

Abstract
Background
Although Mycobacterium tuberculosis isolates are consisted of several different lineages and the epidemiology analyses are usually assessed relative to a particular reference genome, M. tuberculosis H37Rv, which might introduce some biased results. Those analyses are essentially based genome sequence information of M. tuberculosis and could be performed in sillico in theory, with whole genome sequence (WGS) data available in the databases and obtained by next generation sequencers (NGSs). As an approach to establish higher resolution methods for such analyses, whole genome sequences of the M. tuberculosis complexes (MTBCs) strains available on databases were aligned to construct virtual reference genome sequences called the consensus sequence (CS), and evaluated its feasibility in in sillico epidemiological analyses.
Results
The consensus sequence (CS) was successfully constructed and utilized to perform phylogenetic analysis, evaluation of read mapping efficacy, which is crucial for detecting single nucleotide polymorphisms (SNPs), and various MTBC typing methods virtually including spoligotyping, VNTR, Long sequence polymorphism and Beijing typing. SNPs detected based on CS, in comparison with H37Rv, were utilized in concatemer-based phylogenetic analysis to determine their reliability relative to a phylogenetic tree based on whole genome alignment as the gold standard. Statistical comparison of phylogenic trees based on CS with that of H37Rv indicated the former showed always better results that that of later. SNP detection and concatenation with CS was advantageous because the frequency of crucial SNPs distinguishing among strain lineages was higher than those of H37Rv. The number of SNPs detected was lower with the consensus than with the H37Rv sequence, resulting in a significant reduction in computational time. Performance of each virtual typing was satisfactory and accorded with those published when those are available.
Conclusions
These results indicated that virtual CS constructed from genome sequence data is an ideal approach as a reference for MTBC studies.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1368-9) contains supplementary material, which is available to authorized users.
Keywords: Mycobacterium tuberculosis, Consensus sequence, Virtual typing, Phylogenetic analysis, SNP concatemer
Background
Tuberculosis is one of the most prevalent and deadly bacterial infections affecting humans, with almost 9 million new cases worldwide and more than 1.4 million deaths in 2010 [1]. It has been estimated that 310000 patients newly diagnosed with pulmonary tuberculosis in 2011 were infected with multidrug-resistant (MDR) bacteria [2], with 9% of these patients, living in 84 countries, having extensively drug-resistant (XDR) tuberculosis [3]. Moreover, the World Health Organization (WHO) has estimated that 350000 of the 1.4 million deaths per year from tuberculosis are associated with HIV coinfection [4].
A variety of molecular typing methods have been used to classify M. tuberculosis strains for epidemiological studies, including assessment of the presence of the IS6110 restriction fragment length polymorphism (RFLP) [5], spoligotyping, analysis of mycobacterial interspersed repetitive unit-variable number tandem repeats (MIRU-VNTR) [6] and large sequence polymorphisms (LSPs) [7,8]. Their target sequences are mobile elements (e.g. IS6110), repetitive sequences (e.g. spoligotyping and MIRU-VNTR) and relatively long sequence polymorphisms (at least 7 bp), with many strains belonging to unrelated lineages possessing these DNA elements in common. According to the SpolDB4 of the international online spoligotype database (http://www.pasteur-guadeloupe.fr/tb/bd_myco.html), clinical strains of M. tuberculosis obtained worldwide can be classified into 10 major groups [9]. Although it is useful to identify clonal lineage on the global scale, the discriminatory power of this method may not be sufficient for evaluation of genetically closely related isolates, including those from areas of tuberculosis outbreak. According to the SpolDB4, all M. tuberculosis strains belonging to the Beijing family, predominantly from Far-East Asia [10], share the same spoligotype patterns [9]. Over 70% of the clinical strains isolated in Japan were found to belong to the Beijing family [11]. Similarly, MIRU-VNTR genotyping [6,12], based on the typing of 12 MIRU loci, has become a global standard in the epidemiological typing of M. tuberculosis. Since its first use, many investigators have tried to find an ideal combination to further distinguish among genetically closely related strains [13,14]. This has led to the formulation of optimized sets, including a 15-locus system as the new standard for routine epidemiological discrimination and a 24-locus system as a high-resolution tool for phylogenetic studies [14]. Although these 15- and 24-locus VNTR locus systems have been utilized for first-line typing, they are insufficient in distinguishing among closely related strains of the Beijing family to define deep phylogenetic structures [15]. LSPs have been utilized to determine whether lineages of M. tuberculosis were associated with specific human populations [7,8]. Utilizing LSPs, MTBCs could be divided into at least 6 phylogeographical lineages, each associated with specific, sympatric human populations. Taken together, that conventional molecular typing methods for MTBC are limited in distinguishing among strain subtypes.
This limitation may be overcome by next generation whole genome sequencing (WGS) for genome-based epidemiology [16]. WGS is becoming the ultimate tool for diagnosing and typing pathogens, and has amplified the impact of molecular diagnostics on clinical microbiology [17]. The potential of WGS-based MTBC genotyping has started to be explored [18]. In phylogenetic analysis based on WGS data, sequence reads are mapped to a reference genome, usually H37Rv, and single nucleotide polymorphisms (SNPs) are detected and concatenated to generate an artificial genome sequence representing each isolate. This approach has been shown to be robustness and of high resolution [19-21].
In comparative genome or phylogenetic analysis, generated genomes or WGS data are compared to results from reference genomes. The M. tuberculosis strain H37Rv was the first sequenced in its entirety [22] and has been utilized extensively as the reference genome in these investigations. In these comparisons, the sequences of one or several newly sequenced genomes are compared to the sequence of a reference genome. If the reference genome used in these analyses does not contain a gene or marker possessed by the newly sequenced genome, this method cannot be used to determine the evolutionary fate of the genetic context of the latter.
The evolution of MTBC genomes has several unique features. Although several mycobacterium phages have been reported [23,24], they were found to evolve through mutation without acquiring any external genetic traits. This feature differs strikingly from that observed in conventional drug resistance bacteria such as Pseudomonas aeruginosa and Escherichia coli [25]. The nature of its evolution makes the MTBC genome highly stable, and the genetic diversity of MTBCs obtained in clinical settings is essentially restricted in SNPs and indels. Generally, mobile elements such as transposons, phages and plasmids are omitted from phylogenetic analysis because their rate of evolution differs from that of the intrinsic chromosome. MTBCs, however, are composed of several lineages, and the development of an artificial reference sequence of the entire MTBC genome, containing sequence information from all lineages, would be extremely useful. Although some SNPs and/or indels may be omitted or ignored when using a particular genome sequence of a real isolate as a reference, these SNPs and/or indels would not be missed in comparative genomics, especially in phylogenetic research.
In this study, we constructed a consensus whole MTBC genome sequence from the sequence data of 19 MTBC strains isolated and characterized through April 2012. For proof of concept, we compared the consensus and H37Rv genomes as reference standards for phylogenetic analysis, SNP detection and secondary analysis of WGS data. In addition, we found that spoligotype, VNTR type, LSP classification, Beijing type and antibiotic susceptibility profiles could be determined simultaneously, resulting in a virtual diagnosis in the absence of actual experiments except for WGS.
Results
Construction and features of a virtual M. tuberculosis consensus genome
We constructed two types of M. tuberculosis consensus genome sequences, one consisting of 13 M. tuberculosis strains and the M. bovis BCG, M. africanum and M. canettii strains shown in Table 1, and the other consisting only of the 13 M. tuberculosis strains. In this study, the former was used for further analysis, unless otherwise specified. Public databases such as DNA Data Bank of Japan do not accept virtual sequence for the registration. Thus, CSs constructed in this study are available as the supplemental data (Additional files 1, 2 and 3).
Table 1.
