the most important M. tuberculosis virulence factors, particularly in Beijing strains, such as GC1237, which can have unusual pDIMs and PGLs that have been equated with hypervirulence and an ability to modulate cytokine responses [52,53].
In our DC experiments, mutations known to be associated with a loss of pDIM production (Rv2929 to Rv2942) [44] were strongly associated with fitness cost both 3 days (p = 5 × 10−7), and 7 days (p = 8 × 10−12) after the DCs were infected (Figure 3 and Additional file 4). By day 7, all the genes of the pDIM loci [44] except drrA had a fitness cost when mutated, as well as much of the Rv0096-Rv0101 loci, another region associated with pDIM production and virulence in mice [54]. This correlates with reports of pDIM mutants being attenuated in both mice [55] and in macrophages [56], phenotypes that have been associated with an inability to prevent phagosomal acidification [56]. Observations by Tailleux et al. [32] that the pDIM region was upregulated during infection of DCs, further supporting a role for pDIMs in DCs.
Mutation of many of the genes unique to the production of PGL also had a fitness cost (pkS15/1, Rv2957-2959c [57] and Rv2962c) at day 3 and day 7 suggesting that PGL, at least in our GC1237 strain, has a role in M. tuberculosis survival in DCs (Additional file 4). This is in contrast to the situation observed in macrophages infected with H37Rv, where PGL associated attenuation was only observed in pDIM-minus strains [56]. Whether this anomaly represents a difference between DC and macrophages, or whether it is associated with strain-specific difference in PGL profiles is difficult to ascertain.
Lipids containing mycolic acids: Cord factor and sulfolipids. Trehalose dimycolate (TDM or cord factor) and trehalose monomycolate (TMM) are another group of well characterised M. tuberculosis virulence factors. Cells with lowered amounts of TMM and TDM are less able to inhibit lysosomal fusion in macrophages [58] and less able to grow in macrophages [59]. In this study we observed that TDM is also an important virulence factor in DCs (Additional file 4).
As cord factor is an essential component of the mycobacterial cell wall [60] many of the genes involved in its synthesis will not be present in the library. However, some mutants that alter the amount, or change the structural details, are viable. One such group of genes is the Ag85 complex, fbpABC, which are all partially redundant and yet all had fitness costs in DCs (Additional file 4). These genes are involved in the transfer of mycolic acid from one TMM to another, to generate TDM and release trehalose [60]. It has been previously shown that mutating fbpA and fbpC produces M. tuberculosis that contains less TDM than wild type strains, that are less able to evade killing within macrophages, and that are more susceptible to reactive oxygen (ROS) and reactive nitrogen (RNI) species [61]. In other studies fbpA and fbpC have been shown to be less able to modulate the expression of MHC-II in DCs and are less able to restrict T-cell activation [60,61]. Mutants of fbpD, which has no TMM/TDM phenotype and is thought not to be a mycolate transferase [62], did not have fitness costs in DCs in our study.
Mutations in genes that modify the structure of the mycolic acids that are bound to the trehalose of TMM and TDM also had fitness costs in DCs (Additional file 4). The acylating genes papA3 [63] and pks4, and the mycolic acid transporter, mmpl10 [64] all had fitness costs after both 3 and 7 days in DCs. Mutantions that alter or abolish the methyl, keto and cyclopropyl modifications of these mycolic acids (mmaA1 [65], mmaA3 [66], mmaA4 [67,68], umaA [69]) also were less fit in DCs (mma2 and cma2 partially complement one another [65] and did not have a fitness cost when mutated). These mutations are known to have altered virulence characteristics; mmaA1, mmaA3, mmaA4 and umaA being highly attenuated and hyper-inflammatory in macrophages and in murine infection models [66,67,70].
Sulfolipids, are another group of acylated trehaloses that are abundant in the mycobacterial cell wall [71]. The ability of purified sulfolipids to modify cellular responses is well established [72,73] but there are fewer reports of a direct role in virulence. Gilmore [74] observed a decrease in intracellular growth in macrophages in sulfolipid minus mutants, while Passemer et. al. [56] also noted a loss of virulence in macrophages but only in pDIM minus M. tuberculosis strains. In our DC studies the mutations of the genes involved in sulfolipid synthesis (Rv3820-Rv3826) [75-77]] all had fitness costs (p = 0.008 and 0.007, after 3 and 7 days post-infection, respectively) as did the sulfolipid transporter, mmpl8 [76], despite the production of pDIMs (Figure 3 and Additional file 4). Gene nutrient interaction analysis also identified cysteine/methionine metabolism as important for survival in DCs (p = 0.019 after 7 days; Figure 3 and Additional file 4). Although this may be associated with a requirement for amino acid synthesis, this seems unlikely as it is thought that methionine is available to M. tuberculosis in vivo [78]. However, a functional sulphur metabolism would be required for the supply of sulphur from such amino acid substrates to sulfolipid synthesis and this may in part explain the fitness costs associated with cysteine/methionine metabolism. Taken together, these data clearly show that in DCs, sulfolipids have an important and as yet, not fully elucidated, role even when pDIMs are present.
Lipids: PIM, LM and LAM. Phosphatidylinositol mannosides (PIM), lipomannan (LM) and lipoarabinomannans (LAM) are also essential components of the M. tuberculosis cell wall and so genes required for their synthesis [79,80] will not be present in the library. Both PIM and LAM have been implicated in survival in macrophages, by amongst other mechanisms, the inhibition of phagolysosomal fusion [81], and the modulation of both cytokine responses and of the phagosomal maturation process [25]. Much of this activity is thought to require LAM with an intact mannose cap [82], although cap-less mutants (∆Rv1635c) have been reported to have an unaltered wild-type phenotype in murine macrophages and in vivo [83]. In our human DC experiments, mutants predicted to result in a non-mannose capped LAM (Rv2181, Rv1635c, Rv1565c) all had a fitness cost both 3 days and 7 days post-infection (Additional file 4). This is consistent with studies showing that M. tuberculosis binding to human DCs involves interaction of manLAM with the receptor DC-SIGN [29,84].
Cholesterol side chain catabolism contributes to fitness in DCs
Cholesterol is both an important nutrient for intracellular M. tuberculosis and a critical part of the host cell’s response to M. tuberculosis [85]. As such, the catabolism of cholesterol has multiple functions during macrophage infection. It provides propionyl-coA precursors for the synthesis of pDIM [86,87] and the sulfolipid, SL-1 [88] via methyl malonyl-coA. It can be used to supply carbon to central metabolism of M. tuberculosis as succinate and pyruvate [86]. Finally, the degradation of host cholesterol can be involved in the modulation of intracellular trafficking and immune signalling [89-91]. Prior to this present study, it was not known which of these functions, if any, were important during DC infection, although cholesterol catabolism genes and the associated kstR regulon [92] were known to be upregulated [32]. Our data provides evidence for the importance of cholesterol catabolism during DC infection (p = 0.05, Figure 2c and Figure 3), an interpretation confirmed by the gene-nutrient interaction analysis (Additional file 5). However fitness costs were mainly associated with mutants of the mce4 cholesterol transporting operon [47](p = 0.0026 after 7 days), genes involved in early steps in cholesterol catabolism eg choD, Rv1106c, kstD, and genes involved in side chain degradation [48,93] (Figure 3, Additional file 4, Biocyc pathway p = 0.0002 after 7 days), notably the igr region [94,95]. Fitness costs were not apparent for mutations in genes involved in sterol ring degrading pathways, such as hsaA-G (although several of these are known to have only minimal effects on growth in cholesterol e.g. hsaEFG [48], and the genes of the kstR2 regulon [46,96].
The fitness costs associated with disruption of cholesterol side chain degradation pathways and the importance of mycolic acid synthesis during DC infection together suggests that a critical function of cholesterol degradation is to supply propionyl-CoA for the synthesis of pDIM and SL-1. Both which are virulence factors that we have shown are important for survival in DCs. However, an absolute requirement for cholesterol derived carbon seems unlikely, as the genes required for the release of carbon from the sterol component of cholesterol [48] did not have fitness costs. There is however evidence for a role for cholesterol catabolism in modulating the DC response to M. tuberculosis. Consistent with previous findings in macrophages, our data indicates a fitness cost for mutations in choD. ChoD is an extracellular cholesterol oxidase that when mutated does not abolish the M. tuberculosis ability to grow on cholesterol, but does attenuate M. tuberculosis in macrophages via inhibition of TLR2 signalling [91] and reduction of iNOS and ROS responses [89].
