

Abstract
Allogeneic blood and marrow transplantation (allo-BMT) is an effective immunotherapeutic treatment that can provide partial or complete remission for patients with hematological malignancies. Mature donor T cells in the donor inoculum play a central role in mediating graft-versus-tumor (GVT) responses by destroying residual tumor cells that persist after conditioning regimens. Alloreactivity towards minor histocompatibility antigens (miHA), which are varied tissue-related self-peptides presented in the context of major histocompatibility complex (MHC) molecules on recipient cells, some of which may be shared on tumor cells, is a dominant factor for the development of GVT. Potentially, GVT can also be directed to tumor-associated antigens or tumor-specific antigens that are more specific to the tumor cells themselves. The full exploitation of allo-BMT, however, is greatly limited by the development of graft-versus-host disease (GVHD), which is mediated by the donor T cell response against the miHA expressed in the recipient’s cells of the intestine, skin, and liver. Because of the significance of GVT and GVHD responses in determining the clinical outcome of patients, miHA and tumor antigens have been intensively studied, and one active immunotherapeutic approach to separate these two responses has been cancer vaccination after allo-BMT. The combination of these two strategies has an advantage over vaccination of the patient without allo-BMT because his or her immune system has already been exposed and rendered unresponsive to the tumor antigens. The conditioning for allo-BMT eliminates the patient’s existing immune system, including regulatory elements, and provides a more permissive environment for the newly developing donor immune compartment to selectively target the malignant cells. Utilizing recent technological advances, the identities of many human miHA and tumor antigenic peptides have been defined and are currently being evaluated in clinical and basic immunological studies for their ability to produce effective T cell responses. The first step towards this goal is the identification of targetable tumor antigens. In this review, we will highlight some of the technologies currently used to identify tumor antigens and anti-tumor T cell clones in hematological malignancies.
Keywords: Minor histocompatibility antigens, Tumor-associated antigens, Tumor-specific antigens, Tumor vaccination, Blood and marrow transplantation, Hematological malignancy
INTRODUCTION
Adoptive T cell therapy in the form of allogeneic blood and marrow transplantation (allo-BMT) has proven to be one of the few curative treatments for a number of drug-resistant hematological malignancies [1,2]. To date, the gold standard of immunotherapy used in the treatment of patients with acute myeloid leukemia (AML), lymphoma, chronic lymphocytic leukemia (CLL), chronic myeloid leukemia (CML), and multiple myeloma (MM) is the administration of donor lymphocytes along with, or at some time after, allo-BMT (in the form of donor lymphocyte infusions) [3]. The broad donor-derived CD4+ and CD8+ T cell repertoire targeting a diversity of undefined (allogeneic) tumor antigens is exploited in this setting [1,3,4].
Allo-BMT permits partial or complete remission in a significant percentage of patients. Although mature donor T cells in the donor inoculum facilitate patient immune reconstitution and mediate graft-versus-tumor (GVT) responses by destroying residual tumor cells that persist after conditioning regimens [5–7], the full exploitation of this clinical intervention is greatly limited by the development of graft-versus-host disease (GVHD). This primary complication of allo-BMT is due to the donor T cell allogeneic response against tissue antigens in the intestine, skin, and liver, which may or may not be shared by tumor cells [8–10]. Therefore, even in a fully major histocompatibility complex (MHC)-matched (in humans, HLA-matched) transplantation settings, the alloreactivity towards recipient cells is the key factor for the development of both GVT and GVHD effects. Unfortunately, the uncoupling of these two events to improve transplantation outcomes has yet to be achieved in a consistent and efficient manner. GVT can also be directed to tumor-specific target antigens that are not expressed by normal host tissues. Therefore, to reduce the development of GVHD and potentiate the GVT response, one active immune approach in the clinic has been the use of cancer vaccination after allo-BMT [11]. This combined strategy has an advantage over mere vaccination of the patient without allo-BMT, for the immune system of the patient has already been exposed and, hence, has become unresponsive to the tumor antigens, whereas the newly developing donor immune compartment can be educated to selectively target malignant cells. The first step towards this goal is the identification of targetable tumor antigens [12,13]. In this review, we will highlight some of the technologies currently used to identify tumor antigens and antitumor T cell clones in hematological malignancies.
