

Abstract
Objective
Increased peripheral metabolism of cortisol may explain compensatory ACTH-dependent adrenal steroidogenesis and hence hyperandrogenism in polycystic ovary syndrome (PCOS). Previous studies have described an increased 5α-reduction of cortisol or impaired regeneration of cortisol by 11β-HSD1 in PCOS. However, these observations may be confounded by obesity. Moreover, the relationship between alterations in cortisol metabolism and the extent of adrenal androgen hyper-secretion in response to ACTH has not been established. This study aimed to examine the association between cortisol metabolism and ACTH-dependent adrenal hyperandrogenism in PCOS, independently of obesity.
Design
We compared 90 PCOS women (age 18-45 yr) stratified by adrenal androgen responses to ACTH1-24 and 45 controls matched for age and body weight.
Methods
PCOS women were stratified as normal responders-NR, intermediate responders-IR, and high responders-HR to 250 μg ACTH1-24: NR (n= 27) had androstenedione and DHEA responses within 2 SD of the mean in controls; IR (n= 43) had DHEA responses >2 SD above controls; HR (n= 20) had both androstenedione and DHEA responses >2 SD above controls.
Results
All groups were similar for age, body weight, and body fat distribution. Basal testosterone, androstenedione, and 5α-dihydrotestosterone plasma levels were similarly elevated among the three groups of PCOS compared with controls, whereas basal DHEA-S was higher in HR (2.8±1.2 μg/mL) and IR (2.4±1.1 μg/mL) than in NR (1.8±0.8 μg/mL) and controls (1.7±0.6 μg/mL). The HR group had the lowest basal plasma cortisol levels (101±36 ng/mL versus IR 135±42 ng/mL, NR 144±48 ng/mL, and controls 165±48 ng/mL; all P< 0.01), but the greatest cortisol response to ACTH1-24 (Δ(60-0)cortisol 173±60 ng/mL versus IR 136±51 ng/mL, NR 114±50 ng/mL, and controls 127±50 ng/mL; all P< 0.01), and the highest urinary excretion of total and 5β-reduced cortisol metabolites (eg 5β-tetrahydrocortisol/cortisol ratio 25.2±15.3 versus IR 18.8±10.7, NR 19.7±11.4, and controls 17.2±13.7; all P< 0.05). There were no differences in urinary excretion of 5α-reduced cortisol metabolites or in 5α-dihydrotestosterone/testosterone ratio between groups.
Conclusions
Adrenal androgen excess in PCOS is associated with increased inactivation of cortisol by 5β-reductase that may lower cortisol blood levels and stimulate ACTH-dependent steroidogenesis.
Keywords: 5α-reductase, 5β-reductase, adrenal hyperandrogenism, polycystic ovary syndrome, obesity
Introduction
The polycystic ovary syndrome (PCOS) affects between 5-7% of women. Although the ovary is the principal source of androgen excess in most of these patients, between 40-70% also demonstrate elevated circulating levels of adrenal androgens, particularly DHEA-S (1-3). The mechanism of adrenal androgen excess in these patients remains poorly understood. One hypothesis is that adrenal hyperandrogenism in PCOS arises from an exaggerated secretory response of the adrenal cortex to ACTH (4). This theory is supported by the close correlation between the responsiveness of adrenal androgens (i.e. androstenedione and dehydroepiandrosterone; DHEA) to ACTH and circulating DHEA-S levels (4). The mechanism leading to the increased responsiveness to ACTH for androstenedione and DHEA in PCOS remains to be determined. It has been postulated that this condition may be secondary to a compensatory overactivation of the hypothalamic-pituitary-adrenal (HPA) axis in response to increased metabolic clearance of cortisol. Similarly to what has been suggested in simple obesity (5), in PCOS increased cortisol metabolism might increase CRF and ACTH secretion by decreasing the HPA axis negative feedback signal, thus maintaining normal cortisol levels at the expense of increased ACTH-dependent steroidogenesis, and therefore of adrenal hyperandrogenism.
Most metabolism of cortisol in humans is by enzymes in the liver (A-ring reductases) and kidney (11β-hydroxysteroid dehydrogenase type 2; 11β-HSD2). The metabolic clearance rate of cortisol is also influenced by the extent of regeneration of cortisol from inactive cortisone, by 11β-HSD1, mainly in the liver (6).
A-ring reductases catalyse 5α- or 5β-reduction of several steroids, converting cortisol to inactive metabolites and testosterone to 5α- or 5β-dihydrotestosterone (DHT). 5β-DHT is inactive but its 5α-isomer (5α-DHT) is the most potent natural androgen. In humans, two isoenzymes of 5α-reductase are encoded by separate genes. 5α-Reductase type 1 enzyme is found in non-genital skin, liver, brain, muscle and to a lesser extent in stromal cells of adipose tissue. 5α-Reductase type 2 is predominantly expressed in male accessory reproductive structures, such as the prostate, epididymis and seminal vesicles, and genital skin (7). 5β-Reductase is expressed mainly in liver and at lower levels in testis and colon (8).
In cohorts of patients with PCOS, lower ratios of cortisol/cortisone metabolites (9) or higher 5α-reduced cortisol metabolites (10-12) in urine have been reported, suggesting reduced 11β-HSD1 activity (9), or increased 5α-reductase activity (10-12), respectively. However, these results may be confounded by coexistent obesity, since obesity in men and women is associated with impaired liver 11β-HSD1 activity (13, 14) and increased urinary excretion of A-ring reduced cortisol metabolites, particularly 5α-reduced metabolites (15). Moreover, the relationship between alterations in cortisol metabolism and the extent of adrenal androgen hyper-secretion or increased responsiveness to ACTH has not been established.
The aim of the present study was to characterize whether exaggerated adrenal androgen responses to exogenous ACTH in PCOS are related to altered peripheral cortisol metabolism, independently of obesity. To achieve this we stratified PCOS patients according to androstenedione and DHEA response to ACTH.