Genes that prevent phagosomal maturation in macrophages also have a fitness cost in DCs
M. tuberculosis residing in both DCs and macrophages are contained within phagosomes, whose development has been arrested at an early stage in the endocytic pathway [29]. However macrophage and DC phagosomes do differ in other respects: DC mycobacterium-containing phagosomes undergo only limited acidification and have more restricted connectivity to other parts of the endocytic system [29,31]. Although we do not assay for a phagosome maturation phenotype in this study, previous studies have demonstrated that in general M. tuberculosis mutants with an abrogated ability to inhibit macrophage phagosomal maturation are also attenuated for intracellular survival [34]. We compared our intra-DC fitness data with published data-sets generated using genome-wide screens for mutants that are defective in their ability to inhibit murine macrophage phagosome maturation [34,97,98]. We observed that many of these phagosome-control mutants also had reduced fitness in DCs at both 3 and 7 days post-infection (p = 0.058 /0.0027 [34,97] and p = 0.016/0.0023 [34,97] respectively) (Figure 3 and Additional file 4). Similarly, many genes recognised individually as involved in modulating phagosomal maturation e.g. sapM [99], PE_PGRS62 [100], PE_PGRS30 [101], eis [102] also had fitness costs. Finally, many of the virulence associated functions and structures such as cell-wall lipids and LAM [103], that we have already identified as important for fitness in these DCs are known to be involved in preventing phagosomal maturation, properties which may in part explain the intra-DC fitness costs of mutating these genes. We conclude that despite the difference between DC and macrophage phagosomes, control of phagosome-lysosome fusion remains as important for M. tuberculosis fitness in human DCs as it is in macrophages and is modulated using similar mechanisms.
A novel role for ESX transport systems in DC
There are five ESX/type VII secretion systems/regions in the M. tuberculosis genome, of which two are essential for in vitro growth on microbiological medium (ESX-3 and ESX-5) [104,105]. We show that mutations, particularly in the pore-forming components [49], of the other three ESX systems (ESX-1, ESX-2 and ESX-4) all had fitness costs for growth in human DCs (p = 0.0007, 0.0005 and 0.042 respectively after 7 days, Figure 3 and Additional file 4). ESX-1 is the best characterised of these systems and is involved in the export of the classic virulence factors, ESAT-6, CFP-10 and many Esp proteins. ESX-2 and ESX-4 are less well studied and until now have not been recognised as virulence determinants [106].
Unexpectedly, the intra-DC fitness cost associated with loss of the ESX-1 secretion apparatus were not found to be associated with mutants of the canonical ESX-1 substrates, ESAT-6 and CFP10 (Figure 3 and Additional file 4). This discrepancy, which has been observed previously in mice [107,108], implies an additional function for the ESX-1 system that is important for survival and virulence in DCs. Work by Joshi et al. [109] with an eccA1 mutant of M. marinum supports such a conclusion, with evidence that ESX-1 is not only required for the export of ESAT-6 type proteins, but also for the export of enzymes involved in the synthesis of mycolic acids, pDIMs and PGLs e.g. Pks13, KasB, KasA, MmaA4, Pks5, Mas, Pks15/1, PpsD, and PpsE. This links with our findings that production of wild-type amounts of these lipids is important for survival in DCs. Many PPE genes are also thought to be exported by ESX systems [49,110]. Indeed, in M. marinum ESX-5 has been shown to export many of the PPE proteins [111,112]. These PPE genes are defined by a proline-glutamate-glutamate motif, but may have varying roles in M. tuberculosis physiology and virulence and remain poorly characterized (for a review see Akhter et al., 2012 [113]). This study shows strikingly that many of these PPE proteins are important for fitness in the human DC with over half of the annotated PPE mutants lost from the library by day 7 (p = 1.5 × 10−6), including many of the PPE genes chromosomally and functionally associated with ESX-5 in M. marinum (PPE28, PPE29, PPE30, PPE31, PPE32 and PPE33) [111]. It seems reasonable to extrapolate that in M. tuberculosis, ESX-5 may also secrete these PPE proteins and potentially many of the numerous other PPE proteins that we have shown are important for intra-DC virulence. Thus we believe ESX-5 is an important virulence factor in M. tuberculosis.
M. tuberculosis responses to oxidative and nitrosative stress
Human DCs are thought to produce less ROS than macrophages in response to M. tuberculosis infection [32,114] and are more resistant to ROS associated apoptosis. M. tuberculosis resistance to ROS is mediated via a range of genes and cellular components, such as the cell wall, which scavenges free radicals, and protein and DNA repair systems, which are actively involved in withstanding ROS stress [115]. Many genes associated with ROS resistance do have fitness costs in DCs (Additional file 4): genes of methionine/cysteine metabolism (as identified by gene-nutrient interaction analysis) [116]; glutamyl-cysteine ligase; Rv1806 to Rv1809 (PPE20, PPE30-33) [117]; as well as many genes involved in the synthesis of the cell wall components such as LAM, PGL and mycoseric acid. However it is difficult to distinguish a ROS specific role for many of these pleiotropic genes. Specific evidence for significant ROS associated fitness costs in DCs is also difficult to equate with the lack of fitness cost for genes such as those of the oxidized guanine (GO) DNA repair system [118], lsr2 [88], ahpC [119], or mycothiol pathways [120,121], making predictions as to the role of ROS in M. tuberculosis infection of DCs unclear, but suggesting that it does not represent a dominant antimicrobial mechanism.
The role of nitrogen radicals in human DCs is also not well established. Murine DCs are known to produce a nitrosative burst upon M. tuberculosis infection, but there is less evidence for an equivalent response in humans [32,122]. We find that many of the genes that have been associated with resistance to reactive nitrogen in macrophages are also important during DC infection e.g. fbiC, Rv2115c, Rv2097 [123], the nucleotide excision repair system encoded by uvrABCD1D2 [124] and nitrate reductase associated genes (narG, narH, narI, narK1, narK2, narX, nirB, p = 0.0021, Figure 3 and Additional file 4). In macrophages, nitrate reductase is important for many of the M. tuberculosis responses to reactive nitrogen. An active nitrate reductase in the presence of NO, has been shown to promote M. tuberculosis growth [125], to be a source of nitrite (which could then acts as signal molecule for M. tuberculosis) [122] or potentially disrupt NO signalled apoptosis [126]. Without nitrate reductase M. tuberculosis is more susceptible to both acid [127] and peroxide stress [128]. Our data imply that NO, or at least nitrate, is indeed present in the phago-lysosome of human DCs. This requirement for nitrate reductase is unlikely to be associated with resistance to acid (at least in DCs) as has been described for macrophages [127] but more likely relates to its other growth promoting roles such as providing an alternative electron acceptor, making available a source of metabolic nitrogen, or as a mechanism of preventing apoptosis.
Conclusions
The ability to survive within DCs is integral to the ability of M. tuberculosis to manipulate and on occasions usurp the host's immune response. As such the genetic requirements for these processes represent important M. tuberculosis virulence components. By using high-throughput analysis of transposon libraries we have produced a comprehensive genome-scale assessment of the genetic requirements of a clinical M. tuberculosis W-Beijing isolate, GC1237, for survival in DCs. Such an approach connects gene and function, allowing analysis at multiple scales: from single loci, regulon, metabolic pathway, and even whole cell, rather than on the gene-by-gene basis produced by more conventional gene knockout strategies involving one or two mutants. Such a strategy has been used previously with the laboratory strain M. tuberculosis H37Rv, to successfully describe the genes required for a range of virulence related environments: in vitro growth [48]; infection of mice [37]; survival in murine macrophage, human cell lines and murine bone marrow derived macrophages [21]; and for the ability to inhibit phagosome maturation [22,34,97,98].
Our data shows that the survival of M. tuberculosis within human DCs requires a large number of genes (see Figure 4 for a summary), many of which have previously been recognised as also being important during the invasion and survival of macrophages. Perhaps the best characterised of these are the genes involved in the synthesis, transport and modification of cell wall lipids. These lipids are clearly very important to the ability of M. tuberculosis to survive within DCs. Our data demonstrates that not only are the well-established virulence associated lipids important such as pDIM and cord factor, but also lipids that had previously only been ascribed secondary roles such as PGL and sulfolipids [56]. These secondary lipids have long been recognised as modulators of macrophages when added exogenously [74], or associated with modulating cytokine production [52,53] and virulence in mice, but have not previously been shown to have a direct effect on intracellular virulence of M. tuberculosis [56] and may be especially important during DC infection. The apparent requirement for M. tuberculosis GC1237 to have functional cholesterol catabolism and sulphur metabolism when in DCs may also be linked to the supply of the metabolites required for the synthesis of these cell wall lipids, although roles in nutrition or immune modulation cannot be ruled out.