CLASSIFICATION OF TUMOR ANTIGENS
Tumor antigens are classified according to their distribution as tumor-specific antigens (TSA), which are only expressed by the tumor, or as tumor-associated antigens (TAA), which can also be found in other normal cell types [11,13–16]. Unique tumor antigens, on the other hand, are those of patient-restricted expression whereas shared antigens are commonly present across various samples of the same histologic subtype of malignancy and on different tumor types, but not in normal tissues, except for testis and placenta. Although shared antigens constitute an ideal group to develop broadly applicable cancer vaccines, the identification of unique TSA has the potential to develop into highly effective personalized immunotherapeutic interventions. Other classifications that stem from the combinations of these different types of antigens are as follows.
Unique TSA
These antigens result from somatic point mutations induced by carcinogens and, therefore, occur in a single tumor of one patient; thus, they represent a bone fide TSA not capable of being expressed by any normal tissue. Importantly, unique TSA have the potential to elicit more effective antitumor vaccine responses than shared antigens because of their resistance to immunoselection, particularly if the mutated protein is critical for the preservation of neoplastic cells. For a thorough review of identified unique tumor antigens, see Parmiani et al. [14].
Shared TSA
These antigens are expressed in different tumors but not in healthy tissues. The most prominent antigens among this group are the cancer-testis family of antigens including MAGE [17,18], BAGE, LAGE, GAGE, and NY-ESO-1 [19–22], which in normal tissues are restricted only to the testis and placenta but can be found in MM, breast, ovarian, and head and neck cancers [23,24].
Shared TAA
This category of antigens, although not tumor specific, is overexpressed in different types of tumors. Examples of these antigens are human telomerase reverse transcriptase [25], survivin [26,27], proteinase-3 [28,29], Wilms tumor gene-encoded transcription factor-1 (WT1) [30,31], mucin-1 [32], and preferentially expressed antigen of melanoma (PRAME) [33], which can be found only at very low levels in healthy tissues, such as the adrenal glands, ovaries, and endometrium. Shared TAA have been considered as potential targets for cancer immunotherapy. Arai et al. [34] demonstrated that CD8+ cytotoxic T cell (CTL) clones specific for human telomerase reverse transcriptase peptides exerted cytotoxicity against leukemia cells in an HLA-A24–restricted manner, while sparing HLA-A24− leukemia cells or HLA-A24−normal cells. PRAME is known to contain at least 4 different HLA-A*0201-restricted epitopes (PRA100–108, PRA142–151, PRA300–309, and PRA425–433) [35] recognized by CTLs [36]. Reports of PRAME antigen expression range from 47% to 70% of AML patients [37,38]. Proteinase-3 is also overexpressed in AML and CML [28,39,40] and WT1 is overtly present in several different types of leukemia [41]. In fact, WT1 ranked in the top 20 antigens with suggestive high therapeutic functionality according to the National Cancer Institute report on the prioritization of cancer antigens for acceleration of translational research [42]. WT1 is also among the most advanced targets for AML immunotherapy, as reflected by the relatively large number of WT1-targeted vaccine trials for AML patients [43]. In those patients, multiple WT1 CTL epitopes have been recognized as immunogenic (including WT137–45, WT1126–134, WT1187–195, and WT1235–243) [43]. WT1 was also found to induce WT1-specific CD4+ helper T cell immunity in patients with AML through active immunization [44]. Mucin-1, an epithelial mucin present in a number of solid tumors, can also be found in MM cell lines and primary tumors [45–49]. For an in-depth review of tumor antigens recognized by T cells, and in particular those pertinent to hematological malignancies, see Novellino et al. [50], Borrello et al. [11], and Anguille et al. [41].