Materials and Methods
Subjects
We investigated 90 unmedicated women with PCOS, aged 18-45 yr, and 45 controls recruited from the general population and comparable for age and weight. PCOS women had at least two of: (i) chronic oligoanovulation (luteal serum progesterone below 2 ng/mL (16)); (ii) hirsutism (Ferriman-Gallwey score ≥ 8 (17) or elevated serum total and free testosterone levels (18)); (iii) polycystic ovarian morphology at ultrasound, according to the Rotterdam consensus conference criteria (19). Hyperprolactinemia, Cushing’s syndrome, congenital adrenal hyperplasia and androgen secreting tumors were excluded by laboratory analysis (20). Controls had normal ovaries by ultrasound, no signs of hyperandrogenism and regular ovulatory mentrual cycles (progesterone levels ≥ 8 ng/mL during the luteal phase (16)). Women were classified as normal-weight if body mass index values (BMI) was > 18 kg/m2 and ≤ 25 kg/m2, overweight if BMI was > 25 kg/m2 and ≤ 30 kg/m2 and obese if BMI was > 30 kg/m2 (21). Twenty-eight (31%) PCOS were normal-weight, 10 (11%) were overweight and 52 (58%) were obese. Within controls, 15 (33%) were normal-weight, 5 (11%) were overweight and 25 (56%) were obese. None of the subjects included in the study had thyroid dysfunction, abnormal prolactin levels, cardiovascular, renal or liver diseases on clinical examination and routine laboratory findings. The protocol was approved by the local ethics committee and written informed consent was obtained.
Assessment protocol
Anthropometric data (height, weight, and waist circumference), systolic and diastolic blood pressure, and fat mass and fat-free mass by bioimpedence (Akern-BIA, Pontassieve, Florence, Italy) were obtained from each subject, as previously described (22). The number of menses in the previous 6 months was recorded. Studies were performed between d 5 and 10 of the menstrual cycle, or randomly during severe oligomenorrhea or amenorrhea, after excluding pregnancy by appropriate testing. All subjects completed a 24-h urine collection and attended at 0800-0830h after overnight fast. Basal blood samples for hormonal (total testosterone, androstenedione, 17OH-progesterone, DHEA-S, 5α-DHT, sex hormone binding globulin-SHBG, cortisol) and metabolic (glucose, insulin, total cholesterol, HDL-cholesterol, triglycerides, ALT, AST) determinations were collected before a 75g oral glucose tolerance test (OGTT) with blood samples taken after 30, 60, 90, 120 and 180 min for glucose determination and after 60, 120 and 180 min for insulin determinations. On a second day, an ACTH1-24 stimulation test (Synacthen, 250 μg i.v. at 0800-0830h) was performed, and samples for cortisol, DHEA, androstenedione, and 17OH-progesterone determinations were obtained at baseline and 60 minutes after stimulation. Samples were immediately chilled on ice and centrifuged; 24-h urine and serum were stored at −20 C and plasma at −80 C.
Biochemical assays in plasma
The assays employed for biochemical measurements have been reported elsewhere (23, 24) except 5α-DHT which was measured by an HPLC-RIA method (25).
The free androgen index (FAI) was calculated as the ratio between total testosterone and SHBG (26). Insulin resistance was estimated using the Quantitative Insulin-Sensitivity Check Index (QUICKI) and the Insulin Sensitivity Index during the OGTT (ISI) (27, 28).
Urine excretion of cortisol and its metabolites
5β-Tetrahydrocortisol (THF), 5α-THF, 5β-tetrahydrocortisone (THE), cortols, cortolones, and cortisone were measured in 24-h urine by electron impact gas chromatography-mass spectrometry (GC-MS) with minor modifications from a previously described method (29). Briefly, a urine aliquot was equilibrated with Internal Standards (11-epiTHF, 11α-hydrocortisone). The steroids were purified by Sep-Pak C18 extraction, digested with β-glucoronidase, reextracted and converted to HDMS-TSIM derivatives. The samples were injected into a GC-MS (Agilent: GC 6890 – MS 5973) in Selected Ion Mode. Cortisol and its metabolites were quantified by the ratio of metabolite: Internal Standard area against standard curves for each steroid, included in every assay batch. Accuracy and precision were tested in every batch by measuring cortisol in a reference plasma sample (DG KG, Referenzistitut fur Bioanalitic Geschaftsstelle, Im Muhlenbachb 52a D-53127 Bonn). Total cortisol excretion was calculated from the sum of 5β-THF, 5α-THF, 5β-THE, cortols and cortolones (30). Relative 5α- and 5β-reduction of cortisol was assessed by Ulick’s A-ring reduction quotients, 5α-THF/cortisol, 5β-THF/cortisol, and 5β-THE/cortisone (31). The balance of 5β- and 5α-reductases was also assessed by the ratio 5β-THF/5α-THF (15). The balance between 11β-HSD1 and 11β-HSD2 activities in all tissues was assessed as the ratio of (5α-THF+5β-THF)/5β-THE (29, 32). Renal 11β-HSD2 activity was assessed as urinary cortisol/cortisone ratio (29).
Statistical analysis
Data are shown as means ± standard deviation (SD). Glucose and insulin response to the OGTT was expressed as area under the curve (AUC), which was calculated by the trapezoidal method. Hormone response to ACTH1-24 stimulation test was calculated by the difference between 60min and basal concentrations (Δ(60-0)). Arbitrary cut-off values used to stratify responses of androstenedione and DHEA to ACTH1-24 in PCOS women was calculated from the mean+2 SD of Δ(60-0) of androstenedione and DHEA in controls. The data were compared among the groups by analysis of variance (ANOVA). Simple correlation analyses were used to relate adrenal androgens, cortisol metabolism and insulin sensitivity within PCOS. The impact of obesity (as BMI and % fat mass) on these correlations was tested by multiple correlation analyses.
Statistical analyses were performed on SPSS/PC+ version 8 (Chicago, IL, USA). Two-tailed P values < 0.05 were considered statistically significant.
Results
Stratification of PCOS women according to the response of adrenal androgens to ACTH
PCOS women were classified into three groups (normal responders-NR, intermediate responders-IR, high responders-HR) according to the response of androstenedione and DHEA to ACTH1-24 compared with the group of controls. NR (n= 27) had androstenedione and DHEA responses to ACTH1-24 within 2 SD of the mean in controls; IR (n= 43) had DHEA response >2 SD of controls but androstenedione response within 2 SD of controls; HR (n= 20) had both androstenedione and DHEA responses >2 SD of controls (Figure 1A, 1B). None of the PCOS women evaluated had an isolated androstenedione response >2 SD of controls.
Figure 1.