Figure 4.
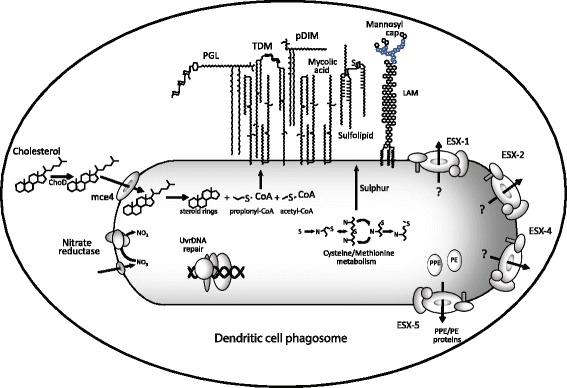
Summary of the major cellular components and processes that are associated with fitness costs during M. tuberculosis survival within DC.
In macrophages, cell wall lipids act, in part, by modulating the phagosomal environment. Despite differences in the physiology and cell biology of macrophage and DC, mycobacterial modulation of the phagosome remains important in DCs, with similarities between genes required for survival in DCs and groups of genes identified as required for phagosomal modulation in macrophages. The ESX-1 secretion system is well established as a fundamental virulence mechanism of M. tuberculosis, being involved in host-pathogen interactions including phagosome escape and cytotoxicity. Here, we demonstrate a role during DC infection for not only ESX-1 but also ESX-2 and ESX-4, and a requirement for PPE genes, which implicates ESX-5 as also having a direct role in mycobacterial pathogenesis. The functions of the abundant and characteristic PPE gene/protein family have remained elusive but we show, for the first time, that the intra-DC environment represents a powerful non-redundant selection for their maintenance in the genome. We also identify important roles for sulphur and nitrogen metabolism. Cysteine/methionine metabolism is an important fitness determinant, possibly related to resistance to reactive species or to the production of sulfolipids. Nitrate reductase was also important during survival within DCs, and again the mechanism is unclear, but resistance to reactive nitrogen intermediates, nitrogen acquisition, or a role in intercellular signalling would seem likely candidates.
Clearly, many M. tuberculosis intracellular virulence mechanisms are common to both macrophages and DCs. This may reflect the close relationship between the myeloid DC and the macrophage which represent different but related cell types within a single mononuclear phagocytic system. However, we also identify novel DC virulence mechanisms that are important in human DC infection but which are either absent or have been overlooked in macrophages. Further investigations into the mechanisms that underlie the importance of these functions during infection are required to further our understanding of the nature of the inter-relationship between host and pathogen during pathogenesis.
Methods
M. tuberculosis culture and library construction
M. tuberculosis GC1237, a W-Beijing strain [36] was routinely grown at 37°C on Middlebrook 7H11 plates containing 10% OADC and 0.2% glycerol or in 7H9 media containing 10% ADC, 0.2% glycerol and 0.05% Tween 80®. M. tuberculosis were enumerated by plating 10 μl drops of serially diluted culture onto 7H11 plates. M. smegmatis mc2 were grown in Nutrient Broth II with 0.2% glycerol and 0.05% Tween 80® or on solid Nutrient Broth II media with 0.2% glycerol. The temperature sensitive phage, phAE87 [129] containing pMycoMar [130] was prepared by adding an aliquot of late log phase M. smegmatis to Nutrient Broth II top agar with an appropriate number of phage such that the resultant plaques were touching. After 3 days at 30°C the top agar was scraped from the plates and shaken with an equal volume of MP buffer (150 mM NaCl, 50 mM Tris, 10 mM MgSO4, 2 mM CaCl2, pH 7.4), the preparation was centrifuged, the supernatant passed through a 0.22 μM filter, and the phage enumerated. The M. tuberculosis library was prepared by washing an M. tuberculosis GC1237 culture, of approximate OD600 1.0 with an equal volume of MP buffer and resuspending in 0.1 volumes of phAE87 at an multiplicity of infection (MOI) of between 10–100, incubating at 37°C for 4 h and plating on 7H11 media containing kanamycin at 30 μg ml−1. The resultant library was scraped from the plates and stored at −80°C in 10% glycerol.
Generation of monocyte derived dendritic cells
Monocytes were isolated from leukocyte cones (National Health Service Blood and Transplant, UK), from 5 different donors. For each, the cones were diluted with PBS containing 2 mM EDTA, layered onto HistoPacque®-1077, centrifuged at 400 × g for 30 min and allowed to come to rest without braking. Monocytes were purified from the resulting buffy coat using CD14 Microbeads (Miltenyi, Bisley, UK) as recommended by the manufacturers. Approximately 1 x 107 monocytes were differentiated into DCs in 75 cm2 flasks incubated at 37°C with 5% CO2. DCs were grown in RPMI 1640 containing GlutaMax™ (Invitrogen, Paisley, UK), supplemented with 10% Fetal Calf Serum (FCS) and 10 ng ml−1 GM-CSF (PeproTech, London, UK) and 20 ng ml−1 IL-4 (PeproTech). After 6 days the differentiated cells were washed three times in PBS by centrifugation, resuspended in fresh growth media and 3 × 106 cells per well cultured in 6 well tissue culture plates.
Flow Cytometry
DCs were washed in FACS buffer (PBS containing 5 mM EDTA, 1% FCS, 0.05% sodium azide). The cells were resuspended in FITC or PE conjugated antibody for CD14, CD83, CD86, CD1a or appropriate isotype controls diluted 1:100 or 1:20 with FACS buffer. The cells were incubated on ice for 30 min and then washed twice in FACS buffer, before resuspending in 4% paraformaldehyde in PBS. Samples were run on FACS Canto™ (BD Biosciences, Oxford, UK) recording 10,000 events.
Infection of dendritic cells with M. tuberculosis
Each DC preparation was infected with a previously frozen aliquot of the M. tuberculosis library cultured for 24 h in 7H9 with ADC but without Tween 80®. Broths were centrifuged and resuspended in DC growth media, and passed multiple times through a 26G needle to disperse clumps. The inocula concentration was estimated by measuring optical density at 600 nM and diluted in DC growth media to give a MOI of 5. The M. tuberculosis were added to the DCs and the infection incubated at 37°C for 4 h. Control wells contained either no M. tuberculosis or no DCs.
After the infection, M. tuberculosis that were not internalized were removed by washing the wells three times with DC growth medium. Planktonic DCs in these first washes were then separated from the bacteria by combining the supernatants, and centrifuging at 500 × g for 5 min three times, each time removing the supernatant and resuspending the cell pellet in fresh DC growth medium. The supernatants were combined and the “extracellular” M. tuberculosis recovered by centrifuging at 3500 × g for 5 min and enumerated by plating on 7H11 and counting colony forming units.
The intracellular fraction was prepared by resuspending the planktonic cell pellet from the extracellular preparation in 0.1% Triton X-100® and adding this back to the adherent cells remaining in the well. This lysate was centrifuged at 3500 × g for 5 min, the bacterial pellet resuspended in an appropriate volume, enumerated and the remainder spread on plates to recover the library for DNA purification and TnSeq analysis (see below). For DC counts, the growth media was removed from a well and centrifuged at 500 × g to give a cellular pellet. Versene™ (Invitrogen) was added to the well and the plate incubated at 37°C for 10 min. The Versene™ was removed and used to resuspend the cellular pellet, and live and dead cells counted using a Trypan Blue exclusion assay. Infections were sampled immediately after washing (4 hours), after 3 days and after 7 days. Recovered libraries were scraped from the plates and stored at −80°C in 10% glycerol.