Mycobacterium strains used in this study
| Species | Strain name | GenBank | Country | Genome inversion points | Comments |
|---|---|---|---|---|---|
| M.tuberculosis | CCDC5079 | CP001642.1 | China | Drug-susceptible isolate belonging to the Beijing family. | |
| CCDC5180 | CP001642.1 | China | Multidrug-resistant clinical isolate. | ||
| CDC1551 | AE000516.2 | USA | |||
| CTRI-2 | CP002992.1 | Russia | |||
| Erdman | AP012340.1 | USA | isolated from sputum samples of patients | ||
| F11 | CP000717.1 | South Africa | Predominant strain in South African epidemic | ||
| H37Ra | CP000611.1. | China | An avirulent strain derived from its virulent parent strain H37 | ||
| H37Rv | AL123456.2 | USA | |||
| KZN605 | CP001976.1 | South Africa | 1. between 932051 and 932052. 2. between 3479594 and 3459595 | Extensively drug-resistant clinical isolate | |
| KZN1435 | CP001658.1 | South Africa | 1. between 931985 and 931986. 2. between 3479865 and 3479866 | Multidrug-resistant clinical isolate | |
| KZN4207 | CP001662.1 | South Africa | 1. between 932007 and 932008. 2. between 3476553 and 3476554 | Drug-susceptible clinical isolate | |
| RGTB327 | CP003233.1 | India | isolated from sputum samples of patients | ||
| RGTB423 | CP003234.1 | India | isolated from sputum samples of patients | ||
| M. bovis BCG | Mexico | CP002095.1 | Mexico | ||
| Moreau RDJ | AM412059.2 | Brazil | Brazilian vaccine strain | ||
| Pasteur | AM408590.1 | France | |||
| Tokyo 172 | AP010918.1 | Japan | |||
| M. africanum | GMO41182 | FR878060.1 | Gambia | ||
| M. canettii | CIPT 140010059 | HE572590.1 | France |
Although the concept of a consensus genome has long been recognized [26-30], no consensus genome had been determined for M. tuberculosis, largely due to difficulties in aligning these relatively large sized genomes. Despite its relatively stable and conserved genome, some M. tuberculosis strains have inversions and rearranged regions in comparison with H37Rv, making alignment more difficult. To overcome the inversions and rearrangements in M. tuberculosis strains, we performed genome rearrangement analyses using publicly available software Mauve [31], finding that the genomes of M. tuberculosis KZN605, KZN1435 and KZN4207 have large inversions, of 0.93–3.46Mbp (Table 1). IS6110 insertion sequences are located immediately adjacent to the flanking regions of all the inversion points, suggesting that the genome rearrangements observed in these 3 strains were driven by the insertion sequence. Manual correction of these inversions generated artificial KZN605, KZN1435 and KZN4206 genome sequences (KZN605_m, KZN1435_m and KZN4206_m, respectively), with the latter used to align the MTBC genomes. These procedures and employing a very fast and publicly available multiple sequence alignment software, MAFFT [32] allowed the successful alignment of the genomes of 19 MTBC strains and 13 M. tuberculosis strains. Merging the alignment results were carried out using sequence editing commercial software, Genetyx, although any software, which can handle multiple alignment data, should be suitable for the purpose. Two types of CSs were prepared with handling SNPs as majority rule or ambiguity rule. In this study, ambiguity sequence was used for further analysis. Insertion sequences derived from strain specific regions were all included in the CSs to increases the amount of sequence information.
The MTBC CS consists of one chromosome of 4991559 bp, with an average GC content of 64.8%. This genome was approximately 0.6 Mbp longer, because all sequence data from all strains used were merged into one sequence, and had a slightly lower GC content than the elements of the 19 strains used to construct the consensus genome. This artificially merged CS was intended for use as a reference genome in analyses of MTBC. Thus, instead of CDS extraction followed by annotation, homology analysis based on the corresponding gene sequences of M. tuberculosis H37Rv, which is extensively used as a reference genome, was performed to annotate the corresponding region in CS. Each region was assigned based on its CDS locus_tag (the Rv numbers), repeat regions, and rRNA and tRNA of H37Rv. All locus_tags of H37Rv were reflected in CS. This annotation resulted in features based on 4395 CDS (annotated as misc_features in CS) and repeat regions according with those H37Rv. Public databases do not accept virtual sequences. Thus the completely annotated sequence in addition to the alignments is available as additional data (Additional file 3).
Genome wide comparisons of M. tuberculosis strains have been performed extensively and repeatedly [33-35]. SNP concatenation is of the state of art methodology extensively used to analyze the phylogenetic relationship of bacterial genome [20,36]. We analyzed SNPs and indels in the CS reference genome in comparison with H37Rv to update fundamental information about polymorphism found in MTBC (Additional file 4: Tables S1 and Additional file 5 Table S2). The number of SNPs was higher in M. canettii than in the other mycobacteria (26068 vs <4000) (Table 2), suggesting a potential erroneous analysis when compared with a particular MTBC strain, such as H37Rv. For the analysis of indel, we chosen >5 bp length as the cut off of indels because indels shorter than 5 bp are overlapped many strains and detection of some them are highly depend on the alignment parameters. As reported previously, 152 of the 305 indels were located in the genes encoding the PE-PGRS and PPE family proteins, while 74 were located in intergenic regions (Additional file 5: Table S2). The position, length, annotation and MTBC strains of all SNPs and indels are shown for further applications, such as searches for lineage specific markers.
Table 2.
Efficacy of SNP calling using the consensus sequence or H37Rv as the reference genome by comparing the number of SNPs detected
| i) individual strains | |||
|---|---|---|---|
| Species | Strain | vs H37Rv | vs consensus sequence |
| M. tuberculosis | CCDC5079 | 632 | 198* |
| CCDC5180 | 378 | 70* | |
| CDC1551 | 543 | 41* | |
| CTRI-2 | 140 | 15* | |
| Erdman | 570 | 104* | |
| F11 | 343 | 129* | |
| H37Ra | 44 | 38* | |
| H37Rv | 12 | ||
| KZN605 | 16 | 9 | |
| KZN1435 | 12 | 2 | |
| KZN4207 | 26 | 0 | |
| RGTB327 | 1242 | 127* | |
| RGTB423 | 2410 | 145* | |
| M. bovis BCG | Mexico | 13 | 0 |
| Moreau RDJ | 136 | 22* | |
| Pasteur | 20 | 0 | |
| Tokyo 172 | 56 | 76* | |
| M. africanum | GMO41182 | 876 | 152* |
| M. canettii | CIPT 140010059 | 24425* | 560 |
| Total SNP found in only one strain | 31882 | 1700 | |
| ii) group comparison | |||
| four BCG strains | 1040 | 52* | |
| three KZN strains | 121 | 15 | |
| All SNP at least found in one strain | 37589 | 3429 | |
Based on SNP calls using MUMmer [58], the number of SNPs called uniquely in individual strains, and in groups of BCG and KZN strains, was determined. *The number of SNPs detected using the H37Rv and consensus sequences as reference were compared for each strain (i) or group of strains (ii) using Fisher's exact test, with significant differences indicated with asterisks (p < 0.0001).
Comparison of the performance of the consensus sequence (CS) and H37Rv as the reference genome in the phylogenetic analyses
To show that CS was superior to the sequence of a particular strain in phylogenetic analysis in preparation of SNP concatemers, we first constructed phylogenetic trees based on concatenated SNP sites from the virtual consensus and H37Rv genome sequences. Two tree construction methods based on a maximum-likelihood (PhyML, [37]) and Bayesian MCMC (BEAST, [38] were used for the validation each other. Most probable trees were selected based on three methods; approximate likelihood-ratio test (aLRT) statistics [39] implemented in PhyML and combination of 9 statistical analysis implemented in CONSEL [40] in the maximum-likelihood methods, and 95% highest posterior density (HPD) in Bayesian MCMC. Tree topology was tested based on statistical methods implemented in CONSEL, and tree distance was quantified using Robinson–Foulds metric [41] implemented in treedist in the PHYLIP package. When compared with the phylogenetic tree based on whole genome alignment of MTBC (Figure 1a) as the gold standard, the tree based on SNP concatemers from CS (Figure 1b) showed essentially the same topology as the maximum-likelihood phylogenetic tree chosen by aLTR statics, whereas the tree based on the H37Rv sequence showed different positioning of RGTB327 and RGTB423 (Figure 1c). In Bayesian MCMC phylogeny chosen by 95% highest posterior density (HPD), compared with the phylogenetic tree based on whole genome alignment of MTBC (Figure 2a), the tree based on SNP concatemers from CS showed different positioning of CDC1551, while that from H37Rv showed different positioning of RGTB327, Erdman, CCDC5079, CCDC5180, H37Rv and H37Ra (Figure 2c). The same tendency was observed in phylogenetic trees chosen by combination of 9 statistical analyses in maximum-likelihood with bootstrapping (Additional file 6: Figure S1 and Additional file 7: Table S3). In all three trees chosen by different statistical methods, distance between tree based on whole genome alignment and SNP concatemers derived from CS was always smaller than that based on whole genome alignment and SNP concatemers derived from H37Rv (Table 3). These results indicated that SNP concatemers based on different reference sequences behave differently in phylogenetic analysis, emphasizing the critical importance of selecting the proper reference sequence, and CS is superior to H37Rv when it is used as the reference sequence in phylogenetic analysis of MTBC. We also compared the computational times required by these analyses (Table 4). Use of CS as the reference markedly reduced the times required for both the maximum-likelihood (5 vs 35 hr with bootstrapping) and Bayes MCMC (2 vs 22 hr) methods without a critical deterioration in tree topology when compared with whole genome alignment (Figures 1 and 2). The time difference observed could be explained essentially by two parameters, the size and quality of alignments. Alignment length based on whole genome, SNPs derived from CS and SNPs derived from H37Rv is 5.0 Mbp, 21,425 bp, and 52,203 bp, respectively, and H37Rv based SNP concatemers contain biased SNPs (see below). Reduced size and better quality of alignment seems to contribute the reduction of computational time.
Figure 1.