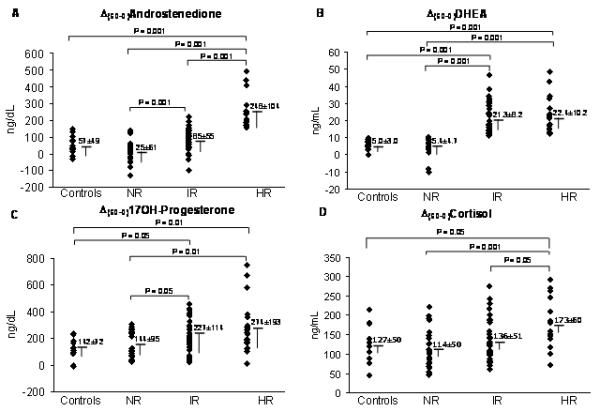
Response of androgens and cortisol to ACTH1-24 (Δ(60-0)) in PCOS normal responders-NR, intermediate responders-IR and high responders-HR, and in controls shown as individual values (scatter plot) and means ± SD.
P< 0.001 for comparison in androstenedione and DHEA, P= 0.025 for comparison in 17OH-Progesterone, and P= 0.001 for comparison in cortisol among the three groups of PCOS (NR, IR, HR) by one-way ANOVA.
To convert to SI units, multiply DHEA by 3.467 (result in nmol/L) and cortisol by 27.59 (result in nmol/L).
The response of 17OH-progesterone to ACTH1-24 was also higher in IR and HR compared with NR and controls (Figure 1C).
Anthropometric parameters (including prevalence of normal-weight, overweight and obesity) and systolic and diastolic blood pressure did not differ among NR, IR, HR and controls (Table 1). Hirsutism score was similarly higher, and number of menses in the previous 6 months similarly lower, in all the PCOS groups compared with controls (Table 1). There were no differences in plasma glucose or triglycerides, but the HR group had the highest total cholesterol and insulinAUC during the OGTT, and the lowest insulin sensitivity scores (QUICKI and ISI) and HDL-cholesterol (Table 1).
Table 1.
Clinical, hormonal and metabolic characteristics in women with PCOS, stratified according to the response of adrenal androgens to ACTH in normal responders-NR, intermediate responders-IR and high responders-HR, and in healthy controls
| Variables | PCOS | ||||
|---|---|---|---|---|---|
| NR (n=27) | IR (n=43) | HR (n=20) | P valuea | Controls (n=45) | |
| Age (yrs) | 26.3±6.0 | 23.8±4.6 | 26.8±6.5 | 0.09 | 23.7±10.3 |
| Body weight (kg) | 84.7±25.4 | 78.1±18.1 | 87.2±20.8 | 0.231 | 79.5±22.6 |
| Body mass index (kg/m2) | 31.7±8.7 | 30.0±7.0 | 32.6±7.6 | 0.447 | 30.0±8.3 |
| ≤25 kg/m2 | 8 (31%) | 14 (33%) | 6 (30%) | 0.436b | |
| >25-≤30 kg/m2 | 2 (7%) | 5 (11%) | 3 (15%) | ||
| >30 kg/m2 | 17 (62%) | 24 (56%) | 11 (55%) | ||
| Waist circumference (cm) | 93.1±18.3 | 88.7±14.9 | 96.4±18.3 | 0.239 | 91.4±20.4 |
| Fatty mass (%) | 63.6±10.0 | 67.8±11.6 | 60.4±10.3 | 0.323 | 60.2±9.9 |
| Free fatty mass (%) | 39.9±11.1 | 32.2±11.6 | 36.8±10.3 | 0.308 | 39.8±9.9 |
| Systolic bood pressure (mmHg) | 123±17 | 128±11 | 123±14 | 0.216 | 128±14 |
| Diastolic blood pressure (mmHg) | 77±13 | 78±10 | 79±9 | 0.799 | 79±7 |
| Hirsutism (Ferriman-Gallwey score) | 10.0±6.9*** | 12.1±6.9*** | 11.4±7.7*** | 0.576 | 2.2±1.0 |
| Menses (n° of in the previous 6 months) | 3.7±2.3*** | 3.8±2.3*** | 3.5±2.1*** | 0.876 | 6.0±0.2 |
| Fasting 0800-0830h plasma: | |||||
| Total testosterone (ng/mL) | 0.66±0.26*** | 0.67±0.18*** | 0.70±0.26*** | 0.818 | 0.48±0.16 |
| FAI | 2.44±1.09*** | 2.99±1.56*** | 2.95±1.50*** | 0.282 | 1.51±0.87 |
| Androstenedione (ng/dL) | 382±163*** | 373±138*** | 401±119*** | 0.799 | 201±57 |
| 17OH-progesterone (ng/dL) | 157±77 | 156±107 | 158±138 | 0.480 | 127±85 |
| DHT (ng/mL) | 0.22±0.13*** | 0.21±0.09*** | 0.22±0.14*** | 0.983 | 0.16±0.07 |
| SHBG (nmol/L) | 31.4±15.6* | 28.6±17.9* | 27.9±13.3* | 0.706 | 39.9±18.4 |
| 5α-DHT/Total testosterone | 0.39±0.47 | 0.34±0.16 | 0.36±0.26 | 0.775 | 0.34±0.14 |
| Oral glucose tolerance test: | |||||
| GlucoseAUC (mg/dL.min−1) | 19,865±4,545 | 19,953±3,663 | 19,562±3,109 | 0.944 | 21,913±3,817 |
| InsulinAUC (μIU/mL.min−1) | 12,348±8,328 | 12,126±11,650 | 16,875±12,606* | 0.354 | 9,318±5,988 |
| QUICKI | 0.341±0.050 | 0.350±0.052 | 0.332±0.030* | 0.320 | 0.361±0.042 |
| ISI | 5.90±5.37 | 7.11±6.30 | 5.34±4.39* | 0.477 | 8.47±5.69 |
| Total-cholesterol (mg/dL) | 179.4±30.0 | 174.2±35.9 | 191.0±43.0* | 0.525 | 169.0±19.9 |
| HDL-cholesterol (mg/dL) | 55.3±16.6 | 53.7±14.3 | 46.4±13.6 #, §, * | 0.061 | 56.4±15.2 |
| Triglycerides (mg/dL) | 102.8±50.3 | 86.6±46.8 | 88.7±36.7 | 0.449 | 97.4±51.5 |
| ALT (U/L) | 20.7±3.8 | 22.1±7.8 | 24.0±8.4 | 0.292 | 19.1±5.2 |
| AST (U/L) | 22.4±8.2 | 25.6±17.1 | 30.8±23.3 | 0.243 | 21.2±10.5 |
Data are means ± SD.