DNA purification and preparation
DNA was extracted from recovered libraries from three of the infections. An aliquot of each recovered library was added to a lysing matrix B tube (MPBiomedicals, Loughborough, UK) containing 25:24:1 phenol:chloroform:isoamyl alchohol and TE, and mechanically disrupted in a Fastprep™ FP120 (MPBiomedicals) at setting 4 for 30 sec. The preparation was centrifuged at 12,000 × g for 5 min, and the supernatant washed twice with chloroform, and precipitated with × 0.7 volumes isopropanol and × 0.1 volumes 3 M sodium acetate. DNA was resuspended in water and sheared by sonicating 3 times for 30 sec each (1 sec on/1 sec off) with a VibraCell® VC300 and a 6 mm probe. Sheared DNA was repaired by incubating with 100 U μl−1 T4 DNA polymerase (Promega, Southampton, UK), 50 mU μl−1 Klenow (Promega), 100 mU μl−1 polynucleotide kinase (New England Biolabs, Hitchin, UK), 2 mM dNTPs all in × 1 T4 DNA ligase buffer (Promega) for 30 min at 20°C. Repaired DNA was purified using QIAquick™ columns (Qiagen, Crawley, UK), and end labelled with A nucleotides by incubating with 0.25 U μl−1 Klenow, 0.2 mM dATP in × 1 Klenow buffer (Promega) for 30 min at 37°C and purified on MiniElute™ PCR purification kits.
Transposon amplification
A linker was generated by annealing oligonucleotides linker1 and linker2 (Additional file 3: Table S2a) at equimolar concentrations in 50 mM NaCl, 5 mM Tris, 0.5 mM EDTA, pH 8 by heating the mix to 95°C for 2 min and then cooling for approximately 1 h to 20°C. The linker was annealed to the sheared genomic DNA at eqimolar ratios of fragment ends using T4 DNA ligase incubated at 16°C for 18 h. The fragments were PCR amplified with 0.2 μM PCR-linker primer and either 0.2 μM primer PCR-MarinerA or PCR-MarinerB (Additional file 3: Table S1), using 0.02 U μl−1 KOD Hot Start DNA polymerase (Novagen, Feltham, UK), ×1 KOD buffer, 0.2 mM dNTPs and 1.5 mM MgCl2. Conditions were 94°C for 2 min, followed by 5 cycles of 95°C for 30 sec, 69°C for 10 sec, 70°C for 30 sec, followed by another 5 cycles with an annealing temperature of 67°C, followed by 20 cycles with an annealing temperature of 65°C. Products sized between 250–500 bp were cut from an agarose gel, cleaned with QIAquick columns, and equal amounts of each PCR product (MarinerA and MarinerB) combined and amplified with primers PCR2-linker and PCR2-Mariner as before but with cycle conditions of 94°C for 2 min, followed by 9 cycles of 95°C for 30 sec, 57°C for 10 sec and 70°C for 30 sec. Fragment sizes were assessed and products quantified with a BioAnalyzer 2100 (Agilent). The indexed products from different samples were combined, spiked with 50% PhiX and sequenced on an Illumina® 2000 HiSeq™ machine, with a single index and a single read of 100 cycles.
Sequence analysis and statistical analysis
Sequence reads were assigned to samples according to their indices and aligned to the GC1237 genome using Bowtie2 [131]. Transposon TA junctions were identified from sequence reads that contained a fragment of GC1237 genome preceded by the terminal-end of the transposon sequence (5’ CGGGGACTTATCAGCCAACCTGT, 2 mismatches were permitted). Mutant fitness was determined by comparing transposon frequencies between time points (between 4 h and 3 days, and 4 h and 7 days) using a method based upon the logistic distribution of the difference of two extreme value random variables [132]. This approach depends upon the difference in length of the two longest transposon-free runs at two time points with null hypothesis of zero difference. Values for the three repeats were combined using Fisher’s combined probability test as reviewed by Loughin, 2004 [133], and a value of p < 0.05 was considered to represent a significant loss of fitness. Correlations between the library at different time points were assessed using the Spearman’s rank order coefficient. The significance of gene group enrichments was determined using the Mann–Whitney U test (Wilcoxon rank-sum test).
Accumulation curves
To determine the library coverage the number of unique TA sites mutated was plotted against the number of insertions sequenced. Asymptotes were calculated by fitting Michaelis-Menten curves to the plots.
Gene Nutrient Interactions
Gene-nutrient interaction (GNI) analysis, as developed by Idit et al. [50], aims to describe the relationship between gene essentiality and the presence or absence of nutrients in the growth environment. Using the approach, the 101 nutrients from the GSMN-TB M. tuberculosis metabolic model [51] were randomly sampled to generate 1.17 × 106in silico growth media. Flux balance analysis was carried for each growth media to identify essential genes. A gene was considered essential if the drop in the growth rate after its knockout was more than 20 %. Positive/negative GNI were identified when a nutrient was present/absent in a significantly number of media under which the gene was essential, as estimated by calculating a p-value based on the hypergeometric distribution. The calculation above has been implemented as an extension to Surrey FBA software [134] and is available on request.
Availability of supporting data
All raw sequence read files have been submitted to the Sequence Read Archive of the National Center for Biotechnology Information under SRA Study accession, SRP057496.
Acknowledgements
The authors would also like to thank Richard Talbot of Ark Genomics, Edinburgh for technical advice regarding sequencing; Amine Namouchi and Brigitte Gicquel of the Institut Pasteur, Paris for providing the genome of M. tuberculosis GC1237; and Rachel Butler of the University of Surrey for support using the flow cytometer. This work was supported by EraSysBioPlus TB-HOST-NET grant BB/I00453X/1 and Wellcome Trust grant WT090242MA.
Abbreviations
- DC
Dendritic cells
- GNI
Gene nutrient interaction
- LAM
Lipoarabinomannans
- LM
Lipomannan
- MOI
Multiplicity of infection
- Mtb
Mycobacterium tuberculosis
- PBMC
Peripheral blood monocytes
- PBS
Phosphate buffered saline
- PCR
Polymerase chain reaction
- pDIM
Phthiocerol dimycolates
- PGL
Phenolic glycolipids
- PIM
Phosphatidylinositol mannosides
- ROS
Reactive oxygen species
- TCA
Tricarboxylic acid cycle
- TDM
Trehalose dimycolate
- TMM
Trehalose monomycolate
Additional files
An accumulation curve of the transposon insertion sites in the M. tuberculosis library.
Flow cytometry data of surface markers of differentiated PBMCs.
A list of primers used in this study.
An Excel file containing p values associated with the fitness during survival in DC, arranged by functional group.
An Excel file containing the results of gene nutrient-interaction analysis.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
TAM carried out the experiments and contributed to the planning, data analysis, results interpretation and writing. HW contributed to the data analysis and computer simulations. AMK contributed to the planning, data analysis and computer simulations. GRS contributed the planning, results interpretation and writing. All authors read and approved the final manuscript.
Contributor Information
Tom A Mendum, Email: t.mendum@surrey.ac.uk.
Huihai Wu, Email: h.wu@surrey.ac.uk.
Andrzej M Kierzek, Email: a.kierzek@surrey.ac.uk.
Graham R Stewart, Email: g.stewart@surrey.ac.uk.