Minor Histocompatibility Antigens (miHA)
In the context of an MHC-matched allo-BMT, alloreactive CD8+ and CD4+ donor T cells can also be directed at non–MHC-encoded polymorphic peptides known as miHA, presented by both MHC class I and class II molecules [51,52] on allogeneic host cells. Many of these miHA are encoded by allelic genes that can differ between patient and donor because of single nucleotide polymorphisms (SNPs). The target molecules involved in the GVT response can be any of the TSA or TAA described above, as well as tissue- and tumor-specific miHA. Donor T cells have the advantage of recognizing all of these target antigens in an immunologically permissive environment, whereas in the patient, T cell responses are generated only against TSA and TAA and are subject to tolerizing mechanisms. Conceptually, in the allo-BMT setting, all tumor antigens presented in the context of MHC that are recognized by donor but not host T cells are miHA.
In 1978, Korngold and Sprent demonstrated that transfer of bone marrow cells containing T cells into lethally irradiated MHC-matched recipient mice caused GVHD, suggesting that miHAs were the main target for eliciting this disease [53]. The use of miHA as tumor targets after allo-BMT derives from the notion that some of these antigens are exclusively expressed on normal and malignant host hematopoietic cells permitting, hypothetically, the separation of GVT and GVHD pathological responses [54,55]. In some cases, miHA can also be considered as TAA in that they may be overexpressed in tumor cells in comparison to the rest of the hematopoietic compartment. The contribution of miHA in GVT has been evaluated mainly by isolating CTL with tumor lytic capability and studying their effect on normal host hematopoietic cells or nonhematopoietic fibroblast cells. Later on in this review, we will discuss work by the present authors using other technological approaches aimed at identifying and separating tumor versus tissue reactive T cell clones using spectratype analysis and T cell receptor (TCR) sequencing.
HA-1 and HA-2 constitute the first 2 miHA identified to be solely expressed on hematopoietic cells [54,56], including progenitor cells. Both of these antigens have been found to be expressed in all leukemia and MM cells [57,58]. Subsequently, a number of other hematopoietic miHA have been identified, including HB-1, an acute B-lymphoblastoid-leukemia-related antigen [59], and proliferation-associated nuclear element 1 gene [60,61], a B cell CLL–related antigen. Although ubiquitous, the ATP-dependent, interferon-responsive gene (ADIR) is also highly expressed in activated hematopoietic cells, including MM and various solid tumors [62]. LRH1, encoded by the P2X5 gene, is hematopoietic specific and expressed in leukemic cells and their CD34+ progenitors [63]. A number of other miHA encoded by ubiquitously expressed genes appear to be preferentially expressed in activated hematopoietic cells and malignant cells (see Table 1 in references [13,64]). A retrospective analysis on the impact of a panel of 17 immunogenic miHAs including HA-1, HA-2, HA-8, ATP-dependent, interferon-responsive, proliferation-associated nuclear element 1, LRH1, SP110, ECGF, and ACC2, in patients who received a partial T cell–depleted HLA-identical allo-BMT, revealed that in sibling transplantations, mismatches in one or more of the studied autosomal-encoded miHA resulted in an improved relapse-free survival rate, especially in MM patients [65].
Of note, mismatches in individual miHA, including HA-1, HA-2, and HA-8, have been associated with increased GVHD occurrence and lower relapse rates [66], although other studies could not confirm these results [67]. The adoptive transfer of miHA-specific CTLs selected on the basis of recognition of recipient hematopoietic cells but not skin fibroblasts has also unexpectedly been associated with GVHD. Likewise, in a murine model of BMT, infusion of tumor-specific CTLs identified by CDR3-size spectratype analysis was shown to induce a significant GVT response, but the same tumor-reactive Vβ family, which initially showed no hematopoietic alloreactivity was ultimately the causal entity of gut pathology in recipient mice, when administered at higher dosages [68]. Taken together, the results from these clinical trials and murine models suggest that responses to target tissue–related miHA are complex and may vary not only amongst different individuals but also between tissue types.