Comparison among the three groups of PCOS (NR, IR, HR) by one-way ANOVA;
comparison by two-tailed Fisher’s exact test.
P< 0.05,
P< 0.001 for post-hoc comparison between NR, IR or HR and controls.
P< 0.05 for post-hoc comparison between HR and NR.
P< 0.05 for post-hoc comparison between HR and IR.
FAI, free androgen index; DHT, dihydrotestosterone; SHBG, sex hormone binding globulin; AUC, area under the curve of the oral glucose tolerance test, calculated by the trapezoidal rule.
To convert to SI units, multiply total testosterone by 0.0347 (result in nmol/L), androstenedione by 0.0349 (result in nmol/L), 17-hydroxyprogesterone by 0.0303 (result in nmol/L), DHT by 3.467 (result in nmol/L), glucose by 0.0555 (result in mmol/L), insulin by 6.945 (result in pmol/L), Total- and HDL-cholesterol by 0.0259 (result in mmol/L) and triglycerides by 0.0113 (result in mmol/L)
Basal plasma levels of testosterone (total and free), androstenedione and 5α-DHT were similarly higher, and SHBG similarly lower in all three groups of PCOS compared with controls (Table 1). However, basal 17OH-progesterone and 5α-DHT/total testosterone ratio did not differ between any groups (Table 1).
Basal DHEA-S was higher in IR and HR groups compared with NR and controls (Figure 2A).
Figure 2.
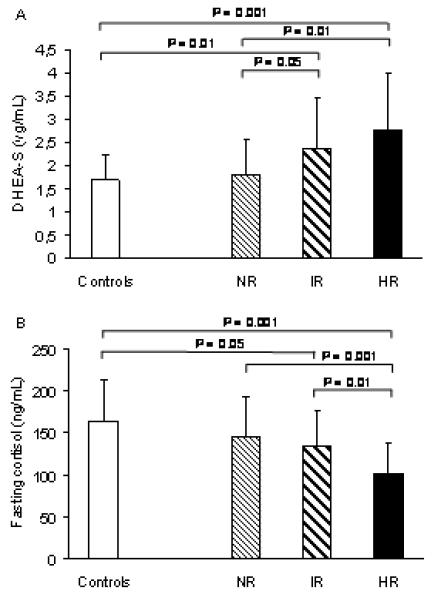
Fasting plasma DHEA-S and cortisol levels in PCOS normal responders-NR, intermediate responders-IR and high responders-HR, and in controls. Data are means ± SD.
P= 0.004 for comparison in DHEA-S and P= 0.003 for comparison in cortisol among the three groups of PCOS (NR, IR, HR) by one-way ANOVA.
To convert to SI units, multiply DHEA-S by 2.741 (result in μmol/L).
Cortisol and its metabolism
The HR group had the lowest fasting plasma cortisol levels (Figure 2B), but the greatest response of cortisol to ACTH1-24 (Figure 1D). Both the HR and IR groups had higher total cortisol metabolite excretion (5β-THF + 5α-THF + 5β-THE + cortols + cortolones) than controls (Table 2), which was attributable to increased excretion of 5β-THF and 5β-THE. Both the HR and IR groups had higher absolute excretion rates of 5β-THF and 5β-THE levels than controls, although only the HR group had 5β-THF and 5β-THE excretion higher than the NR group. In contrast, 5α-THF excretion did not differ between any groups (Table 2).
Table 2.
24-h Urine cortisol metabolites in women with PCOS, stratified according to the response of adrenal androgens to ACTH in normal responders-NR, intermediate responders-IR and high responders-HR, and in healthy controls
| Variables | PCOS | ||||
|---|---|---|---|---|---|
| NR (n=27) | IR (n=43) | HR (n=20) | P valuea | Controls (n=45) | |
| 5β-THF (μg/d) | 1592±904 | 1830±1026* | 2013±803 #, * | 0.046 | 1593±637 |
| 5α-THF (μg/d) | 2536±1921 | 2971±2543 | 2653±2340 | 0.732 | 2344±1407 |
| 5β-THE (μg/d) | 3292±1631 | 4136±3012* | 4224±1703 #, * | 0.050 | 3197±1783 |
| Total (μg/d) b | 11329±5866 | 13459±8250* | 13236±5708* | 0.453 | 10418±6155 |
| Cortisol/cortisone | 0.57±0.22 | 0.65±0.29 | 0.53±0.21 | 0.138 | 0.57±0.26 |
| (5α-THF + 5β-THF)/ 5β-THE | 1.24±0.46** | 1.35±0.75* | 1.06±0.36*** | 0.233 | 1.61±0.53 |
Data are means ± SD.
Comparison among the three groups of PCOS (NR, IR, HR) by one-way ANOVA.
Total cortisol metabolites = 5β-THF + 5α-THF + 5β-THE + cortols + cortolones.
Data are means ± SD.
P< 0.05,
P< 0.01,
P< 0.001 for post-hoc comparison between NR, IR, or HR and controls.
P< 0.05 for post-hoc comparison between HR and NR.
The A-ring reduction quotients reflecting 5β-reductase activity (5β-THF/cortisol and 5β-THE/cortisone) were higher in HR compared with IR, NR and controls, but they did not differ between the IR and NR groups (Figure 3A, 3C). In contrast, the quotient reflecting 5α-reductase activity (5α-THF/cortisol) did not differ between groups (Figure 3B). Similarly, the 5β-THF/5α-THF ratio, that reflects the balance of 5β- and 5α-reductases, was higher in HR than in IR, NR and controls, but it was similar between the IR and NR groups (Figure 3D). The ratio of cortisol/cortisone, reflecting renal 11β-HSD2 activity did not differ between groups (Table 2) but the (5α-THF+5β-THF)/5β-THE ratio was similarly lower in all PCOS groups compared with controls, consistent with a lower 11β-HSD1 activity in PCOS (Table 2).
Figure 3.