References
- 1.Dye C, Williams BG. The Population Dynamics and Control of Tuberculosis. Science. 2010;328:856–861. doi: 10.1126/science.1185449. [DOI] [PubMed] [Google Scholar]
- 2.Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, Cho SN, Via LE, Barry III. Neutrophils Are the Predominant Infected Phagocytic Cells in the Airways of Patients With Active Pulmonary TB. CHEST Journal. 2010;137:122–128. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolf AJ, Linas B, Trevejo-Nuñez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. Mycobacterium tuberculosis Infects Dendritic Cells with High Frequency and Impairs Their Function In Vivo. The Journal of Immunology. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Romo GS, Pedroza-Gonzalez A, Lambrecht BN, Aquilar-Leon D, Estrada-Garcia I, Hernandez-Pando R, Flores-Romo L. Mycobacterium tuberculosis manipulates pulmonary APCs subverting early immune responses. Immunobiology. 2013;218:393–401. doi: 10.1016/j.imbio.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 5.Segal AW, Geisow M, Garcia R, Harper A, Miller R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature. 1981;290:406–409. doi: 10.1038/290406a0. [DOI] [PubMed] [Google Scholar]
- 6.Persson YA, Blomgran-Julinder R, Rahman S, Zheng L, Stendahl O. Mycobacterium tuberculosis-induced apoptotic neutrophils trigger a pro-inflammatory response in macrophages through release of heat shock protein 72, acting in synergy with the bacteria. Microbes and Infection. 2008;10:233–240. doi: 10.1016/j.micinf.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Hedlund S, Persson A, Vujic A, Che KF, Stendahl O, Larsson M. Dendritic cell activation by sensing Mycobacterium tuberculosisG€“induced apoptotic neutrophils via DC-SIGN. Human Immunology. 2010;71:535–540. doi: 10.1016/j.humimm.2010.02.022. [DOI] [PubMed] [Google Scholar]
- 8.Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, Allen RD, Gluck SL, Heuser J, Russell DG. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science. 1994;263:678–681. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- 9.Russell DG, Dant J, Sturgill-Koszycki S. Mycobacterium avium- and Mycobacterium tuberculosis-containing vacuoles are dynamic, fusion-competent vesicles that are accessible to glycosphingolipids from the host cell plasmalemma. J Immunol. 1996;156:4764–4773. [PubMed] [Google Scholar]
- 10.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 11.Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beste DJ, Noh K, Niedenfuhr S, Mendum TA, Hawkins ND, Ward JL, Beale MH, Wiechert W, McFadden J. 13C-flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular Mycobacterium tuberculosis. Chem Biol. 2013;20:1012–1021. doi: 10.1016/j.chembiol.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doz E, Rose S, Nigou J, Gilleron M, Puzo G, Erard F, Ryffel B, Quesniaux VF. Acylation determines the toll-like receptor (TLR)-dependent positive versus TLR2-, mannose receptor-, and SIGNR1-independent negative regulation of pro-inflammatory cytokines by mycobacterial lipomannan. J Biol Chem. 2007;282:26014–26025. doi: 10.1074/jbc.M702690200. [DOI] [PubMed] [Google Scholar]
- 15.Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol. 2010;8:296–307. doi: 10.1038/nrmicro2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butler RE, Brodin P, Jang J, Jang MS, Robertson BD, Gicquel B, Stewart GR. The balance of apoptotic and necrotic cell death in Mycobacterium tuberculosis infected macrophages is not dependent on bacterial virulence. PLoS One. 2012;7 doi: 10.1371/journal.pone.0047573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–2020. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 18.Forrellad MA, Klepp LI, Gioffre A, Sabioy Garcia J, Morbidoni HR, de la Paz Santangelo M, Cataldi AA, Bigi F. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4:3–66. doi: 10.4161/viru.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosas-Magallanes V, Stadthagen-Gomez G, Rauzier J, Barreiro LB, Tailleux L, Boudou F, Griffin R, Nigou J, Jackson M, Gicquel B, Neyrolles O. Signature-tagged transposon mutagenesis identifies novel Mycobacterium tuberculosis genes involved in the parasitism of human macrophages. Infect Immun. 2007;75:504–507. doi: 10.1128/IAI.00058-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Molecular Microbiology. 1999;34:257–267. doi: 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- 21.Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci USA. 2005;102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart GR, Patel J, Robertson BD, Rae A, Young DB. Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog. 2005;1:269–278. doi: 10.1371/journal.ppat.0010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mihret A, Mamo G, Tafesse M, Hailu A, Parida S. Dendritic Cells Activate and Mature after Infection with Mycobacterium tuberculosis. BMC Research Notes. 2011;4:247. doi: 10.1186/1756-0500-4-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. The Journal of Immunology. 1997;159:635–643. [PubMed] [Google Scholar]
- 25.Geijtenbeek T, Van Vliet SJ, Koppel EA, Sanchez-Hernandez M, Vandenbroucke-Grauls CM, Appelmelk B, Van Kooyk Y. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med. 2003;197:7. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hava DL, van der Wel N, Cohen N, Dascher CC, Houben D, León L, Agarwal S, Sugita M, van Zon M, Kent SC, et al. Evasion of peptide, but not lipid antigen presentation, through pathogen-induced dendritic cell maturation. Proceedings of the National Academy of Sciences. 2008;105:11281–11286. doi: 10.1073/pnas.0804681105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demangel C, Bertolino P, Britton WJ. Autocrine IL-10 impairs dendritic cell (DC)-derived immune responses to mycobacterial infection by suppressing DC trafficking to draining lymph nodes and local IL-12 production. European Journal of Immunology. 2002;32:994–1002. doi: 10.1002/1521-4141(200204)32:4<994::AID-IMMU994>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 28.Buettner M, Meinken C, Bastian M, Bhat R, St-Ýssel E, Faller G, Cianciolo G, Ficker J, Wagner M, RÝllinghoff M, Stenger S. Inverse Correlation of Maturity and Antibacterial Activity in Human Dendritic Cells. The Journal of Immunology. 2005;174:4203–4209. doi: 10.4049/jimmunol.174.7.4203. [DOI] [PubMed] [Google Scholar]
- 29.Tailleux L, Neyrolles O, Honoré-Bouakline S, Perret E, Sanchez FO, Abastado JP, Lagrange PH, Gluckman JC, Rosenzwajg M, Herrmann JL. Constrained Intracellular Survival of Mycobacterium tuberculosis in Human Dendritic Cells. The Journal of Immunology. 2003;170:1939–1948. doi: 10.4049/jimmunol.170.4.1939. [DOI] [PubMed] [Google Scholar]
- 30.Bodnar KA, Serbina NV, Flynn JL. Fate of Mycobacterium tuberculosis within murine dendritic cells. Infect Immun. 2001;69:800–809. doi: 10.1128/IAI.69.2.800-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romagnoli A, Etna MP, Giacomini E, Pardini M, Remoli ME, Corazzari M, Falasca L, Goletti D, Gafa V, Simeone R, et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy. 2012;8:1357–1370. doi: 10.4161/auto.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tailleux L, Waddell SJ, Pelizzola M, Mortellaro A, Withers M, Tanne A, Castagnoli PR, Gicquel B, Stoker NG, Butcher PD, et al. Probing host pathogen cross-talk by transcriptional profiling of both Mycobacterium tuberculosis and infected human dendritic cells and macrophages. PLoS ONE. 2008;3 doi: 10.1371/journal.pone.0001403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. M. tuberculosis and M. leprae Translocate from the Phagolysosome to the Cytosol in Myeloid Cells. Cell. 2007;129:1287–1298. doi: 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 34.Pethe K, Swenson DL, Alonso S, Anderson J, Wang C, Russell DG. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc Natl Acad Sci USA. 2004;101:13642–13647. doi: 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walburger A, Koul A, Ferrari G, Nguyen L, Prescianotto-Baschong C, Huygen K, Klebl B, Thompson C, Bacher G, Pieters J. Protein Kinase G from Pathogenic Mycobacteria Promotes Survival Within Macrophages. Science. 2004;304:1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 36.Caminero JA, Pena MJ, Campos-Herrero MI, Rodriguez JC, Garcia I, Cabrera P, Lafoz C, Samper S, Takiff H, Afonso O, et al. Epidemiological evidence of the spread of a Mycobacterium tuberculosis strain of the Beijing genotype on Gran Canaria Island. Am J Respir Crit Care Med. 2001;164:1165–1170. doi: 10.1164/ajrccm.164.7.2101031. [DOI] [PubMed] [Google Scholar]