In 2009, The Translational Research Working Group of the National Cancer Institute specified a number of criteria for determining the suitability of a given tumor antigen for therapeutic application. The following characteristics were evaluated and prioritized in descending order to determine the “ideal” cancer antigen: (1) therapeutic function, (2) immunogenicity, (3) role of the antigen in oncogenicity, (4) specificity, (5) expression level and percent of antigen-positive cells, (6) stem cell expression, (7) number of patients with antigen-positive cancers, (8) number of antigenic epitopes, and (9) cellular location of antigen expression. Although there was no assessment of miHA using this prioritization analysis, it was concluded that none of the 75 tumor antigens studied fit all the criteria of the “ideal” cancer antigen. Nevertheless, 46 antigens were reported as immunogenic in clinical trials and 20 antigens had suggestive clinical efficacy in the “therapeutic function” category [42].
Approaches to Identify Tumor Antigens
The characterization of a tumor-specific CTL epitope from the human melanoma antigen MAGE-1 was reported in 1992 by Boon et al. [17,18]. Since this discovery, a number of technological advances have led to a great increase in the number of recognized TAA. The first strategy used to identify tumor antigens was peptide elution. This approach is based on high-liquid performance chromatography paired with mass spectrometric sequencing of the miHA peptide eluted from the cell surface of MHC molecules [62,69–72]. This technique, despite being very successful, has only yielded positive results in identifying HLA class I–presented miHA. In addition to peptide elution, forward or “T cell-to-antigen”–based strategy and reverse immunology are two major strategies that have been recently used in the identification of tumor antigens and CD8+ and CD4+ T cell epitopes contained in these tumor-specific proteins.
Forward Immunology Methods
Broadly speaking, these methods are characterized by the isolation of tumor-reactive T cells generated from an autologous peripheral blood mononuclear cell coculture with tumor cells or from individuals who underwent transplantation demonstrating a clinical response to donor lymphocyte infusions after allo-BMT, followed by the subsequent identification of the antigens that elicited a T cell response. For a review of immunogenic scenarios conducive for the identification of tumor antigens in this forward manner, see Kawakami et al. [73]. Forward immunological techniques and their variations can be applied to identify miHA presented in the context of either MHC-I or MHC-II.
After the isolation and expansion of CTLs, these lines can be used to isolate the tumor-specific cDNA that encodes the recognized CTL epitope by screening of cDNA expression libraries (Figure 1) derived from the tumor or Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines from members of CEPH families (Jean Dausset-Centre d’Etude du Polymorphisme Humain [CEPH]; http://www.cephb.fr/en/cephdb/), followed by genetic linkage analysis [74]. Genetic linkage analysis identifies the genomic locus of the miHA by pair-wise correlation of the miHA phenotype of large CEPH families with thousands of genetic markers identified in their genomes. Alternatively, as miHA are associated with common polymorphisms within the human population, they can also be identified using genetic association studies. This alternative genetic approach uses the extensive linkage disequilibrium found within the human genome to efficiently localize the target loci, based on recent advances of large-scale genotyping technologies and the assets of the International HapMap Project (www.hapmap.org). Using this approach, Spaapen et al. [75] identified a CD19-encoded miHA, presented by MHC class II molecules by correlating the miHA phenotypes of 23 CEPH individuals with the SNP genotypes derived from HapMap. Subsequently, the authors also identified the miHA recognized by the 1GF5 CD4+ T cell clone, isolated from a patient with MM undergoing a strong GVT response associated with acute GVHD, using another derivate of forward techniques, a zygosity-genotype correlation analysis with embedded HapMap SNP genotypes from the “Utah residents with ancestry from northern and western Europe” (known as CEU population) [76].
Figure 1.