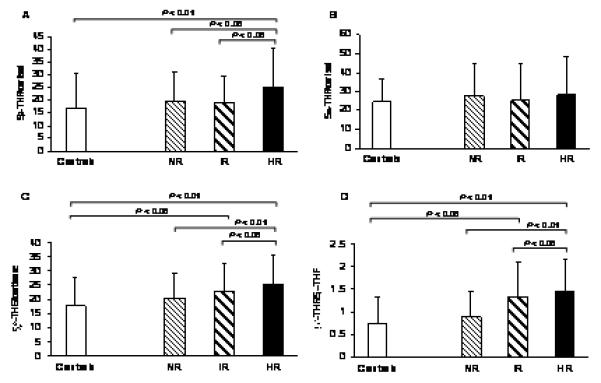
Relative 5β- (5βTHF/cortisol, 5βTHE/cortisone) and 5α- (5αTHF/cortisol) reduction of cortisol and the balance of 5β- and 5α-reductases (5βTHF/5αTHF) in PCOS normal responders-NR, intermediate responders-IR and high responders-HR, and in controls. Data are means ± SD.
P= 0.049, P= 0.053, P= 0.825, and P= 0.044 for comparison in 5βTHF/cortisol, 5βTHE/cortisone, 5αTHF/cortisol, and 5βTHF/5αTHF levels, respectively, among the three groups of PCOS (NR, IR, HR) by one-way ANOVA.
Correlations between the response of adrenal androgens to ACTH, cortisol metabolism, DHEA-S and insulin sensitivity
Given the variability of hyperandrogenism even within the IR and HR groups (Figure 1), and the inevitably arbitrary cut-offs used to stratify these groups, simple correlation analyses were undertaken in all the PCOS women considered together. The response of androstenedione and DHEA to ACTH1-24 were negatively (P< 0.001) correlated with fasting cortisol levels (Figure 4A, B) but positively correlated with cortisol response to ACTH1-24 (r= 0.33, P= 0.002 and r= 0.22, P= 0.042, respectively). The androgen response to ACTH1-24 was also positively correlated with DHEA-S fasting levels (r= 0.22, P= 0.037 for androstenedione; r= 0.27, P= 0.016 for DHEA), whereas it was not correlated with insulinAUC during the OGTT (r= 0.19, P= 0.113 for androstenedione; r= 0.02, P= 0.857 for DHEA), nor with QUICKI (r= −0.11, P= 0.312 for androstenedione; r= 0.03, P= 0.773 for DHEA) or ISI (r= −0.06, P= 0.604 for androstenedione; r= −0.03, P= 0.772 for DHEA). Both androstenedione and DHEA responses to ACTH1-24 were positively correlated with 5βTHE/cortisone (r= 0.30, P= 0.003 and r= 0.24, P= 0.023, respectively), whereas only the response of DHEA was significantly positively correlated with 5βTHF/5αTHF (r= 0.24, P= 0.023). The androgen response to ACTH1-24 was not correlated with 5βTHF/cortisol (r= 0.16, P= 0.138 for androstenedione; r= 0.20, P= 0.063 for DHEA), nor with 5αTHF/cortisol (r= 0.03, P= 0.819 for androstenedione; r= 0.05, P= 0.676 for DHEA), or with the (5α-THF+5β-THF)/5β-THE ratio (r= −0.16, P= 0.142 for androstenedione; r= −0.11, P= 0.327 for DHEA).
Figure 4.
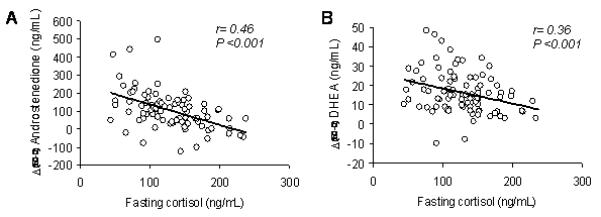
Relationship between androstenedione or DHEA response to ACTH1-24 and fasting plasma cortisol levels.
Both 5βTHE/cortisone and 5βTHF/cortisol were negatively correlated with fasting cortisol levels (r= −0.28, P= 0.035 and r= −0.29, P= 0.031, respectively), but were not correlated with DHEA-S (r= −0.01, P= 0.905 and r= −0.04, P= 0.745, respectively). 5βTHE/cortisone, but not 5βTHF/cortisol, was significantly positively correlated with insulinAUC during the OGTT (r= 0.32, P= 0.007), and negatively with QUICKI (r= 0.31, P= 0.004) and ISI (r= 0.26, P= 0.015). 5βTHF/5αTHF ratio also correlated positively with insulinAUC (r= 0.26, P= 0.032).
These relationships were maintained when BMI or % fat mass were included in multiple regression analyses (data not shown).
Discussion
In this study we used the adrenal androgen (androstenedione and DHEA) responses to exogenous ACTH to stratify PCOS patients in order to examine predictors of adrenal androgen excess. In the ‘high responder’ group, where both androstenedione and DHEA responses were >2 SD of controls, we found abnormalities of adrenal steroid secretion and metabolism which were distinct both from healthy controls and from PCOS patients with normal responses to ACTH. Similar, but less striking, differences were observed in a group of ‘intermediate responders’, where DHEA response was >2 SD of controls, whereas androstenedione response was within 2 SD of controls. Specifically, the ‘high responders’ had evidence of chronic activation of the HPA axis, with an exaggerated cortisol and 17OH-progesterone response to ACTH. However, morning plasma cortisol values were decreased in this group. In addition, this group was characterized by an elevated 24h urinary cortisol metabolite excretion attributable to a selective increase in urinary excretion of 5β-reduced cortisol metabolites, which suggests enhanced 5β-reductase activity. Based on these findings we infer that the activation of the HPA axis in ‘high responder’ group may represent a compensatory response to enhanced peripheral metabolism of cortisol by 5β-reductase, particularly in the liver, thereby explaining higher adrenal androgen and cortisol responses to ACTH as well as lower than normal fasting cortisol concentrations.