- 37.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fortsch D, Rollinghoff M, Stenger S. IL-10 converts human dendritic cells into macrophage-like cells with increased antibacterial activity against virulent Mycobacterium tuberculosis. J Immunol. 2000;165:978–987. doi: 10.4049/jimmunol.165.2.978. [DOI] [PubMed] [Google Scholar]
- 39.Rivero-Lezcano OM, Gonzalez-Cortes C, Reyes-Ruvalcaba D, Diez-Tascon C. CCL20 is overexpressed in Mycobacterium tuberculosis-infected monocytes and inhibits the production of reactive oxygen species (ROS) Clin Exp Immunol. 2010;162:289–297. doi: 10.1111/j.1365-2249.2010.04168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olakanmi O, Kesavalu B, Abdalla MY, Britigan BE. Iron acquisition by Mycobacterium tuberculosis residing within myeloid dendritic cells. Microbial Pathogenesis. 2013;65:21–28. doi: 10.1016/j.micpath.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Ryan R, O'Sullivan M, Keane J. Mycobacterium tuberculosis infection induces non-apoptotic cell death of human dendritic cells. BMC Microbiology. 2011;11:237. doi: 10.1186/1471-2180-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abdalla H, Srinivasan L, Shah S, Mayer-Barber KD, Sher A, Sutterwala FS, Briken V. Mycobacterium tuberculosis Infection of Dendritic Cells Leads to Partially Caspase-1/11-Independent IL-1β and IL-18 Secretion but Not to Pyroptosis. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0040722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, Rubin EJ. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cox JS, Chen B, McNeil M, Jacobs WR., Jr Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 45.Simeone R, Huet G, Constant P, Malaga W, Lemassu A, Laval F, Daffe M, Guilhot C, Chalut C. Functional characterisation of three o-methyltransferases involved in the biosynthesis of phenolglycolipids in Mycobacterium tuberculosis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0058954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kendall SL, Burgess P, Balhana R, Withers M, Ten Bokum A, Lott JS, Gao C, Uhia-Castro I, Stoker NG. Cholesterol utilization in mycobacteria is controlled by two TetR-type transcriptional regulators: kstR and kstR2. Microbiology. 2010;156:1362–1371. doi: 10.1099/mic.0.034538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohn WW, van der Geize R, Stewart GR, Okamoto S, Liu J, Dijkhuizen L, Eltis LD. The actinobacterial mce4 locus encodes a steroid transporter. J Biol Chem. 2008;283:35368–35374. doi: 10.1074/jbc.M805496200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Griffin J-E, Pandey A-K, Gilmore S-A, Mizrahi V, McKinney J-D, Bertozzi C-R, Sassetti C-M. Cholesterol Catabolism by Mycobacterium tuberculosis Requires Transcriptional and Metabolic Adaptations. Chemistry & Biology. 2012;19:218–227. doi: 10.1016/j.chembiol.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Houben EN, Korotkov KV, Bitter W. Take five - Type VII secretion systems of Mycobacteria. Biochim Biophys Acta. 1843;2014:1707–1716. doi: 10.1016/j.bbamcr.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 50.Diamant I, Eldar YC, Rokhlenko O, Ruppin E, Shlomi T. A network-based method for predicting gene-nutrient interactions and its application to yeast amino-acid metabolism. Mol Biosyst. 2009;5:1732–1739. doi: 10.1039/B823287N. [DOI] [PubMed] [Google Scholar]
- 51.Beste DJ, Hooper T, Stewart G, Bonde B, Avignone-Rossa C, Bushell ME, Wheeler P, Klamt S, Kierzek AM, McFadden J. GSMN-TB: a web-based genome-scale network model of Mycobacterium tuberculosis metabolism. Genome Biol. 2007;8:R89. doi: 10.1186/gb-2007-8-5-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, Kaplan G, Barry CE., 3rd A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–87. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 53.Huet G, Constant P, Malaga W, Laneelle MA, Kremer K, van Soolingen D, Daffe M, Guilhot C. A lipid profile typifies the Beijing strains of Mycobacterium tuberculosis: identification of a mutation responsible for a modification of the structures of phthiocerol dimycocerosates and phenolic glycolipids. J Biol Chem. 2009;284:27101–27113. doi: 10.1074/jbc.M109.041939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dhar N, McKinney JD. Mycobacterium tuberculosis persistence mutants identified by screening in isoniazid-treated mice. Proc Natl Acad Sci U S A. 2010;107:12275–12280. doi: 10.1073/pnas.1003219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sirakova TD, Dubey VS, Cynamon MH, Kolattukudy PE. Attenuation of Mycobacterium tuberculosis by disruption of a mas-like gene or a chalcone synthase-like gene, which causes deficiency in dimycocerosyl phthiocerol synthesis. J Bacteriol. 2003;185:2999–3008. doi: 10.1128/JB.185.10.2999-3008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Passemar C, Arbués A, Malaga W, Mercier I, Moreau F, Lepourry L, Neyrolles O, Guilhot C, Astarie-Dequeker C. Multiple deletions in the polyketide synthase gene repertoire of Mycobacterium tuberculosis reveal functional overlap of cell envelope lipids in host-pathogen interactions. Cellular Microbiology. 2013;16:195–213. doi: 10.1111/cmi.12214. [DOI] [PubMed] [Google Scholar]
- 57.Perez E, Constant P, Lemassu A, Laval F, Daffe M, Guilhot C. Characterization of three glycosyltransferases involved in the biosynthesis of the phenolic glycolipid antigens from the Mycobacterium tuberculosis complex. J Biol Chem. 2004;279:42574–42583. doi: 10.1074/jbc.M406246200. [DOI] [PubMed] [Google Scholar]
- 58.Indrigo J, Hunter RL, Jr, Actor JK. Cord factor trehalose 6,6'-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology. 2003;149:2049–2059. doi: 10.1099/mic.0.26226-0. [DOI] [PubMed] [Google Scholar]
- 59.Armitige LY, Jagannath C, Wanger AR, Norris SJ. Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infect Immun. 2000;68:767–778. doi: 10.1128/iai.68.2.767-778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Role of the Major Antigen of Mycobacterium tuberculosis in Cell Wall Biogenesis. Science. 1997;276:1420–1422. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 61.Katti MK, Dai G, Armitige LY, Rivera Marrero C, Daniel S, Singh CR, Lindsey DR, Dhandayuthapani S, Hunter RL, Jagannath C. The Delta fbpA mutant derived from Mycobacterium tuberculosis H37Rv has an enhanced susceptibility to intracellular antimicrobial oxidative mechanisms, undergoes limited phagosome maturation and activates macrophages and dendritic cells. Cell Microbiol. 2008;10:1286–1303. doi: 10.1111/j.1462-5822.2008.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puech V, Guilhot C, Perez E, Tropis M, Armitige LY, Gicquel B, Daffe M. Evidence for a partial redundancy of the fibronectin-binding proteins for the transfer of mycoloyl residues onto the cell wall arabinogalactan termini of Mycobacterium tuberculosis. Mol Microbiol. 2002;44:1109–1122. doi: 10.1046/j.1365-2958.2002.02953.x. [DOI] [PubMed] [Google Scholar]
- 63.Hatzios SK, Schelle MW, Holsclaw CM, Behrens CR, Botyanszki Z, Lin FL, Carlson BL, Kumar P, Leary JA, Bertozzi CR. PapA3 is an acyltransferase required for polyacyltrehalose biosynthesis in Mycobacterium tuberculosis. J Biol Chem. 2009;284:12745–12751. doi: 10.1074/jbc.M809088200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodriguez JE, Ramirez AS, Salas LP, Helguera-Repetto C, Gonzalez-y-Merchand J, Soto CY, Hernandez-Pando R. Transcription of genes involved in sulfolipid and polyacyltrehalose biosynthesis of Mycobacterium tuberculosis in experimental latent tuberculosis infection. PLoS One. 2013;8 doi: 10.1371/journal.pone.0058378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barkan D, Rao V, Sukenick GD, Glickman MS. Redundant function of cmaA2 and mmaA2 in Mycobacterium tuberculosis cis cyclopropanation of oxygenated mycolates. J Bacteriol. 2010;192:3661–3668. doi: 10.1128/JB.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuan Y, Zhu Y, Crane DD, Barry Iii CE. The effect of oxygenated mycolic acid composition on cell wall function and macrophage growth in Mycobacterium tuberculosis. Molecular Microbiology. 1998;29:1449–1458. doi: 10.1046/j.1365-2958.1998.01026.x. [DOI] [PubMed] [Google Scholar]
- 67.Dao DN, Sweeney K, Hsu T, Gurcha SS, Nascimento IP, Roshevsky D, Besra GS, Chan J, Porcelli SA, Jacobs WR. Mycolic acid modification by the mmaA4 gene of M. tuberculosis modulates IL-12 production. PLoS Pathog. 2008;4:e1000081. doi: 10.1371/journal.ppat.1000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boissier F, Bardou F, Guillet V, Uttenweiler-Joseph S, Daffe M, Quemard A, Mourey L. Further insight into S-adenosylmethionine-dependent methyltransferases: structural characterization of Hma, an enzyme essential for the biosynthesis of oxygenated mycolic acids in Mycobacterium tuberculosis. J Biol Chem. 2006;281:4434–4445. doi: 10.1074/jbc.M510250200. [DOI] [PubMed] [Google Scholar]