Schematic representation of a forward immunology method for the identification of miHA as tumor targets. Panel (A-1): Identification and expansion of T cell clone with strong GVT/GVHD reactivity. Panel (A-2): The reactive T cell clone is then scanned against a cDNA library with EBV-LCL from CEPH families to identify the cDNA clone eliciting cytotoxic activity. Genetic linkage analysis is performed on this cDNA clone to identify genes encoding the potential tumor miHA. Panel (B): Preparation of cDNA library from patient-derived tumor cells or from EBV-LCL from CEPH families. Current methods for the production and expansion of T cell clones are discussed in more detail in the “Production of bulk T cells as a tool for discovery of TAA” section of this review.
Whole genome association scans is another forward approach that allows for high-throughput identification of miHA [77], where third-party EBV-B cell lines selected for their coexpression of pertinent HLA molecules, are genotyped for more than 1 million SNPs. The miHA are then identified by analysis of association between T cell recognition of these EBV-transformed lymphoblastoid cell lines and individual SNP genotypes measured in these lines [77,78], while recognition of nonhematopoietic fibroblasts and other stromal cells is ruled out to ensure that the T cell clone exclusively reacts against the hematopoietic (ie, of tumor origin) compartment [75].
All of these approaches, however, are limited by the low affinity interaction of the TCR with their specific MHC-peptide complex and the technical difficulties associated with library cloning platforms. Although yet to be applied to the identification of new miHA, Siewert et al. identified target antigens of CD8+ T cells using combinatorial libraries coding for short peptides and a single-cell detection system with HLA-A*0201 MHC molecules presenting influenza matrix protein (flu58–66) peptides [79]. The MHC class I cDNA was cotransfected along with a plasmid-coded combinatorial nonamer peptide library into COS-7 cells, which allowed antigen processing and presentation for T cell recognition. For screening, a reporter T cell hybridoma cell line was cotransfected with the specific CD8 TCR α and β chains and the super green fluorescence protein (sGFP) reporter gene under the transcriptional control of the nuclear factor of activated T cells enhancer. To identify and isolate an antigenic peptide, a single nuclear factor of activated T cells–activated TCR expressing T cell hybridoma cell that recognized the correct antigenic peptide expressing COS-7 cell during coculture was subsequently isolated and the antigenic peptide plasmid was cloned to determine the precise sequence of the peptide [79].
Reverse Immunology Methods
An alternative strategy used in the identification of tumor antigens is reverse immunology, in which the prediction of miHA on hematopoietic cells constitute the starting point and peptide candidates are subsequently screened for their capacity to induce a T cell–specific response [80,81] (Figure 2). In silico analysis uses prediction algorithms, such as SYFPEITHI [82] (http://www.syfpeithi.de/), SNP-derived epitope prediction program [83], which is based on SYFPEITHI, “BIMAS” (http://bimas.dcrt.nih.gov/molbio/hla_bind/), and TEPITOPEpan [84,85] (http://www.biokdd.-fudan.edu.cn/Service/TEPITOPEpan/) to determine peptides with putative strong binding to HLA-I and HLA-II molecules. However, because peptide-HLA binding affinity and proteolytic cleavage also play key roles in determining the biological feasibility of miHA, the vast majority of T cell responses detected using this original reverse manner approach were directed against epitopes that are not naturally processed and presented [86,87]. Recent technological advances from the HLA-associated peptidome of hematopoietic cells by mass spectrometric analysis (HLA peptidomics) SNP databases, MHC-tetramer technology, and multiparametric flow cytometric analysis were instrumental in the identification of eluted peptide candidates that can undergo HLA-restricted processing and presentation [80]. Likewise, proteasomal cleavage and transporter associated with antigen processing transport efficiency has been combined with reverse computational approaches to optimize the selection of candidate epitopes [88]. Feldhahn et al. described a different approach for large-scale detection of tissue-specific miHA [89] that uses netMHCpan, a high-throughput computational method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence [90].
Figure 2.