We also found that ‘high responder’ as well as ‘intermediate responder’ PCOS groups had higher fasting DHEA-S levels than ‘normal responder’ and controls. Since increased levels of DHEA-S are commonly referred as an index of adrenal androgen overproduction in PCOS (33), we can infer that an exaggerated secretory response of adrenal androgens to ACTH may be at the basis of adrenal hyperandrogenism in PCOS. This theory, in agreement with previous reports (4), is supported by the close positive correlation between the responsiveness of androstenedione and DHEA to ACTH and circulating DHEA-S levels, observed within the PCOS cohort. However, we can also infer that only when there is an overall hyper-responsiveness of the adrenal cortex to ACTH (i.e. increased response of androstenedione, DHEA, 17OH-progesterone, and cortisol), as observed in the ‘high responders’, are circulating levels of DHEA-S increased; this is consistent with the adrenal hyperandrogenism, being sustained by an increased inactivation of cortisol by 5β-reductase. Low fasting cortisol levels may be considered an additional marker reflecting increased cortisol metabolism.
Surprisingly, at variance of previous studies, we did not find evidence of increased excretion of 5α-reduced cortisol metabolites in PCOS when compared with controls (10-12, 34). The finding of normal 5α-reductase activity in PCOS is further emphasized by the lack of any difference in 5α-DHT/total testosterone ratio in blood between PCOS and controls. The discrepancy with previous reports may be partially explained by the relatively low number of subjects enrolled in the other studies (10-12, 34), since there is considerable interindividual variation in urinary steroid excretion rates. Indeed, no significant difference in 5α-reductase activity between PCOS and controls was found in another study in which a larger population was screened (9). Another potential confounding factor, not fully explored in the other studies, is the impact of obesity, since we suspect that enhanced peripheral clearance of cortisol by 5α-reductase only occurs in PCOS as a manifestation of co-existent obesity (13). In our study the prevalence of normal-weight, overweight and obesity was similar between PCOS and controls, so that any confounding effect of body weight on urinary metabolites reflecting 5α-reductase activity was avoided. Additionally, no relationship of the secretory response of adrenal androgens to ACTH with 5α-reduced cortisol metabolite excretion was observed within the PCOS group. This result, together with the finding of similar values of both 5α-reduced cortisol metabolites and 5α-DHT/total testosterone ratio among the three groups of PCOS, suggests that 5α-reductase activity is not a determinant of the responsiveness of adrenal androgens to ACTH.
The urine (5α-THF+5β-THF)/5β-THE ratio was decreased in all the PCOS groups compared with controls, which is consistent with some (9), but not all (10, 11), previous reports. Since the urinary free cortisol/cortisone ratio (an index of renal 11β-HSD2 activity) was similar to controls, we can attribute the lower THFs/THE ratio of PCOS to an impaired 11β-HSD1 activity, which appears to occur in PCOS irrespective of associated obesity. However, the lack of a significant relationships between adrenal hyperandrogenism and the urine (5α-THF+5β-THF)/5β-THE ratio suggests that it is unlikely that variations in 11β-HSD1 are an important determinant of ACTH-dependent adrenal hyperandrogenism in PCOS.
However, these data are probably not conclusive, since there is evidence that the urinary THFs/THE ratio is an inadequate indicator of 11β-HSD1 activity in the presence of altered A-ring reductase activities. For example, one study in men showed that high liver fat content was associated with increased 5β-reductase activity and with reduced urinary THFs/THE ratios, despite there being no change in conversion of cortisone to cortisol on first pass metabolism by 11β-HSD1 (35). More specific measurement of regeneration of cortisol from cortisone will be required to understand the role of 11β-HSD1, and its functional variability, in PCOS.
The mechanisms of up-regulation of 5β-reductase in the ‘high responder’ PCOS group is unclear, since published information about the regulation of 5β-reductase is rather sketchy. It may be that hyperinsulinaemia is involved since insulin sensitising agents ameliorate the increase in 5β-reductase mRNA and activity which occurs in the liver of obese rats (36). Indeed, we found a positive correlation between 5β-reductase activity and insulin levels in our cohort of women with PCOS, independently of body composition. Previous studies in rats described programmed regulation of 5β-reductase activity by androgens, since testosterone treatment of female rats at birth increased the activity of 5β-reductase after ovariectomy in adulthood (37). However, in our studies the elevation of testosterone was similar in the three PCOS groups. Finally, a selective increase in urinary excretion of 5β-reduced cortisol and cortisone metabolites has been demonstrated previously in otherwise healthy men with high liver fat content (35). Unfortunately, we did not assess fat accumulation in the liver in the present study, but in PCOS women we did not find any correlation between serum transaminases (ALT and AST) and 5β-reductase indices.
The consequences of increased 5β-reductase activity may include not only increased peripheral metabolism of cortisol and compensatory activation of the HPA axis, but also metabolic disturbances, because 5β-reductase is involved in cholesterol and bile acid metabolism (6). Accordingly, the ‘high responder’ PCOS women appear to have a more severe dysmetabolic pattern, with lower plasma HDL-cholesterol levels and hyperinsulinaemia. It has been proposed that insulin may be responsible for both adrenal and ovarian hyperandrogenism in PCOS (38). During a euglycemic hyperinsulinemic clamp ACTH-stimulated steroidogenesis was potentiated in hyperandrogenic women (39), with a disproportionate rise in 17OH-pregnenolone, 17OH-progesterone and DHEA, rather than cortisol, progesterone and androstenedione, suggestive of inhibition of 17,20-lyase activity (39). Similar results were obtained in men (40). However, in our data exaggerated secretory response of androstenedione and DHEA to ACTH was not associated with a disproportionate 17OH-progesterone response to ACTH, suggesting that 17,20-lyase activity is not altered. Moreover, no association of androstenedione and DHEA response to ACTH and insulin levels was found in our cohort of women with PCOS, which does not suggest that insulin is a key driver for adrenal androgen production in PCOS. However, whether very high insulin concentrations, as are obtained during a clamp study, may dysregulate 17,20-lyase activity in PCOS, as previously suggested (39, 40) cannot be excluded by our data.