- 69.McAdam RA, Quan S, Smith DA, Bardarov S, Betts JC, Cook FC, Hooker EU, Lewis AP, Woollard P, Everett MJ, et al. Characterization of a Mycobacterium tuberculosis H37Rv transposon library reveals insertions in 351 ORFs and mutants with altered virulence. Microbiology. 2002;148:2975–2986. doi: 10.1099/00221287-148-10-2975. [DOI] [PubMed] [Google Scholar]
- 70.Dubnau E, Chan J, Raynaud C, Mohan VP, Lanéelle M-A, Yu K, Quémard A, Smith I, Daffé M. Oxygenated mycolic acids are necessary for virulence of Mycobacterium tuberculosis in mice. Molecular Microbiology. 2000;36:630–637. doi: 10.1046/j.1365-2958.2000.01882.x. [DOI] [PubMed] [Google Scholar]
- 71.Middlebrook G, Coleman CM, Schaefer WB. Sulfolipid from virulent tubercle bacilli. Proceedings of the National Academy of Sciences. 1959;45:1801–1804. doi: 10.1073/pnas.45.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang L, Goren MB, Holzer TJ, Andersen BR. Effect of Mycobacterium tuberculosis-derived sulfolipid I on human phagocytic cells. Infection and Immunity. 1988;56:2876–2883. doi: 10.1128/iai.56.11.2876-2883.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pabst MJ, Gross JM, Brozna JP, Goren MB. Inhibition of macrophage priming by sulfatide from Mycobacterium tuberculosis. J Immunol. 1988;140:634–640. [PubMed] [Google Scholar]
- 74.Gilmore SA, Schelle MW, Holsclaw CM, Leigh CD, Jain M, Cox JS, Leary JA, Bertozzi CR. Sulfolipid-1 biosynthesis restricts Mycobacterium tuberculosis growth in human macrophages. ACS Chem Biol. 2012;7:863–870. doi: 10.1021/cb200311s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumar P, Schelle MW, Jain M, Lin FL, Petzold CJ, Leavell MD, Leary JA, Cox JS, Bertozzi CR. PapA1 and PapA2 are acyltransferases essential for the biosynthesis of the Mycobacterium tuberculosis virulence factor sulfolipid-1. Proc Natl Acad Sci U S A. 2007;104:11221–11226. doi: 10.1073/pnas.0611649104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Converse SE, Mougous JD, Leavell MD, Leary JA, Bertozzi CR, Cox JS. MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc Natl Acad Sci U S A. 2003;100:6121–6126. doi: 10.1073/pnas.1030024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lynett J, Stokes RW. Selection of transposon mutants of Mycobacterium tuberculosis with increased macrophage infectivity identifies fadD23 to be involved in sulfolipid production and association with macrophages. Microbiology. 2007;153:3133–3140. doi: 10.1099/mic.0.2007/007864-0. [DOI] [PubMed] [Google Scholar]
- 78.McAdam RA, Weisbrod TR, Martin J, Scuderi JD, Brown AM, Cirillo JD, Bloom BR, Jacobs WR., Jr In vivo growth characteristics of leucine and methionine auxotrophic mutants of Mycobacterium bovis BCG generated by transposon mutagenesis. Infect Immun. 1995;63:1004–1012. doi: 10.1128/iai.63.3.1004-1012.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.DeJesus MA, Zhang YJ, Sassetti CM, Rubin EJ, Sacchettini JC, Ioerger TR. Bayesian analysis of gene essentiality based on sequencing of transposon insertion libraries. Bioinformatics. 2013;29:695–703. doi: 10.1093/bioinformatics/btt043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fukuda T, Matsumura T, Ato M, Hamasaki M, Nishiuchi Y, Murakami Y, Maeda Y, Yoshimori T, Matsumoto S, Kobayashi K, et al. Critical roles for lipomannan and lipoarabinomannan in cell wall integrity of mycobacteria and pathogenesis of tuberculosis. MBio. 2013;4:e00472–00412. doi: 10.1128/mBio.00472-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mishra AK, Driessen NN, Appelmelk BJ, Besra GS. Lipoarabinomannan and related glycoconjugates: structure, biogenesis and role in Mycobacterium tuberculosis physiology and host-pathogen interaction. FEMS Microbiol Rev. 2011;35:1126–1157. doi: 10.1111/j.1574-6976.2011.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Welin A, Winberg ME, Abdalla H, Sarndahl E, Rasmusson B, Stendahl O, Lerm M. Incorporation of Mycobacterium tuberculosis lipoarabinomannan into macrophage membrane rafts is a prerequisite for the phagosomal maturation block. Infect Immun. 2008;76:2882–2887. doi: 10.1128/IAI.01549-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Afonso-Barroso A, Clark SO, Williams A, Rosa GT, Nobrega C, Silva-Gomes S, et al.: Lipoarabinomannan mannose caps do not affect mycobacterial virulence or the induction of protective immunity in experimental animal models of infection and have minimal impact on in vitro inflammatory responses. Cell Microbiol 2013;15:660-74. [DOI] [PubMed]
- 84.Pitarque S, Herrmann JL, Duteyrat JL, Jackson M, Stewart GR, Lecointe F, Payre B, Schwartz O, Young DB, Marchal G, et al. Deciphering the molecular bases of Mycobacterium tuberculosis binding to the lectin DC-SIGN reveals an underestimated complexity. Biochem J. 2005;392:615–624. doi: 10.1042/BJ20050709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ouellet H, Johnston JB, de Montellano PR. Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol. 2011;19:530–539. doi: 10.1016/j.tim.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee EJ, Pontes MH, Groisman EA. A bacterial virulence protein promotes pathogenicity by inhibiting the bacterium's own F1F0 ATP synthase. Cell. 2013;154:146–156. doi: 10.1016/j.cell.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jain M, Petzold CJ, Schelle MW, Leavell MD, Mougous JD, Bertozzi CR, Leary JA, Cox JS. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci U S A. 2007;104:5133–5138. doi: 10.1073/pnas.0610634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Colangeli R, Haq A, Arcus VL, Summers E, Magliozzo RS, McBride A, Mitra AK, Radjainia M, Khajo A, Jacobs WR, Jr, et al. The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. Proc Natl Acad Sci U S A. 2009;106:4414–4418. doi: 10.1073/pnas.0810126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klink M, Brzezinska M, Szulc I, Brzostek A, Kielbik M, Sulowska Z, Dziadek J. Cholesterol oxidase is indispensable in the pathogenesis of Mycobacterium tuberculosis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0073333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nguyen DH, Taub DD. Inhibition of chemokine receptor function by membrane cholesterol oxidation. Exp Cell Res. 2003;291:36–45. doi: 10.1016/s0014-4827(03)00345-8. [DOI] [PubMed] [Google Scholar]
- 91.Bednarska K, Kielbik M, Sulowska Z, Dziadek J: Cholesterol Oxidase Binds TLR2 and Modulates Functional Responses of Human Macrophages. Mediators Inflamm. 2014:498395. [DOI] [PMC free article] [PubMed]
- 92.Kendall SL, Withers M, Soffair CN, Moreland NJ, Gurcha S, Sidders B, Frita R, Ten Bokum A, Besra GS, Lott JS, Stoker NG. A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol Microbiol. 2007;65:684–699. doi: 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Slayden RA, Jackson M, Zucker J, Ramirez MV, Dawson CC, Crew R, Sampson NS, Thomas ST, Jamshidi N, Sisk P, et al. Updating and curating metabolic pathways of TB. Tuberculosis. 2013;93:47–59. doi: 10.1016/j.tube.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chang JC, Harik NS, Liao RP, Sherman DR. Identification of Mycobacterial genes that alter growth and pathology in macrophages and in mice. J InfectDis. 2007;196:788–795. doi: 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- 95.Thomas ST, VanderVen BC, Sherman DR, Russell DG, Sampson NS. Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J Biol Chem. 2011;286:43668–43678. doi: 10.1074/jbc.M111.313643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Casabon I, Zhu SH, Otani H, Liu J, Mohn WW, Eltis LD. Regulation of the KstR2 regulon of Mycobacterium tuberculosis by a cholesterol catabolite. Mol Microbiol. 2013;89:1201–1212. doi: 10.1111/mmi.12340. [DOI] [PubMed] [Google Scholar]