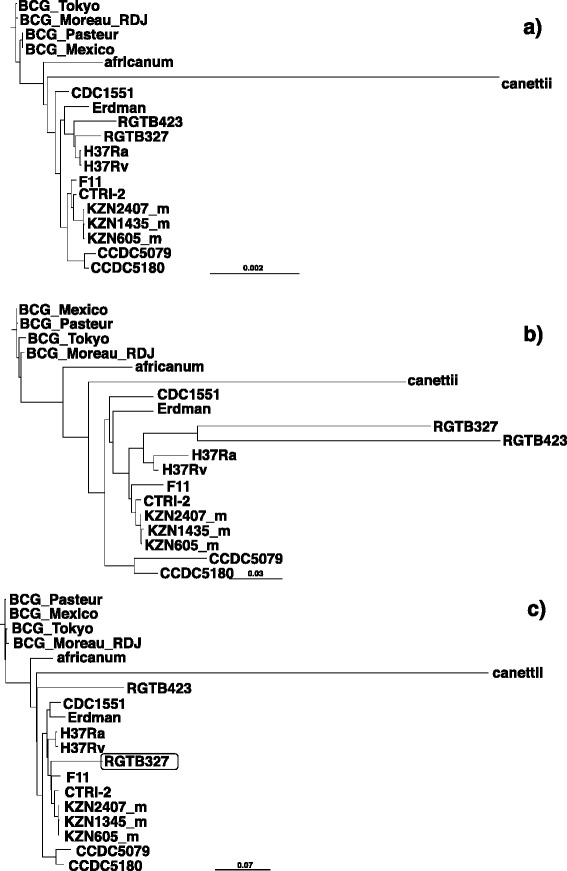
Maximum-likelihood phylogenies based on whole genome and SNP concatenated sequence alignment. Phylogenetic trees based on whole genome sequences (a), SNP concatemers using CS as reference (b) and SNP concatemers using the H37Rv genome sequence as reference (c) were constructed using PhyML 3.0 [39]. Most probable trees were selected based on aLTR statics implemented in PhyML [39]. Isolates, clustered into different positions compared with the phylogenetic tree based on the whole genome sequences of M. tuberculosis strains RGTB327 and RGTB423, are indicated in the squares. For the KZN series, inversion-corrected sequences were used for the alignment and marked “_m”. aLTR statics values for each branch are shown. In Figure 1a, clusters of lineage 4 and 2 are indicated in the squares.
Figure 2.

Bayesian post-probable phylogenies based on whole genome and SNP concatenated sequence alignment. Description of data: Phylogenetic trees based on whole genome sequence (a), SNP concatemers using the consensus genome sequence as reference (b) and SNP concatemers using H37Rv genome as reference were constructed using BEAST [38]. All relevant parameters reached an effective sample size of >100, indicating good convergence of the chains. For each branch, 95% highest posterior density is shown with good support. Isolates, clustered into different positions compared with the phylogenetic tree based on the whole genome sequences of M. tuberculosis are indicated in the squares.
Table 3.
Distance analysis among phylogenic trees constructed based on maximum-likelihood and Bayesian MCMC methods
| Phylogenetic analysis | vs CS | vs H37Rv | ||
|---|---|---|---|---|
| Unrooted | Rooted | Unrooted | Rooted | |
| PhyML(aLRT) | 2.88E-01 | 2.88E-01 | 5.95E-01 | 5.95E-01 |
| PhyML(consel selected) | 2.89E-01 | 2.89E-01 | 5.96E-01 | 5.96E-01 |
| BEAST | 1.68E + 03 | 1.20E + 03 | 2.61E + 03 | 1.91E + 03 |
To quantify the branch score distance between trees, Robinson and Fould test [41] implemented in treedist in the Phylip package was utilized. Both of unrooted and rooted scores were calculated.
Table 4.
Computational time for each phylogenetic analysis
| Sequence type | PhyML3 (bootstrapping) | PhyML3 (aLRT) | BEAST ver.1.7 |
|---|---|---|---|
| Whole genome | 34h59min55sec | 2min49sec | 21h40min45sec |
| Consensus sequence | 4h50min9sec | 1min24sec | 1h47min24sec |
| H37Rv | 19h24min38sec | 2min37sec | 2h38min31sec |
For PhyML, two tree selection methods, 100 bootstrappings and aLRT were performed. In the bootstrap analyses, multithreading with 16 CPUs were utilized to reduce the computational time. For BEAST, 10 million samplings were performed for each analysis. Computational times were based on the log file of each analysis.
To obtain further insight on the behavior of SNP concatemers relative to on the different reference sequences, we analyzed the number of SNP called in individual strains using as reference the consensus or the H37Rv genome sequence (Table 5). We observed marked bias in the number of SNPs called in each strain when H37Rv was the reference. The number of SNPs was much higher in M. canettii than in the other strains. Large numbers of SNPs were also present in M. tuberculosis strains RGTB423, RGTB327, CCDC5079, CCDC5180, CDC1551 and Erdman, which behave differently in phylogenetic analyses based on their SNP concatemers (Additional file 7: Figure S1). Differences among strains in the number of SNPs were reduced when CS was used as the reference. Statistical analysis indicated that the rate of detection of SNPs unique to a particular strain was significantly higher using the consensus than using the H37Rv sequence as a reference (Table 2). The only exception was M. canettii, which showed a higher detection rate when compared with H37Rv. The BCG and KZN series, each consists of closely related strains, with individual strains having small numbers of unique SNPs. In comparing the number of detection of SNPs commonly found in BCG or KZN strains (Table 2), we found that detection of SNPs in BCG strains was greater using the consensus than the H37Rv sequence, although no significant difference was observed in detecting SNPs in KZN strains. These results indicated that SNP calling with CS makes possible the better detection of truly unique and crucial SNPs, which discriminate accurately among the strains.
Table 5.
Comparison of Illumina read mapping efficacy using clinical isolates derived from different lineages using Bowtie2 and SAMtools
| i) Comparison of the numbers of mapped and unmapped reads to the H37Rv sequence or consensus sequence | ||||||||
|---|---|---|---|---|---|---|---|---|
| LineAge | H37Rv | Consensus sequence | Subtraction of ratio (%) | |||||
| Mapping stringency* | Local | End to end | Local | End to end | Local | End to end | ||
| (CS minus H37Rv) | ||||||||
| F092 | EAI | mapped | 681561 | 664376 | 684952 | 667250 | ||
| unmapped | 22261 | 39446 | 18870 | 36572 | ||||
| ratio (%) | 96.837 | 94.395 | 97.319 | 94.804 | 0.482 | 0.408 | ||
| J156 | EAI | mapped | 1680156 | 1650866 | 1689673 | 1658917 | ||
| unmapped | 40162 | 69452 | 30645 | 61401 | ||||
| ratio (%) | 97.665 | 95.963 | 98.219 | 96.431 | 0.553 | 0.468 | ||
| F038 | Haarlem, LAM, X etc. | mapped | 1024873 | 997301 | 1029625 | 1000714 | ||
| unmapped | 75113 | 102685 | 70361 | 99272 | ||||
| ratio (%) | 93.171 | 90.665 | 93.603 | 90.975 | 0.432 | 0.310 | ||
| F070 | Haarlem, LAM, X etc. | mapped | 858126 | 840921 | 861393 | 843463 | ||
| unmapped | 27822 | 45027 | 24555 | 42485 | ||||
| ratio (%) | 96.860 | 94.918 | 97.228 | 95.205 | 0.369 | 0.287 | ||
| J073 | Haarlem, LAM, X etc. | mapped | 1534315 | 1503494 | 1537891 | 1504256 | ||
| unmapped | 11979 | 42800 | 8403 | 42038 | ||||
| ratio (%) | 99.225 | 97.232 | 99.457 | 97.281 | 0.231 | 0.049 | ||
| J147 | Haarlem, LAM, X etc. | mapped | 847807 | 836269 | 849747 | 836489 | ||
| unmapped | 11775 | 23313 | 9835 | 23093 | ||||
| ratio (%) | 98.630 | 97.288 | 98.856 | 97.313 | 0.226 | 0.026 | ||
| F081 | other non-Beijing | mapped | 1004912 | 974425 | 1008107 | 976556 | ||
| unmapped | 43978 | 74465 | 40783 | 72334 | ||||
| ratio (%) | 95.807 | 92.901 | 96.112 | 93.104 | 0.305 | 0.203 | ||
| J020 | other non-Beijing | mapped | 1081365 | 1062537 | 1085065 | 1065704 | ||
| unmapped | 11293 | 30121 | 7593 | 26954 | ||||
| ratio (%) | 98.966 | 97.243 | 99.305 | 97.533 | 0.339 | 0.290 | ||
| J027 | other non-Beijing | mapped | 751633 | 741219 | 754861 | 744254 | ||
| unmapped | 5259 | 15673 | 2031 | 12638 | ||||
| ratio (%) | 99.305 | 97.929 | 99.732 | 98.330 | 0.426 | 0.401 | ||
| F022 | Ancestral Beijing | mapped | 1162270 | 1143243 | 1166545 | 1147960 | ||
| unmapped | 26600 | 45627 | 22325 | 40910 | ||||
| ratio (%) | 97.763 | 96.162 | 98.122 | 96.559 | 0.360 | 0.397 | ||
| J090 | Ancestral Beijing | mapped | 490815 | 484340 | 492424 | 486326 | ||
| unmapped | 5153 | 11628 | 3544 | 9642 | ||||
| ratio (%) | 98.961 | 97.655 | 99.285 | 98.056 | 0.324 | 0.400 | ||
| J002 | Ancestral Beijing | mapped | 736473 | 727044 | 739288 | 730539 | ||
| unmapped | 5757 | 15186 | 2942 | 11691 | ||||
| ratio (%) | 99.224 | 97.954 | 99.604 | 98.425 | 0.379 | 0.471 | ||
| J029 | Modern Beijing | mapped | 953792 | 936476 | 957539 | 941221 | ||
| unmapped | 10220 | 27536 | 6473 | 22791 | ||||
| ratio (%) | 98.940 | 97.144 | 99.329 | 97.636 | 0.389 | 0.492 | ||
| F076 | Modern Beijing | mapped | 532526 | 519473 | 534742 | 522431 | ||
| unmapped | 16374 | 29427 | 14158 | 26469 | ||||
| ratio (%) | 97.017 | 94.639 | 97.421 | 95.178 | 0.404 | 0.539 | ||
| J111 | Modern Beijing | mapped | 719693 | 708076 | 722895 | 712304 | ||
| unmapped | 14341 | 25958 | 11139 | 21730 | ||||
| ratio (%) | 98.046 | 96.464 | 98.482 | 97.040 | 0.436 | 0.576 | ||
| ii) Comparison of mappping frequency ratio (%) among the MTBC lineanges | ||||||||
| EAI | Haarlem, LAM, X etc. | other non-Beijing | ||||||
| Haarlem, LAM, X etc. | ns | - | - | |||||
| other non-Beijing | ns | ns | - | |||||
| Beijing | P < 0.05 | ns | ns | |||||
In this analysis CS based on 13 M. tuberculosis strains (Table 1) was used as the consensus sequence. i) After mapping with Bowtie2 [42] against H37Rv or CS, the idxstats command of SAMtools [43] was used to calculate the mapping efficacy (Table 5). In read mapping with Bowtie2, both of local and end-to-end mapping mode were performed, and the other parameters were set with default values. Significant differences in mapping frequencies were assessed using multiple comparisons of proportions tests [44]. For all isolates, the difference between H37Rv and CS as a reference differed significantly (p < 0.0001). For both mapping modes, the ratio of mapped to total reads was calculated, and these values used to calculate differences in mapping frequency between the consensus and H37Rv sequences by simple subtraction.