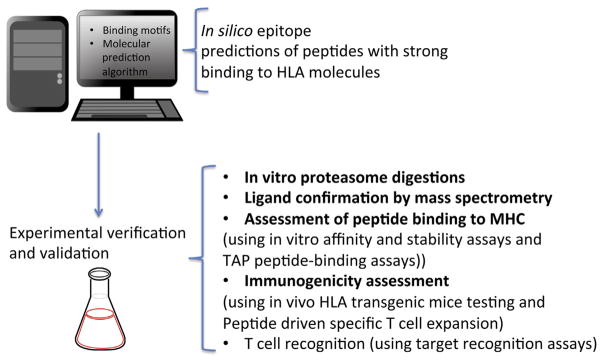
Schematic representation of reversed immunological methods to determine miHA peptides. Steps for determining and validating miHA epitopes eliciting relevant antitumor T cell activity are depicted in this figure. A first phase of computational prediction determines a number of putative epitopes, which are then confirmed experimentally for their capacity to induce an in vivo T cell response.
Innovative technologies and reduced costs of genomic sequencing have opened the door for the identification of tumor-associated genes through whole exome sequencing. Recently, Rosenberg et al. at the National Cancer Institute developed a screening method to identify mutated gene products from patients’ tumors and their potential T cell epitopes that may be recognized by isolated tumor-infiltrated lymphocytes (TILs). Although originally tested in melanoma, this approach could be extrapolated to other cancers, including hematological malignancies. Initially, whole exome sequencing of the tumor was compared to normal patient’s DNA to identify somatic mutations. Candidate mutated T cell epitopes identified in silico using netMHCpan were subsequently synthesized in COS-7 cell lines that were stably transduced to express the appropriate HLA (as described above), and assayed for their recognition by TILs. The adoptive transfer of TILs to patients that were generated after exposure to these identified dominant target epitopes mediated significant and durable tumor regression [91].
PRODUCTION OF BULK T CELLS AS A TOOL FOR DISCOVERY OF TAA
The TCR is a heterodimeric protein, comprising an α and a β chain, that recognizes antigens presented by MHC molecules. Diversity of the TCR repertoire allows the adaptive immune system to protect the body against a vast array of potential pathogens, such as cancer cells and allo-antigens. These chains are somatically rearranged from individual gene segments to create millions of different surface receptors, with the majority of T cells expressing a single productively rearranged TCR α and β chain allele. The identification of tumor-reactive T cells and their TCR usage responsible for mounting a significant anti-tumor response is necessary for the discovery of novel tumor antigens. The generation of a sufficient number of tumor reactive cells is critical for determining their cognate tumor antigens by forward immunological approaches and for their adoptive transfer into patients as immunotherapy.
Unfortunately, the duration of the in vitro selection and expansion of tumor reactive T cells as shown in Figure 1(A-1) frequently leads to T cell exhaustion, reducing their capacity to produce cytokines and proliferate to an extent that makes them unsuitable for use as a tool for antigen discovery. A more feasible approach to generate sufficient number of functional T cells for therapeutic transfer has been the production of TCR-transgenic T cells restricted to a particular tumor epitope [92,93]. This approach uses viral transfer of the genes encoding the TCR α and β chains of identified tumor-specific clones into primary T cells. The advantage of viral transduction facilitates the generation of large amounts of antigen-specific CTLs in several days rather than several weeks, and thereby bypasses the development of proliferative senescence and its concomitant decrease in T cell killing activity.
Vβ CDR-3 size spectratyping and TCR deep sequencing, two techniques aimed at determining the TCR repertoire usage, have opened the possibility of dissociating GVT and GVHD responses by identifying allo-reactive and unique tumor-reactive T cell clones. Our group and others have extensively used spectratyping to predict Vβ families with in vivo allo-reactive potential in both murine models of BMT and clinical samples [94–97]. Using this technique, we identified those families capable of mounting a strong anti-tumor response [68,98] that overlapped only moderately with the induction of GVHD [68]. In particular, our approach has focused on in vitro mixed lymphocyte cultures, which expose donor T cells to hematopoietic allo-antigens from patient-derived peripheral blood mononuclear cells after the conditioning regimen to identify expanding T cell clones [94]. We are currently including mixed lymphocyte cultures of donor T cells stimulated with patient-derived tumor cells (obtained before conditioning) to distinguish unique donor-patient antitumor responses (work in progress). Although spectratype results can be further used to identify the particular TCR clone that is likely driving the expansion of a Vβ family [99], the feasibility for the rapid identification of tumor or GVHD effector T cells is low because of lengthy cloning techniques and the random selection of a limited number of colonies. The advent of next-generation high-throughput TCR sequencing can generate a sequence of tens of millions of TCRs from a single sample within a few days. This methodology uses a multiplex-based PCR method to identify TCRβ chains from genomic DNA, which in turn can be used to pinpoint tumor-reactive T cells [100–102]. Although the conditions for the identification of the TCRα chain are currently being optimized, this technology has the potential to transform the recognition of relevant TCR clones that can then be engineered into primary T cells for downstream immunotherapeutic use [103].