- 97.MacGurn JA, Cox JS. A Genetic Screen for Mycobacterium tuberculosis Mutants Defective for Phagosome Maturation Arrest Identifies Components of the ESX-1 Secretion System. Infection and Immunity. 2007;75:2668–2678. doi: 10.1128/IAI.01872-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brodin P, Poquet Y, Levillain F, Peguillet I, Larrouy-Maumus G, Gilleron M, Ewann F, Christophe T, Fenistein D, Jang J, et al. High Content Phenotypic Cell-Based Visual Screen Identifies Mycobacterium tuberculosis Acyltrehalose-Containing Glycolipids Involved in Phagosome Remodeling. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2005;102:4033–4038. doi: 10.1073/pnas.0409716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thi EP, Hong CJ, Sanghera G, Reiner NE. Identification of the Mycobacterium tuberculosis protein PE-PGRS62 as a novel effector that functions to block phagosome maturation and inhibit iNOS expression. Cell Microbiol. 2013;15:795–808. doi: 10.1111/cmi.12073. [DOI] [PubMed] [Google Scholar]
- 101.Iantomasi R, Sali M, Cascioferro A, Palucci I, Zumbo A, Soldini S, Rocca S, Greco E, Maulucci G, De Spirito M, et al. PE_PGRS30 is required for the full virulence of Mycobacterium tuberculosis. Cell Microbiol. 2012;14:356–367. doi: 10.1111/j.1462-5822.2011.01721.x. [DOI] [PubMed] [Google Scholar]
- 102.Kim KH, An DR, Song J, Yoon JY, Kim HS, Yoon HJ, Im HN, Kim J. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci U S A. 2012;109:7729–7734. doi: 10.1073/pnas.1120251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proceedings of the National Academy of Sciences. 2003;100:5437–5442. doi: 10.1073/pnas.0737613100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng TY, Siddiqi N, Fortune SM, Moody DB, Rubin EJ. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci U S A. 2009;106:18792–18797. doi: 10.1073/pnas.0900589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Di Luca M, Bottai D, Batoni G, Orgeur M, Aulicino A, Counoupas C, Campa M, Brosch R, Esin S. The ESX-5 associated eccB-EccC locus is essential for Mycobacterium tuberculosis viability. PLoS One. 2012;7 doi: 10.1371/journal.pone.0052059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stoop EJ, Bitter W, van der Sar AM. Tubercle bacilli rely on a type VII army for pathogenicity. Trends Microbiol. 2012;20:477–484. doi: 10.1016/j.tim.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 107.Bottai D, Majlessi L, Simeone R, Frigui W, Laurent C, Lenormand P, Chen J, Rosenkrands I, Huerre M, Leclerc C, et al. ESAT-6 Secretion-Independent Impact of ESX-1 Genes espF and espG1 on Virulence of Mycobacterium tuberculosis. Journal of Infectious Diseases. 2011;203:1155–1164. doi: 10.1093/infdis/jiq089. [DOI] [PubMed] [Google Scholar]
- 108.Chen JM, Zhang M, Rybniker J, Basterra L, Dhar N, Tischler AD, Pojer F, Cole ST. Phenotypic profiling of Mycobacterium tuberculosis EspA point mutants reveals that blockage of ESAT-6 and CFP-10 secretion in vitro does not always correlate with attenuation of virulence. J Bacteriol. 2013;195:5421–5430. doi: 10.1128/JB.00967-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Joshi SA, Ball DA, Sun MG, Carlsson F, Watkins BY, Aggarwal N, McCracken JM, Huynh KK, Brown EJ. EccA1, a component of the Mycobacterium marinum ESX-1 protein virulence factor secretion pathway, regulates mycolic acid lipid synthesis. Chem Biol. 2012;19:372–380. doi: 10.1016/j.chembiol.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 110.Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, Bitter W, Brennan MJ, Leclerc C, Batoni G, et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol Microbiol. 2012;83:1195–1209. doi: 10.1111/j.1365-2958.2012.08001.x. [DOI] [PubMed] [Google Scholar]
- 111.Abdallah AM, Verboom T, Weerdenburg EM, van Pittius NC G, Mahasha PW, Jimenez C, Parra M, Cadieux N, Brennan MJ, Appelmelk BJ, Bitter W. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol. 2009;73:329–340. doi: 10.1111/j.1365-2958.2009.06783.x. [DOI] [PubMed] [Google Scholar]
- 112.Daleke MH, Ummels R, Bawono P, Heringa J, Vandenbroucke-Grauls CM, Luirink J, Bitter W. General secretion signal for the mycobacterial type VII secretion pathway. Proc Natl Acad Sci U S A. 2012;109:11342–11347. doi: 10.1073/pnas.1119453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Akhter Y, Ehebauer MT, Mukhopadhyay S, Hasnain SE. The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie. 2012;94:110–116. doi: 10.1016/j.biochi.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 114.Werling D, Hope JC, Howard CJ, Jungi TW. Differential production of cytokines, reactive oxygen and nitrogen by bovine macrophages and dendritic cells stimulated with Toll-like receptor agonists. Immunology. 2004;111:41–52. doi: 10.1111/j.1365-2567.2003.01781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 2009;11:1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Senaratne RH, De Silva AD, Williams SJ, Mougous JD, Reader JR, Zhang T, Chan S, Sidders B, Lee DH, Chan J, et al. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Molecular Microbiology. 2006;59:1744–1753. doi: 10.1111/j.1365-2958.2006.05075.x. [DOI] [PubMed] [Google Scholar]
- 117.Voskuil MI, Schnappinger D, Rutherford R, Liu Y, Schoolnik GK. Regulation of the Mycobacterium tuberculosis PE/PPE genes. Tuberculosis. 2004;84:256–262. doi: 10.1016/j.tube.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 118.Patil AG, Sang PB, Govindan A, Varshney U. Mycobacterium tuberculosis MutT1 (Rv2985) and ADPRase (Rv1700) proteins constitute a two-stage mechanism of 8-oxo-dGTP and 8-oxo-GTP detoxification and adenosine to cytidine mutation avoidance. J Biol Chem. 2013;288:11252–11262. doi: 10.1074/jbc.M112.442566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hillas PJ, del Alba FS, Oyarzabal J, Wilks A, Ortiz De Montellano PR. The AhpC and AhpD antioxidant defense system of Mycobacterium tuberculosis. J Biol Chem. 2000;275:18801–18809. doi: 10.1074/jbc.M001001200. [DOI] [PubMed] [Google Scholar]
- 120.Newton GL, Av-Gay Y, Fahey RC. N-Acetyl-1-D-myo-inosityl-2-amino-2-deoxy-alpha-D-glucopyranoside deacetylase (MshB) is a key enzyme in mycothiol biosynthesis. J Bacteriol. 2000;182:6958–6963. doi: 10.1128/jb.182.24.6958-6963.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Buchmeier NA, Newton GL, Koledin T, Fahey RC. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol Microbiol. 2003;47:1723–1732. doi: 10.1046/j.1365-2958.2003.03416.x. [DOI] [PubMed] [Google Scholar]
- 122.Cunningham-Bussel A, Zhang T, Nathan CF: Nitrite produced by Mycobacterium tuberculosis in human macrophages in physiologic oxygen impacts bacterial ATP consumption and gene expression. Proc Natl Acad Sci. 2013;110:E4256-65. [DOI] [PMC free article] [PubMed]
- 123.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 124.Houghton J, Townsend C, Williams AR, Rodgers A, Rand L, Walker KB, Bottger EC, Springer B, Davis EO. Important role for Mycobacterium tuberculosis UvrD1 in pathogenesis and persistence apart from its function in nucleotide excision repair. J Bacteriol. 2012;194:2916–2923. doi: 10.1128/JB.06654-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jung JY, Madan-Lala R, Georgieva M, Rengarajan J, Sohaskey CD, Bange FC, Robinson CM. The intracellular environment of human macrophages that produce nitric oxide promotes growth of mycobacteria. Infect Immun. 2013;81:3198–3209. doi: 10.1128/IAI.00611-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herbst S, Schaible UE, Schneider BE. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tan MP, Sequeira P, Lin WW, Phong WY, Cliff P, Ng SH, Lee BH, Camacho L, Schnappinger D, Ehrt S, et al. Nitrate respiration protects hypoxic Mycobacterium tuberculosis against acid- and reactive nitrogen species stresses. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cunningham-Bussel A, Bange FC, Nathan CF. Nitrite impacts the survival of Mycobacterium tuberculosis in response to isoniazid and hydrogen peroxide. Microbiologyopen. 2013;2:901–911. doi: 10.1002/mbo3.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bardarov S, Kriakov J, Carriere C, Yu S, Vaamonde C, McAdam RA, Bloom BR, Hatfull GF, Jacobs WR., Jr Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1997;94:10961–10966. doi: 10.1073/pnas.94.20.10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc Natl Acad Sci U S A. 1999;96:1645–1650. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nadarajah S, Kotz S. A generalized logistic distribution. International Journal of Mathematics and Mathematical Sciences. 2005;2005:3169–3174. [Google Scholar]
- 133.Loughin TM. A systematic comparison of methods for combining p-values from independent tests. Computational Statistics & Data Analysis. 2004;47:467–485. [Google Scholar]
- 134.Gevorgyan A, Bushell ME, Avignone-Rossa C, Kierzek AM. SurreyFBA: a command line tool and graphics user interface for constraint-based modeling of genome-scale metabolic reaction networks. Bioinformatics. 2011;27:433–434. doi: 10.1093/bioinformatics/btq679. [DOI] [PubMed] [Google Scholar]