ii) Based on the difference in mapping frequency in 1), the mapping frequencies of MTBC lineages were compared using Mann–Whitney U tests. Combination of Beijing and EAI sequences showed the significan difference (p < 0.05) in mapping frequencies when compared relative to the consensus and H37Rv sequences, the latter belonging to the Haarlem, LAM, X etc. lineage (linage 4).
Comparison of Illumina read mapping efficacy
Mapping of sequence reads from WGS, such as Illumina, to a reference genome sequence is the first and most crucial step in detecting SNPs and indels in isolates of interest, and for subsequent phylogenetic analysis with SNP concatenated sequences. To compare the mapping results using the consensus and H37Rv sequences as the reference, approximately one million 251 bp x 2 pair-end reads were obtained from clinical M. tuberculosis isolates of different MTBC lineages (Tables 5 and 6). About a million reads per isolate were used for the analyses. First, we used Bowite2 [42] and SAMtools [43], which is well established read mapping tool and analysis tool for the resulting mapping, respectively. After mapping with Bowtie against H37Rv or CS, the idxstats command of SAMtools was used to calculate the mapping efficacy (Table 5). Significance tests for multiple comparisons of proportions [44] indicated that each combination of consensus and H37Rv sequences for individual read data in each mapping stringency setting showed significant difference in the proportion of mapped reads (p < 0.0001), with the proportion always higher using CS as the reference. To compare the ratio of reads mapped to unmapped, ratio of mapped reads to total reads (%) in mapping with H37Rv as the reference was subtracted from that of CS. In both of local and end-to-end mapping mode of bowtie, the mapping ratio with CS is better than that of H37Rv.
Table 6.
Comparison of Illumina read mapping efficacy using clinical isolates derived from different lineages using
| Isolate | Lineage | H37Rv | Consensus sequence | Subtraction of ratio (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mapping stringency* | Ambiguity | Medium | Strict | Ambiguity | Medium | Strict | (Consensus-H37Rv) | ||||
| F092 | EAI | mapped | 676219 | 676007 | 674779 | 677079 | 676941 | 676114 | |||
| unmapped | 20275 | 20487 | 21715 | 19415 | 19553 | 20380 | |||||
| ratio (%) | 97.089 | 97.059 | 96.882 | 97.212 | 97.193 | 97.074 | 0.123 | 0.134 | 0.192 | ||
| J156 | EAI | mapped | 1656675 | 1656191 | 1652024 | 1661496 | 1660887 | 1657869 | |||
| unmapped | 37151 | 37635 | 41802 | 32330 | 32939 | 35957 | |||||
| ratio (%) | 97.807 | 97.778 | 97.532 | 98.091 | 98.055 | 97.877 | 0.285 | 0.277 | 0.345 | ||
| F038 | Haarlem, LAM, X etc. | mapped | 985717 | 985256 | 978713 | 986844 | 986318 | 980541 | |||
| unmapped | 43761 | 44222 | 50765 | 42634 | 43160 | 48937 | |||||
| ratio (%) | 95.749 | 95.704 | 95.069 | 95.859 | 95.808 | 95.246 | 0.109 | 0.103 | 0.178 | ||
| F070 | Haarlem, LAM, X etc. | mapped | 847048 | 846879 | 844774 | 847486 | 847257 | 845485 | |||
| unmapped | 21212 | 21381 | 23486 | 20774 | 21003 | 22775 | |||||
| ratio (%) | 97.557 | 97.537 | 97.295 | 97.607 | 97.581 | 97.377 | 0.05 | 0.044 | 0.082 | ||
| J073 | Haarlem, LAM, X etc. | mapped | 1511361 | 1511205 | 1508937 | 1512926 | 1512725 | 1511328 | |||
| unmapped | 25005 | 25211 | 27479 | 23490 | 23691 | 25088 | |||||
| ratio (%) | 98.372 | 98.359 | 98.211 | 98.471 | 98.458 | 98.367 | 0.099 | 0.099 | 0.156 | ||
| J147 | Haarlem, LAM, X etc. | mapped | 835484 | 835319 | 834077 | 835192 | 834976 | 834227 | |||
| unmapped | 14694 | 14859 | 16101 | 14986 | 15202 | 15951 | |||||
| ratio (%) | 98.272 | 98.252 | 98.106 | 98.237 | 98.212 | 98.124 | −0.034 | −0.04 | 0.018 | ||
| F081 | other non-Beijing | mapped | 984775 | 984303 | 981542 | 986321 | 986079 | 983479 | |||
| unmapped | 31067 | 31539 | 34300 | 29521 | 29763 | 32363 | |||||
| ratio (%) | 96.942 | 96.895 | 96.623 | 97.094 | 97.07 | 96.814 | 0.152 | 0.175 | 0.191 | ||
| J020 | other non-Beijing | mapped | 1061748 | 1061460 | 1059770 | 1063613 | 1063346 | 1062167 | |||
| unmapped | 22918 | 23206 | 24896 | 21053 | 21320 | 22499 | |||||
| ratio (%) | 97.887 | 97.861 | 97.705 | 98.059 | 98.034 | 97.926 | 0.172 | 0.174 | 0.221 | ||
| J027 | other | mapped | 741045 | 740904 | 739801 | 742516 | 742329 | 741694 | |||
| non-Beijing | unmapped | 13721 | 13862 | 14965 | 12250 | 12437 | 13072 | ||||
| ratio (%) | 98.182 | 98.163 | 98.017 | 98.377 | 98.352 | 98.268 | 0.195 | 0.189 | 0.251 | ||
| F022 | Ancestral Beijing | mapped | 1140373 | 1140107 | 1137877 | 1145147 | 1144862 | 1143607 | |||
| unmapped | 32673 | 32939 | 35169 | 27899 | 28184 | 29439 | |||||
| ratio (%) | 97.215 | 97.192 | 97.002 | 97.622 | 97.597 | 97.49 | 0.407 | 0.405 | 0.488 | ||
| J090 | Ancestral Beijing | mapped | 2087545 | 2086983 | 2082777 | 2095411 | 2094879 | 2092249 | |||
| unmapped | 47551 | 48113 | 52319 | 39685 | 40217 | 42847 | |||||
| ratio (%) | 97.773 | 97.747 | 97.55 | 98.141 | 98.116 | 97.993 | 0.368 | 0.37 | 0.444 | ||
| J002 | Ancestral Beijing | mapped | 725501 | 725308 | 724182 | 727822 | 727702 | 727147 | |||
| unmapped | 13427 | 13620 | 14746 | 11106 | 11226 | 11781 | |||||
| ratio (%) | 98.183 | 98.157 | 98.004 | 98.497 | 98.481 | 98.406 | 0.314 | 0.324 | 0.401 | ||
| J029 | Modern Beijing | mapped | 935765 | 935598 | 934129 | 939368 | 939185 | 938425 | |||
| unmapped | 21607 | 21774 | 23243 | 18004 | 18187 | 18947 | |||||
| ratio (%) | 97.743 | 97.726 | 97.572 | 98.119 | 98.1 | 98.021 | 0.376 | 0.375 | 0.449 | ||
| F076 | Modern Beijing | mapped | 523546 | 523438 | 522478 | 526300 | 526150 | 525618 | |||
| unmapped | 17480 | 17588 | 18548 | 14726 | 14876 | 15408 | |||||
| ratio (%) | 96.769 | 96.749 | 96.572 | 97.278 | 97.25 | 97.152 | 0.509 | 0.501 | 0.58 | ||
| J111 | Modern Beijing | mapped | 703968 | 703761 | 702412 | 708028 | 707765 | 707024 | |||
| unmapped | 17244 | 17451 | 18800 | 13184 | 13447 | 14188 | |||||
| ratio (%) | 97.609 | 97.58 | 97.393 | 98.172 | 98.135 | 98.033 | 0.563 | 0.555 | 0.639 | ||
| ii) Comparison of mappping frequency ratio (%) among the MTBC lineanges | |||||||||||
| EAI | HaarlemLAM, X etc. | other non-Beijing | |||||||||
| Haarlem,LAM, X etc. | ns | - | - | ||||||||
| non-Beijing | ns | ns | - | ||||||||
| Beijing | P<0.05 | P<0.01 | P<0.05 | ||||||||
In this analysis CS based on 13 M. tuberculosis strains (Table 1) was used as the consensus sequence. i) The effects on mapping efficacy were tested for three combinations of parameters: mismatch cost, insertion cost, deletion cost, matching length and similarity. *Mapping stringency was defined as Ambiguous, with frequencies of mismatch cost, insertion cost, deletion cost, matching length and similarity of 2, 2, 2, 0.5, and 0.8, respectively; Medium, with frequencies of 2, 3, 3, 0.5, and 0.8, respectively; and Strict, with frequencies of 3, 3, 3, 0.5, and 0.95, respectively. Significant differences in mapping frequencies were assessed using multiple comparisons of proportions tests [44]. For all isolates, the difference between H37Rv and CS as a reference differed significantly (p < 0.0001). For each stringency setting, the ratio of mapped to total reads was calculated, and these values used to calculate differences in mapping frequency between the consensus and H37Rv sequences by simple subtraction.