In conclusion, by stratifying patients according to adrenal androgen responses to exogenous ACTH, and matching subjects for body composition, we have clarified the alterations in cortisol metabolism which occur in PCOS. These data suggest a role for increased inactivation of cortisol by 5β-reductase in the pathogenesis of adrenal hyperandrogenism in a subgroup of PCOS women, characterised by increased response of androstenedione, DHEA, 17OH-progesterone and cortisol to ACTH with low circulating basal cortisol levels. The ability to define subgroups in PCOS is important in improving therapy for this heterogeneous condition. Most variables are continuously distributed and any cut-off values are inevitably arbitrary and subject to further investigation, however our comparison between groups stratified by adrenal androgen responses to ACTH was supported by continous correlation analyses. We can speculate that the PCOS group with adrenal hyperandrogenism sustained by an overall hyperresponsiveness of the adrenal cortex to ACTH stimulation may be the most susceptible to existing strategies which enhance negative feedback suppression of the HPA axis, for example with low dose glucocorticoid suppression, or to novel strategies which normalise peripheral cortisol metabolism.
Acknowledgments
Disclosure statement: This work was supported by a Grant from the Sixth EC Program (LSHM-CT-2003-503041).
References
- 1.Carmina E, Koyama T, Chang L, Stanczyk FZ, Lobo RA. Does ethnicity influence the prevalence of adrenal hyperandrogenism and insulin resistance in polycystic ovary syndrome? Am J Obstet Gynecol. 1992;167:1807–1812. doi: 10.1016/0002-9378(92)91779-a. [DOI] [PubMed] [Google Scholar]
- 2.Wild RA, Umstot ED, Andersen RN, Ranney GB, Givens JR. Androgen parameters and their correlation with body weight in one hundred thirty-eight women thought to have hyperandrogenism. Am J Obstet Gynecol. 1983;146:602–605. doi: 10.1016/0002-9378(83)90998-5. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman DI, Klove K, Lobo RA. The prevalence and significance of elevated dehydroepiandrosterone sulfate levels in anovulatory women. Fertil Steril. 1984;42:76–81. doi: 10.1016/s0015-0282(16)47961-6. [DOI] [PubMed] [Google Scholar]
- 4.Azziz R, Black V, Hines GA, Fox LM, Boots LR. Adrenal androgen excess in the polycystic ovary syndrome: sensitivity and responsivity of the hypothalamic-pituitary-adrenal axis. J Clin Endocrinol Metab. 1998;83:2317–2323. doi: 10.1210/jcem.83.7.4948. [DOI] [PubMed] [Google Scholar]
- 5.Walker BR. Activation of the hypothalamic-pituitary-adrenal axis: cause or consequence? GH & IGF Res. 2001;11(Suppl A):S91–95. doi: 10.1016/s1096-6374(01)80015-0. [DOI] [PubMed] [Google Scholar]
- 6.Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1 – a tissue-specific amplifier of glucocorticoid action. Endocrinol. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- 7.Russell DW, Wilson JD. Steroid 5α-reductase: two genes/two enzymes. Ann Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 8.Charbonneau A, The VL. Genomic organization of a human 5beta-reductase and its pseudogene and substrate selectivity of the expressed enzyme. Bioch Bioph Acta. 2001;1517:228–235. doi: 10.1016/s0167-4781(00)00278-5. [DOI] [PubMed] [Google Scholar]
- 9.Rodin A, Thakkar H, Taylor N, Clayton R. Hyperandrogenism in polycystic ovary syndrome: evidence of dysregulation of 11β-hydroxysteroid dehydrogenase. N Engl J Med. 1994;330:460–465. doi: 10.1056/NEJM199402173300703. [DOI] [PubMed] [Google Scholar]
- 10.Stewart PM, Shackleton CH, Beastall GH, Edwards CR. 5α-Reductase activity in polycystic ovary syndrome. Lancet. 1990;335:431–433. doi: 10.1016/0140-6736(90)90664-q. [DOI] [PubMed] [Google Scholar]
- 11.Chin D, Shackleton C, Prasad VK, et al. Increased 5alpha-reductase and normal 11beta-hydroxysteroid dehydrogenase metabolism of C19 and C21 steroids in a young population with polycystic ovarian syndrome. J Pediatr Endocrinol Metab. 2000;13:253–259. doi: 10.1515/jpem.2000.13.3.253. [DOI] [PubMed] [Google Scholar]
- 12.Tsilchorozidou T, Honour JW, Conway GS. Altered cortisol metabolism in polycystic ovary syndrome: insulin enhances 5α-reduction but not the elevated adrenal steroid production rates. J Clin Endocrinol Metab. 2003;88:5907–5913. doi: 10.1210/jc.2003-030240. [DOI] [PubMed] [Google Scholar]
- 13.Rask E, Olsson T, Söderberg S, et al. Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab. 2001;86:1418–1421. doi: 10.1210/jcem.86.3.7453. [DOI] [PubMed] [Google Scholar]
- 14.Stewart PM, Boulton A, Kumar S, Clark PM, Shackleton CH. Cortisol metabolism in human obesity: impaired cortisone→cortisol conversion in subjects with central adiposity. J Clin Endocrinol Metab. 1999;84:1022–1027. doi: 10.1210/jcem.84.3.5538. [DOI] [PubMed] [Google Scholar]
- 15.Andrew R, Phillips DIW, Walker BR. Obesity and gender influence cortisol secretion and metabolism in man. J Clin Endocrinol Metab. 1998;83:1806–1809. doi: 10.1210/jcem.83.5.4951. [DOI] [PubMed] [Google Scholar]
- 16.Nestler JE, Jakubowicz DJ, Evans WS, Pasquali R. Effects of metformin on spontaneous and clomiphene-induced ovulation in the polycystic ovary syndrome. N Engl J Med. 1998;338:1876–1880. doi: 10.1056/NEJM199806253382603. [DOI] [PubMed] [Google Scholar]
- 17.Ferriman D, Gallwey JD. Clinical assessment of body hair growth in women. J Clin Endocrinol Metab. 1961;21:1440–1447. doi: 10.1210/jcem-21-11-1440. [DOI] [PubMed] [Google Scholar]