ii) Based on the difference in mapping frequency in 1), the mapping frequencies of MTBC lineages were compared using Mann–Whitney U tests. The Haarlem, LAM, X and Beijing sequences showed the greatest difference (p < 0.01) in mapping frequencies when compared relative to the consensus and H37Rv sequences.
Based on a number of typing methods, MTBC could be divided into several lineages [7,12,45]. To analyze whether these MTBC lineages influence the read mapping efficacy, we analyzed the values obtained by subtracting the ratio of mapped to total reads number on the H37Rv sequence from that on CS (Table 5 ii). Using the Mann–Whitney U test, we found that isolates from the Beijing family showed significantly greater differences in mapping frequencies between H37Rv and consensus sequences than did isolates from other lineages (P < 0.05), indicating the greater suitability of CS in assessing mapping frequency.
Then we used a commercial software, CLC genomics workbench (CLC bio) to evaluate read mapping efficacy with a different algorism, which is based on Smith and Waterman [46], from Bowtie2. After quality based trimming described in the method section, the sequence reads were mapped to the consensus and H37Rv sequences, and the number of reads mapped and unmapped were compared as did with Bowtie2 and SAMtools. The efficacy of mapping was compared by analyzing three combinations of parameters: mismatch cost, insertion cost, deletion cost, matching length and similarity. Significance tests described above showed that each combination of consensus and H37Rv sequences for individual read data in each mapping stringency setting differed significantly difference in the proportion of mapped reads (p < 0.0001), with the proportion always higher using CS as the reference. The one exception was the isolate J147, which belongs to the Haarlem lineage, as does H37Rv, explaining the higher mapping efficacy found with H37Rv as the reference.
Again, we analyzed whether these MTBC lineages influence the read mapping efficacy in the different methods (Table 6 ii). We found that isolates from the Beijing family showed significantly greater differences in mapping frequencies between H37Rv and consensus sequences than did isolates from other lineages (P < 0.05). The difference was greatest when comparing the Beijing and Haarlem lineages, with the latter being the lineage to which H37Rv belongs (P < 0.01).
The reads failed to map H37Rv specifically and CS specifically were analyzed for their character with contigs constructed by de novo assembling them and BLAST search on the database. Notably, no contigs were created from reads failed to map specifically, indicating the reads were relatively low quality reads to be assembled. BLAST analyses of resulting contigs from reads failed to map H37Rv showed that all contigs had very little identity within the genome sequence of H37Rv as expected, and some of them were MTBC lineage specific while the others were simply missing from genome sequence of H37Rv but not lineage specific.
These results indicate that read mapping of MTBC based on the WGS data is sensitive to both the reference sequence and the MTBC lineage. Our CS provided a better standard for mapping efficacy of different MTBC lineages than did the H37Rv sequence, as well as statistically significant improvements in SNP detection and read mapping, suggesting that CS is a virtually better approach for MTBC research.
Virtual VNTR typing, spoligotyping and LPS typing
We performed VNTR typing of the 19 mycobacterial strains in silico using the MIRU-VNTR 24-loci system [14], which is used commonly in epidemiologic studies of mycobacteria. Based on the reported primer sequence [45], the number of corresponding regions was analyzed in each strain (Figure 3). Of these 19 strains, only the profile for H37Rv was available in the MIRU-VNTRplus database. Comparison of our virtual and actual VNTR profiles of H37Rv showed that 23 of the 24 loci were identical, with one mismatch observed in VNTR3690. This discrepancy may have been due to maintenance of H37Rv stocks in different laboratories [47,48]. When we compared the virtual and actual [49] VNTR profiles of M. tuberculosis strain CTRI-2, we found that 20 of the 24 loci (83%) were identical, whereas the other four loci differed slightly, by one copy per locus (Figure 3). Similarly, the virtual and actual profiles of M. africanum and M. canettii differed in 2 of 24 and 4 of 24 loci, respectively. At present we do not know whether the 2 strains of each species, one tested and the other found in the database, are identical. Thus, these discrepancies may have been due to genetic differences between isolates. Nevertheless, these results suggest that virtual VNTR analysis based on genome sequences is in good agreement with experimental data.
Figure 3.
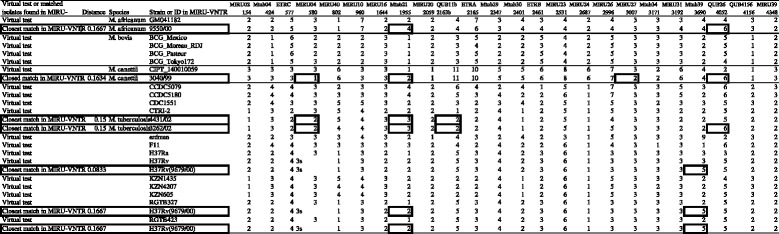
Virtual VNTR profile of each genome and the similarity search in MIRU-VNTR. The number of each VNTR marker in each genome was analyzed based on primer sequences [45]. These numbers were input for VNTR analyses by MIRU-VNTR (http://www.miru-vntrplus.org/). The closest match is presented immediately below the line for the corresponding isolate. Markers differing in number for the analyzed strain and its closest match are indicated in bold boxes.
We also performed spoligotyping in silico, a method based on 43 direct repeat (DR) spacer sequences [50] and commonly used in epidemiological studies of mycobacteria. The virtual spoligotype profile of H37Rv was identical to the actual profile stored in the MIRU-VNTRplus database (Figure 4). Of the 19 strains of mycobacteria tested, 15 were correctly grouped into reasonable lineages, whereas M. canettii, and M. tuberculosis strains H37Ra, RGTB327 and RGTB423 did not yield exact matches. Closer analysis showed that the profile of M. tuberculosis strain RGTB423 differed from that in the MIRU-VNTRplus database by two DR markers, whereas the three other strains differed by one DR marker each.
Figure 4.
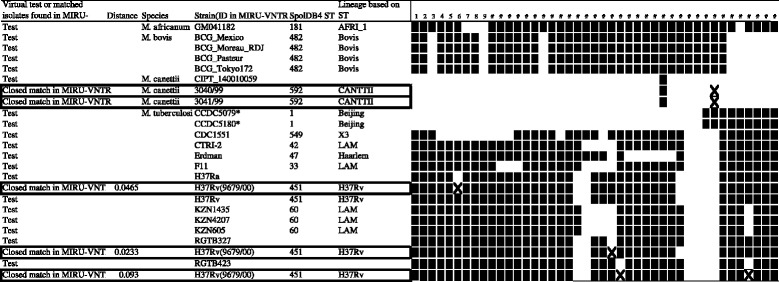
Virtual spoligotyping of each strain and similarity search at MIRU-VNTR. Each strain's genome was analyzed for the presence of absence of each spoligo spacer based on primer sequences [50]. The presence or absence of each spacer input for spoligotyping analysis at MIRU-VNTR (http://www.miru-vntrplus.org/). The closest match by similarity search is shown immediately below the line for the corresponding isolate. Markers differing in presence or absence in the analyzed strain and its closest match are indicated in bold boxes. * reported as Beijing type [56].
Long sequence polymorphisms (LSPs) were introduced to determine whether lineages of M. tuberculosis were associated with specific human populations [7,8]. Utilizing LSPs, MTBC could be divided into at least 6 phylogeographical lineages, each associated with specific, sympatric human populations. LSP analysis was performed on the 19 sequenced genomes of mycobacteria in silico, using primers based on target sequence [8] (Table 7). Fifteen strains were classified as lineage 4 (Euro-American lineage) while CCDC5079 and CCDC5180 were classified lineage 2 (East-Asia lineage) and M. africanum was classified as lineage 6 (West-Africa lineage).
Table 7.
Virtual analyses of LSP and Beijing typing
| Species | Strain name | LSP lineage | Beijing typing | |
|---|---|---|---|---|
| Beijing | Modern/Ancestral | |||
| M. tuberculosis | CCDC5079 | 2 | No | - |
| CCDC5180 | 2 | Yes | Modern | |
| CDC1551 | 4 | No | - | |
| CTRI-2 | 4 | No | - | |
| Erdman | 4 | No | - | |
| F11 | 4 | No | - | |
| H37Ra | 4 | No | - | |
| H37Rv | 4 | No | - | |
| KZN605 | 4 | No | - | |
| KZN1435 | 4 | No | - | |
| KZN4207 | 4 | No | - | |
| RGTB327 | 4 | No | - | |
| RGTB423 | 4 | No | - | |
| M. bovis BCG | Mexico | 4 | No | - |
| Moreau RDJ | 4 | No | - | |
| Pasteur | 4 | No | - | |
| Tokyo 172 | 4 | No | - | |
| M. africanum | GMO41182 | 6 | No | - |
| M. canettii | CIPT 140010059 | 2 | No | - |
Cross-sectional studies in diverse geographic locations have shown epidemiologic associations between Beijing types of M. tuberculosis and increased risks of drug resistance [51]. Beijing typing was originally based on spoligotyping [52], but was later determined by detection of specific SNP [53,54] and PCR analysis of the insertion of IS6110 into specific positions [55]. We analyzed whether the 19 sequenced MTBC genomes could be classified as Beijing type, based on IS6110 insertion between Rv0001 and Rv0002, as shown for the H37Rv genome; or into a modern or ancestral subtype based on IS6110 insertion into the NTF region [55] (Table 7). Of the strains tested, only CCDC5180 was Beijing type. Although spoligotyping indicated that CCDC5079 should also be Beijing type (Figure 4 and [56]), this strain lacks an inserted IS6110 between Rv0001 and Rv0002. Although classification of Beijing type based on IS6110 insertion is limited in classifying MTBC lineages, virtual Beijing typing could be performed using an in silico approach.
Taken together, these results suggest that virtual VNTR, spoligotyping, LSP analysis and Beijing typing in silico can be utilized for epidemiological analysis of mycobacterial strains without the need for PCR amplification and/or hybridization procedures.
Discussion
In this study, we successfully aligned whole genome sequences of 19 MTBC strains by correcting the large rearrangements found in the genomes of KZN605, KZN1435 and KZN4207, and using very fast multiple sequence alignment software, MAFFT [32,57]. This alignment allowed us to create a virtual consensus genome sequence of MTBC, reflecting all genetic information from various lineages. In comparing this CS with that of H37Rv as the reference sequence, we found that CS allowed an unbiased and efficient detection of critical SNPs, distinguishing among the lineages of MTBC. Use of CS as a reference reduced the SNP calling bias, as shown for M. canettii. Moreover, SNP concatemers of MTBC strains based on CS were better able to reproduce a phylogenetic tree based on whole genome alignment than concatemers based on H37Rv. Phylogeny of M. tuberculosis is very closely related the human evolution, and consistent with MTBC displaying characteristics indicative of adaptation to both low and high host densities [20]. Ford at al reported that M. tuberculosis strains from lineage 2 (East Asian lineage and Beijing sublineage) acquire drug resistances in vitro more rapidly than M. tuberculosis strains from lineage 4 (Euro-American lineage) and that this higher rate can be attributed to a higher mutation rate [51]. Thus precise and accurate phylogenetic analysis based on SNP concatemers is becoming a key importance of M. tuberculosis research. Using H37Rv as a reference to call SNP led to some inadequate clustering of M. tuberculosis strain. As shown in Figure 1c, RGTB432 becomes an out-group although it should be placed on the lineage 4 group as in Figure 1a. Thus, adequate reference sequence is not only important for efficient analysis such as mapping of read data from next generation sequencers but also phylogenetic analysis based on SNP concatemers because it directly link to evolution and drug resistance of M. tuberculosis strain. Use of CS also significantly reduced the total number of SNPs detected, decreasing computational time by an order of magnitude. Reduction of computational time is extremely useful when analyzing a large number (more than hundreds) of isolates.
During the construction of CS, we also called SNPs and indels using as reference the H37Rv genome to update information about polymorphisms found in MTBCs. Complete SNP data of individual isolates were presented with position and annotations. As reported previously, about 50% of the indels were located in the genes encoding the PE-PGRS and PPE family of proteins while about 25% were located in intergenic regions. The positions, length, annotation and strains of all SNPs and indels have been reported for further applications, such as exploration of lineage specific gene traits.
Use of CS as a reference also significantly improved the efficacy of short-read mapping of clinical isolates. Use of a particular strain as the reference can affect the mapping results, depending on the lineage of that strain. Testing of isolates of a different lineage from the reference strain can result in the omission of some SNPs and/or indels critical for further analysis. Use of a consensus sequence as a reference would minimize this possibility.
VNTR typing, spoligotyping, LSP analysis and Beijing typing. Thus, technically, it is possible to perform these analyses using sequence data of MTBC strains. To prove this concept, we performed these analyses in silico. Although actual typing data were available for a few of the strains tested, we observed fairly good agreement between actual and expected data. Virtual typing also showed several limitations. For example, Beijing typing classified strain CCDC5079, which belongs to the Beijing family, as non-Beijing type based on IS6110 insertion. Thus, one typing method would not be sufficient to accurately type MTBC isolates. As WGS technologies improve, SNP concatenation would become the ideal typing method. Moreover, we found that in silico analysis using CS was highly reproducible and robust because of its intrinsically objective nature, an objectivity sometimes lacking during actual epidemiological analysis of MTBC. in silico analysis is also labor-saving, since it requires only WGS data.
Our consensus genome sequence does not contain sequence information on several lineages, including lineages 1, 3 and 5 in LPS analysis. This could be a potential shortage because lacks of particular lineages in CS could lead bias in calling SNP as shown in this study. At present, few complete whole genome sequences of these lineages are available in the database. We intend to update our consensus sequence when such information becomes available.
Conclusion
We generated virtual consensus sequences of MTBC from 13 M. tuberculosis and 6 non-tuberculosis strains, and showed that this sequence was superior to the H37Rv sequence as a reference in MTBC research. A completely annotated consensus sequence, relative to the sequence of H37Rv, is available as the additional data. Construction of a web based service integrating the phylogenetic and epidemiological analyses performed in this study is currently underway.
Methods
Multiple genome sequence alignment and construction of a virtual MTBC consensus genome sequence
Whole genome sequences used to construct CSs in this study are detailed in Table 1. Genome sequences of 19 strains available by April 2012 were used: 13 genomes from M. tuberculosis, one each from M. africanum and M. canetti, and four from M. bovis BCG. Genome rearrangement was analyzed by a publicly available software, Mauve [31], with large inversions, hampering genome alignment, observed in M. tuberculosis strains KZN605, KZN1435 and KZN4207 and manually corrected. We constructed two types of consensus whole genome sequences: one containing sequence data from 13 M. tuberculosis strains and the other one containing sequence data from all four species of mycobacteria (19 strains). All sequence data were downloaded from the NCBI databases and aligned using publicly available alignment software, MAFFT version 6 or 7 [32,57] (http://mafft.cbrc.jp/alignment/server/) using our own-build Linux-based server. Consensus sequences were constructed by merging the alignment results using a sequence editing commercial software, Genetyx (Genetyx Inc, Tokyo Japan). Two types of consensus sequences were prepared with handling SNPs as majority rule or ambiguity rule. In this study, ambiguity sequence was used for further analysis.
Annotation of the virtual M. tuberculosis consensus genome
The consensus genome, consisting of an artificially assembled sequence, was annotated manually and edited using a commercial genome sequence editing software, in silico molecular cloning (in silico Biology Co., Kanagawa, Japan). Because CS used for the annotation contains many ambiguous nucleotides and insertion, instead of CDS extraction, homologous regions based on corresponding nucleotide sequences of CDS in H37Rv was determined to extract the corresponding regions in CS, with the assignment of each region based on the locus_tag (the Rv number) of CDS, repeat regions, rRNA and tRNA of H37Rv.