- 18.Gambineri A, Pelusi C, Genghini S, et al. Effect of flutamide and metformin administered alone or in combination in dieting obese women with polycystic ovary syndrome. Clin Endocrinol. 2004;60:241–249. doi: 10.1111/j.1365-2265.2004.01973.x. [DOI] [PubMed] [Google Scholar]
- 19.The Rotterdam ESHRE/ASMR-sponsored PCOS consensus workshop group Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Hum Reprod. 2004;19:41–47. doi: 10.1093/humrep/deh098. [DOI] [PubMed] [Google Scholar]
- 20.Pasquali R, Gambineri A, Biscotti D, et al. Effect of long-term treatment with metformin added to hypocaloric diet on body composition, fat distribution, and androgen and insulin levels in abdominally obese women with and without the polycystic ovary syndrome. J Clin Endocrinol Metab. 2000;85:2767–2774. doi: 10.1210/jcem.85.8.6738. [DOI] [PubMed] [Google Scholar]
- 21.WHO Preventing and Managing the Global Epidemic . Report of a WHO consultation on obesity. World Health Organization; Geneva: 1997. WHO/NUT/NCD/98.1. [PubMed] [Google Scholar]
- 22.Gambineri A, Pelusi C, Manicardi E, et al. Glucose intolerance in a large cohort of mediterranean women with polycystic ovary syndrome. Phenotype and associated factors. Diabetes. 2004;53:2353–2358. doi: 10.2337/diabetes.53.9.2353. [DOI] [PubMed] [Google Scholar]
- 23.Pasquali R, Gambineri A, Anconetani B, et al. The natural history of the metabolic syndrome in young women with the polycystic ovary syndrome and the effect of long-term oestrogen-progestagen treatment. Clin Endocrinol. 1999;50:517–527. doi: 10.1046/j.1365-2265.1999.00701.x. [DOI] [PubMed] [Google Scholar]
- 24.Vicennati V, Pasquali R. Abnormalities of the hypothalamic-pituitary-adrenal axis in nondepressed women with abdominal obesity and relations with insulin resistance: evidence for a central and peripheral alteration. J Clin Endocrinol Metab. 2000;85:4093–4098. doi: 10.1210/jcem.85.11.6946. [DOI] [PubMed] [Google Scholar]
- 25.Boschi S, De Iasio R, Mesini P, et al. Measurement of steroid hormones in plasma by isocratic high performance liquid chromatography coupled to radioimmunoassay. Clin Chim Acta. 1994;231:107–113. doi: 10.1016/0009-8981(94)90260-7. [DOI] [PubMed] [Google Scholar]
- 26.Vermeulen A, Verdonck L, Kaufman JM. A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab. 1999;84:3666–3672. doi: 10.1210/jcem.84.10.6079. [DOI] [PubMed] [Google Scholar]
- 27.Mather KJ, Hunt AE, Steinberg HO, et al. Repeatability characteristic of simple indices of insulin resistance: implications for research applications. J Clin Endocrinol Metab. 2001;86:5457–5464. doi: 10.1210/jcem.86.11.7880. [DOI] [PubMed] [Google Scholar]
- 28.Matsuda M, De Fronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- 29.Best R, Walker BR. Additional value of measurement of urinary cortisone and unconjugated cortisol metabolites in assessing the activity of 11β-hydroxysteroid dehydrogenase in vivo. Clin Endocrinol. 1997;47:231–236. doi: 10.1046/j.1365-2265.1997.2471061.x. [DOI] [PubMed] [Google Scholar]
- 30.Zumoff B, Fukushima DK, Hellman L. Intercomparison of four methods for measuring cortisol production. J Clin Endocrinol Metab. 1974;38:169–175. doi: 10.1210/jcem-38-2-169. [DOI] [PubMed] [Google Scholar]
- 31.Ulick S, Tedde R, Wang JZ. Defective ring A reduction of cortisol as the major metabolic error in the syndrome of apparent mineralcorticoid excess. J Clin Endocrinol Metab. 1992;74:593–599. doi: 10.1210/jcem.74.3.1740492. [DOI] [PubMed] [Google Scholar]
- 32.Palermo M, Shackleton CHL, Mantero F, Stewart PM. Urinary free cortisone and the assessment of 11β-hydroxysteroid dehydrogenase activity in man. Clin Endocrinol. 1996;45:605–611. doi: 10.1046/j.1365-2265.1996.00853.x. [DOI] [PubMed] [Google Scholar]
- 33.Burger HG. Androgen production in women. Fertil Steril. 2002;77(Suppl 4):S3–S5. doi: 10.1016/s0015-0282(02)02985-0. [DOI] [PubMed] [Google Scholar]
- 34.Fassnacht M, Schlenz N, Schneider SB, Wudy SA, Allolio B, Arlt W. Beyond adrenal and ovarian androgen generation: increased peripheral 5α-reductase activity in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:2760–2766. doi: 10.1210/jc.2002-021875. [DOI] [PubMed] [Google Scholar]
- 35.Westerbacka J, Yhi-Järvinen H, Vehkavaara S, et al. Body fat distribution and cortisol metabolism in healthy men: enhanced 5β-reductase and lower cortisol/cortisone metabolite ratios in men with fatty liver. J Clin Endocrinol Metab. 2003;88:4924–4931. doi: 10.1210/jc.2003-030596. [DOI] [PubMed] [Google Scholar]
- 36.Livingstone DEW, McInnes KJ, Walker BR, Andrew R. Increased A-ring reduction of glucocorticoids in obese Zucker rats: effects of insulin sensitization. Obes Res. 2005;13:1523–1526. doi: 10.1038/oby.2005.186. [DOI] [PubMed] [Google Scholar]
- 37.Stenberg A. On the modulating effects of ovaries on neonatal androgen programming of rat liver enzymes. Acta Endocrinol. 1975;78:294–301. doi: 10.1530/acta.0.0780294. [DOI] [PubMed] [Google Scholar]
- 38.Rosenfield RL. Ovarian and adrenal function in polycystic ovary syndrome. Endocrinol Metab Clin North Am. 1999;28:265–293. doi: 10.1016/s0889-8529(05)70070-0. [DOI] [PubMed] [Google Scholar]
- 39.Moghetti P, Castello R, Negri C, et al. Insulin infusion amplifies 17α-hydroxycorticosteroid intermediates response to adrenocorticotropin in hyperandrogenic women: apparent relative impairment of 17,20-lyase activity. J Clin Endocrinol Metab. 1996;81:881–886. doi: 10.1210/jcem.81.3.8772544. [DOI] [PubMed] [Google Scholar]
- 40.Nestler JE, McClanahan MA, Clore JN, Blackard WG. Insulin inhibits adrenal 17,20-lyase activity in man. J Clin Endocrinol Metab. 1992;74:362–367. doi: 10.1210/jcem.74.2.1730815. [DOI] [PubMed] [Google Scholar]