SNP and indel extraction, annotation and characterization
Each genome sequence of MTBC listed in Table 1 was compared with the constructed CS and with the genome sequence of M. tuberculosis H37Rv using a publicly available software, MUMmer 3.0 [58]. SNPs and indels were extracted from each MTBC strain. For SNP analysis, insertions of more than two bp and all deletions were excluded from the resulting data. The resulting files from MUMmer were converted into the VCF format to annotate the SNPs using the a commercial software, CLC genomics workbench (CLC bio). Since the algorithms used to align genome sequences for the construction of the consensus genome (MAFFT) and for the extraction of SNPs and indels (MUMmer) are different, we manually checked the result of indel calls in the MUMmer data, and indels greater than 5 bp were used for manual annotation.
Phylogenetic analysis using SNP concatenated and whole genome sequences
Phylogenetic trees were constructed from SNP and whole-genome sequence alignments. Two methods were used to evaluate robustness: a maximum-likelihood approach, PhyML 3.0 [39] and the Bayesian MCMC framework, BEAST1.7 [38]. For PhyML, GTR and gamma were chosen for a nucleotide substitution model, and tree robustness was evaluated by two methods: approximate likelihood-ratio test (aLTR) [37] implemented in PhyML. Bootstrappings implemented in PhyML was used to generate multiple trees (100 trees for SNP concatemers) for choosing most probable trees based on combination of 9 statistical analyses using CONSEL [40]. Because data size limitation in CONSEL, 40 trees were generated by PhyML for phylogenetic analysis with whole genome alignment.
For BEAST, various combinations of population size change and molecular clock models were compared to find the model that best fit the data. A simple HKY model was used for SNP concatemer based phylogenetic trees, whereas an HKY kappa model was used for whole-genome sequence based phylogenetic trees. MCMC chains were run for 10 million generations, with sampling every 1000th generation. Convergences and Effective Sample Sizes (ESSs) of the estimates were checked using Tracer v1.4 (http://beast.bio.ed.ac.uk/Tracer). Three of the phylogenetic trees we constructed had ESS values greater than 100, suggesting sufficient mixing of the Markov chain. Maximum clade credibility (MCC) trees were created and annotated using TreeAnnotator within the BEAST software package. All analyses were performed on Linux or Windows 7 based computers. Trees were visualized with FigTree (http://tree.bio.ed.ac.uk/software/figtree/). The tree to tree distance were compared using Robinson and Fould test [41] implemented in the treedist program in the Phylip package (http://evolution.genetics.washington.edu/phylip/doc/treedist.html) with both of unrooted and rooted mode. The computation time for each analysis was obtained from the log files. Phylogenetic data related to this study have been registered at TreeBase (http://datadryad.org/(doi:10.5061/dryad.nq070)).
Comparison of Illumina read mapping efficacy
To compare the mapping efficacy using CS as reference with H37Rv, we obtained approximately one million 251 bp x 2 pair-end reads from clinical M. tuberculosis isolates listed in Tables 5 and 6 using MiSeq with Nextera XT library kits (Illumina). The sequence data used have been registered with DNA Data Bank of JAPAN (DDBJ) as accession number DRA001219. First, Read mapping was performed using Bowtie 2 [42] with local mode and end-to-end modes and default parameters. The resultant mapping was analyzed the idxstats coomand of SAMtools [43]. We also analyzed the mapping efficacy using CLC genomics workbench, of which algorithm is based Smith and Waterman [46], after trimming based on base quality (quality score limit = 0.05, removing reads if there are more than 2 ambiguous nucleotides in the reads or less than 15 bp in length). In the analysis with CLC genomics workbench, the influence of three combinations of parameters on mapping was tested: mismatch cost, insertion cost, deletion cost, matching length and similarity. In both analyses, significance of differences in mapping frequencies were assessed using multiple comparisons of proportions tests [44]. We also compared ratio of the number of reads mapped to total read for the two reference sequences, and subtracted values of the mapping frequencies (% mapped reads with consensus sequence minus that of H37Rv) were calculated. The subtracted values were used to compare mapping frequency among the MTBC lineages with Mann–Whitney U tests.
Virtual VNTR typing, spoligotyping, LSPs typing and Beijing typing
Based on the amplification primers for 24-loci VNTR [6], spoligotyping [50], LSPs [7,8], and Beijing typing based on the insertion positions of IS6110 [55], sequence data corresponding to the respective loci or regions were selected. Using the Blast algorithm, these sequence data were used to analyze whether each region is present in the MTBC strains listed in Table 1 [59]. VNTR and spoligotyping results for the isolates or strains in the database were analyzed and compared on the MIRU-VNTR web site (http://www.miru-vntrplus.org/MIRU/index.faces) [45].
Acknowledgements
We thank Mrs. M. Komiya and Mrs. Y. Sakurai for their excellent technical assistance.
This work was supported by a Grant for International Health Research (26A103) to author T.M.A. from the Ministry of Health, Labor, and Welfare, Japan and a Grant for International Health Research (24A103) to author T. K. from the Ministry of Health, Labor, and Welfare, Japan.
Additional files
Consensus sequence of MTBC with SNP handled by majority rule.
Consensus sequence of MTBC with SNP handled by ambiguity rule.
CS with SNP handled by ambiguity rule and annotation based on H37Rv.
SNP distribution among MTBC in comparison with H37Rv. Description of data: SNPs found in each MTBC were summarized with their distribution among the strains and location of the SNPs in the genes.
Indels distribution among MTBC in comparison with H37Rv. Description of data: Indels more than 5 bp in the length found in each MTBC were summarized with their distribution among the strains and location of the SNPs in the genes.
Maximum-likelihood phylogenies based on whole genome and SNP concatenated sequence alignment. Description of data: Phylogenetic trees based on whole genome sequence (a), SNP concatemers using the consensus genome sequence as reference (b) and SNP concatemers using H37Rv genome as reference (c) were constructed using PhyML [37]. Multiple trees (100 trees for a and b, 40 trees for c) were generated by boot-strapping in PhyML analysis, and most probable trees were selected by a combination of 9 statistical methods implemented in CONSEL [40]. Isolates, clustered into different positions compared with the phylogenetic tree based on the whole genome sequences of M. tuberculosis. For the KZN series, inversion-corrected sequences were used for the alignment and marked “_m”.
Probability value (i.e., p-value) to assess the confidence in the selection problem of phylogenetic tree using CONSEL. Description of data: Using PhyML with 100 (for SNP concatemers) or 40 (whole genome (CS) bootstrapping, multiple trees generated were analyzed with CONSEL to select most probable tree. P-values of top 10 trees generated from PhyML are indicated and the most probable trees in the analyses were used to generate Additional file 6: Figure S1. Abbreviation of the statics analyzed in CONSEL; obs: the observed log-likelihood difference. au: the p-value of the approximately unbiased test calculated from the multiscale bootstrap. np: the bootstrap probability calculated from the multiscale bootstrap. bp: the bootstrap probability calculated in the usual manner. kh: the Kishino-Hasegawa test. sh: the Shimodaira-Hasegawa test. wkh: the weighted Kishino-Hasegawa test. wsh: the weighted Shimodaira-Hasegawa test.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KO, M. Kayano and TMA performed the bioinformatic analyses. M. Kato and TMA performed TB genome sequence analyses. TK and TMA supervised the project. TK coordinated the project. KO and TMA prepared the manuscript. All authors read and approved the final manuscript.
Contributor Information
Kayo Okumura, Email: okumura@obihiro.ac.jp.
Masako Kato, Email: mkato@ri.ncgm.go.jp.
Teruo Kirikae, Email: tkirikae@ri.ncgm.go.jp.
Mitsunori Kayano, Email: kayano@obihiro.ac.jp.
Tohru Miyoshi-Akiyama, Email: takiyam@ri.ncgm.go.jp.
References
- 1.Raviglione M, Marais B, Floyd K, Lonnroth K, Getahun H, Migliori GB, et al. Scaling up interventions to achieve global tuberculosis control: progress and new developments. Lancet. 2012;379:1902–13. doi: 10.1016/S0140-6736(12)60727-2. [DOI] [PubMed] [Google Scholar]
- 2.Lawn SD, Mwaba P, Bates M, Piatek A, Alexander H, Marais BJ, et al. Advances in tuberculosis diagnostics: the Xpert MTB/RIF assay and future prospects for a point-of-care test. Lancet Infect Dis. 2013;13:349–61. doi: 10.1016/S1473-3099(13)70008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abubakar I, Zignol M, Falzon D, Raviglione M, Ditiu L, Masham S, et al. Drug-resistant tuberculosis: time for visionary political leadership. Lancet Infect Dis. 2013;13:529–39. doi: 10.1016/S1473-3099(13)70030-6. [DOI] [PubMed] [Google Scholar]
- 4.Marais BJ, Lonnroth K, Lawn SD, Migliori GB, Mwaba P, Glaziou P, et al. Tuberculosis comorbidity with communicable and non-communicable diseases: integrating health services and control efforts. Lancet Infect Dis. 2013;13:436–48. doi: 10.1016/S1473-3099(13)70015-X. [DOI] [PubMed] [Google Scholar]
- 5.Thierry D, Brisson-Noel A, Vincent-Levy-Frebault V, Nguyen S, Guesdon JL, Gicquel B. Characterization of a Mycobacterium tuberculosis insertion sequence, IS6110, and its application in diagnosis. J Clin Microbiol. 1990;28:2668–73. doi: 10.1128/jcm.28.12.2668-2673.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazars E, Lesjean S, Banuls AL, Gilbert M, Vincent V, Gicquel B, et al. High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci U S A. 2001;98:1901–6. doi: 10.1073/pnas.98.4.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci U S A. 2004;101:4871–6. doi: 10.1073/pnas.0305627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:2869–73. doi: 10.1073/pnas.0511240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, et al. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23. doi: 10.1186/1471-2180-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Soolingen D, Qian L, de Haas PE, Douglas JT, Traore H, Portaels F, et al. Predominance of a single genotype of Mycobacterium tuberculosis in countries of east Asia. J Clin Microbiol. 1995;33:3234–8. doi: 10.1128/jcm.33.12.3234-3238.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murase Y, Mitarai S, Sugawara I, Kato S, Maeda S. Promising loci of variable numbers of tandem repeats for typing Beijing family Mycobacterium tuberculosis. J Med Microbiol. 2008;57:873–80. doi: 10.1099/jmm.0.47564-0. [DOI] [PubMed] [Google Scholar]
- 12.Surikova OV, Voitech DS, Kuzmicheva G, Tatkov SI, Mokrousov IV, Narvskaya OV, et al. Efficient differentiation of Mycobacterium tuberculosis strains of the W-Beijing family from Russia using highly polymorphic VNTR loci. Eur J Epidemiol. 2005;20:963–74. doi: 10.1007/s10654-005-3636-5. [DOI] [PubMed] [Google Scholar]
- 13.Le Fleche P, Fabre M, Denoeud F, Koeck JL, Vergnaud G. High resolution, on-line identification of strains from the Mycobacterium tuberculosis complex based on tandem repeat typing. BMC Microbiol. 2002;2:37. doi: 10.1186/1471-2180-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rusch-Gerdes S, Willery E, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2006;44:4498–510. doi: 10.1128/JCM.01392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwamoto T, Yoshida S, Suzuki K, Tomita M, Fujiyama R, Tanaka N, et al. Hypervariable loci that enhance the discriminatory ability of newly proposed 15-loci and 24-loci variable-number tandem repeat typing method on Mycobacterium tuberculosis strains predominated by the Beijing family. FEMS Microbiol Lett. 2007;270:67–74. doi: 10.1111/j.1574-6968.2007.00658.x. [DOI] [PubMed] [Google Scholar]
- 16.Baker S, Hanage WP, Holt KE. Navigating the future of bacterial molecular epidemiology. Curr Opin Microbiol. 2010;13:640–5. doi: 10.1016/j.mib.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bravo LT, Procop GW. Recent advances in diagnostic microbiology. Semin Hematol. 2009;46:248–58. doi: 10.1053/j.seminhematol.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niemann S, Koser CU, Gagneux S, Plinke C, Homolka S, Bignell H, et al. Genomic diversity among drug sensitive and multidrug resistant isolates of Mycobacterium tuberculosis with identical DNA fingerprints. PLoS One. 2009;4:e7407. doi: 10.1371/journal.pone.0007407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364:730–9. doi: 10.1056/NEJMoa1003176. [DOI] [PubMed] [Google Scholar]
- 20.Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45:1176–82. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roetzer A, Diel R, Kohl TA, Ruckert C, Nubel U, Blom J, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 2013;10:e1001387. doi: 10.1371/journal.pmed.1001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 23.Ford ME, Sarkis GJ, Belanger AE, Hendrix RW, Hatfull GF. Genome structure of mycobacteriophage D29: implications for phage evolution. J Mol Biol. 1998;279:143–64. doi: 10.1006/jmbi.1997.1610. [DOI] [PubMed] [Google Scholar]
- 24.Hatfull GF, Sarkis GJ. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol Microbiol. 1993;7:395–405. doi: 10.1111/j.1365-2958.1993.tb01131.x. [DOI] [PubMed] [Google Scholar]
- 25.Rossolini GM, Mantengoli E, Montagnani F, Pollini S. Epidemiology and clinical relevance of microbial resistance determinants versus anti-Gram-positive agents. Curr Opin Microbiol. 2010;13:582–8. doi: 10.1016/j.mib.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Dutilh BE, Huynen MA, Strous M. Increasing the coverage of a metapopulation consensus genome by iterative read mapping and assembly. Bioinformatics. 2009;25:2878–81. doi: 10.1093/bioinformatics/btp377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arenas M, Posada D. Computational design of centralized HIV-1 genes. Curr HIV Res. 2010;8:613–21. doi: 10.2174/157016210794088263. [DOI] [PubMed] [Google Scholar]
- 28.Mlera L, Jere KC, van Dijk AA, O’eill HG. Determination of the whole-genome consensus sequence of the prototype DS-1 rotavirus using sequence-independent genome amplification and 454(R) pyrosequencing. J Virol Methods. 2011;175:266–71. doi: 10.1016/j.jviromet.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Marston DA, McElhinney LM, Ellis RJ, Horton DL, Wise EL, Leech SL, et al. Next generation sequencing of viral RNA genomes. BMC Genomics. 2013;14:444. doi: 10.1186/1471-2164-14-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farrell CM, O’eary NA, Harte RA, Loveland JE, Wilming LG, Wallin C, et al. Current status and new features of the Consensus Coding Sequence database. Nucleic Acids Res. 2013;42(Dabase issue):D865–72. doi: 10.1093/nar/gkt1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479–90. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnold C, Thorne N, Underwood A, Baster K, Gharbia S. Evolution of short sequence repeats in Mycobacterium tuberculosis. FEMS Microbiol Lett. 2006;256:340–6. doi: 10.1111/j.1574-6968.2006.00142.x. [DOI] [PubMed] [Google Scholar]
- 35.Vishnoi A, Roy R, Bhattacharya A. Comparative analysis of bacterial genomes: identification of divergent regions in mycobacterial strains using an anchor-based approach. Nucleic Acids Res. 2007;35:3654–67. doi: 10.1093/nar/gkm209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. 2014;46:279–86. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–52. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 38.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 40.Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–7. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- 41.Robinson DF, Foulds LR. Comparison of phylogenetic trees. Math Biosci. 1981;53:131–47. doi: 10.1016/0025-5564(81)90043-2. [DOI] [Google Scholar]
- 42.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.RYAN TA. Significance tests for multiple comparison of proportions, variances, and other statistics. Psychol Bull. 1960;57:318–28. doi: 10.1037/h0044320. [DOI] [PubMed] [Google Scholar]
- 45.Weniger T, Krawczyk J, Supply P, Niemann S, Harmsen D. MIRU-VNTRplus: a web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res. 2010;38:W326–31. doi: 10.1093/nar/gkq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–7. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 47.Ioerger TR, Feng Y, Ganesula K, Chen X, Dobos KM, Fortune S, et al. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J Bacteriol. 2010;192:3645–53. doi: 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koser CU, Niemann S, Summers DK, Archer JA. Overview of errors in the reference sequence and annotation of Mycobacterium tuberculosis H37Rv, and variation amongst its isolates. Infect Genet Evol. 2012;12:807–10. doi: 10.1016/j.meegid.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 49.Ilina EN, Shitikov EA, Ikryannikova LN, Alekseev DG, Kamashev DE, Malakhova MV, et al. Comparative genomic analysis of Mycobacterium tuberculosis drug resistant strains from Russia. PLoS One. 2013;8:e56577. doi: 10.1371/journal.pone.0056577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45:784–90. doi: 10.1038/ng.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian L, Van Embden JD, Van Der Zanden AG, Weltevreden EF, Duanmu H, Douglas JT. Retrospective analysis of the Beijing family of Mycobacterium tuberculosis in preserved lung tissues. J Clin Microbiol. 1999;37:471–4. doi: 10.1128/jcm.37.2.471-474.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakanishi N, Wada T, Arikawa K, Millet J, Rastogi N, Iwamoto T. Evolutionary robust SNPs reveal the misclassification of Mycobacterium tuberculosis Beijing family strains into sublineages. Infect Genet Evol. 2013;16:174–7. doi: 10.1016/j.meegid.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 54.Filliol I, Motiwala AS, Cavatore M, Qi W, Hazbon MH, Bobadilla del Valle M, et al. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol. 2006;188:759–72. doi: 10.1128/JB.188.2.759-772.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mokrousov I, Ly HM, Otten T, Lan NN, Vyshnevskyi B, Hoffner S, et al. Origin and primary dispersal of the Mycobacterium tuberculosis Beijing genotype: clues from human phylogeography. Genome Res. 2005;15:1357–64. doi: 10.1101/gr.3840605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Chen C, Liu J, Deng H, Pan A, Zhang L, et al. Complete genome sequences of Mycobacterium tuberculosis strains CCDC5079 and CCDC5080, which belong to the Beijing family. J Bacteriol. 2011;193:5591–2. doi: 10.1128/JB.05452-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–98. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 58.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Biossssinf. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]