SUMMARY AND FUTURE DIRECTIONS
The development of clinically efficacious cancer vaccines relies on the identification of targetable tumor antigens. In this review, we summarized some of the major and newest technological advances currently employed in the discovery of these antigens, as well as their limitations. We emphasized the use of these techniques for the recognition of miHA, TAA, and TSA as tumor targets of hematological malignancies in the context of allo-BMT. In particular we reviewed forward methods, which primarily start with identifying the T cell clone(s) responsible for mediating antitumor responses, as well as various modalities of reverse immunological approaches in which prediction of potential antigenic epitopes are screened for their capability of inducing a physiologically plausible GVT reaction. As de novo donor-derived immune cells can be much more readily educated to attack tumor cells, the use of the post–allo-BMT setting offers a unique environment to take advantage of cancer vaccines. We also discussed the techniques for recognition, selection, and expansion of those tumor-specific T cell clones that are necessary to guide the discovery process of tumor antigens. We believe that the recognition of potential tumor antigens, currently being used to generate dendritic cell peptide-loaded vaccines against a particularly relevant miHA, TAA, or TSA [13,24,104], will be streamlined by the capability to sequence the TCR of the responding T cell and to generate bulk quantities of tumor-targeting TCR-transduced T cells for adoptive immunotherapy. The combination of vaccination and TCR engineering will likely be used synergistically in upcoming years to enhance the antitumor response of donor T cells after transplantation and to provide a durable tumor remission, if not complete eradication.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants National Heart, Lung, and Blood Institute R21HL102886 (J.Z.) and NCI RO1CA154244 (R.K).
Footnotes
Financial disclosure: The authors have nothing to disclose.
Conflict of interest statement: There are no conflicts of interest to report.
References
- 1.Zeidan AM, Forde PM, Symons H, et al. HLA-haploidentical donor lymphocyte infusions for patients with relapsed hematologic malignancies after related HLA-haploidentical bone marrow transplantation. Biol Blood Marrow Transplant. 2014;20:314–318. doi: 10.1016/j.bbmt.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luznik L, Fuchs EJ. Donor lymphocyte infusions to treat hematologic malignancies in relapse after allogeneic blood or marrow transplantation. Cancer Control. 2002;9:123–137. doi: 10.1177/107327480200900205. [DOI] [PubMed] [Google Scholar]
- 3.Kolb HJ, Simoes B, Schmid C. Cellular immunotherapy after allogeneic stem cell transplantation in hematologic malignancies. Curr Opin Oncol. 2004;16:167–173. doi: 10.1097/00001622-200403000-00015. [DOI] [PubMed] [Google Scholar]
- 4.Zorn E, Wang KS, Hochberg EP, et al. Infusion of CD4+ donor lymphocytes induces the expansion of CD8+ donor T cells with cytolytic activity directed against recipient hematopoietic cells. Clin Cancer Res. 2002;8:2052–2060. [PubMed] [Google Scholar]
- 5.Alyea EP, DeAngelo DJ, Moldrem J, et al. NCI First International Workshop on The Biology, Prevention and Treatment of Relapse after Allogeneic Hematopoietic Cell Transplantation: report from the committee on prevention of relapse following allogeneic cell transplantation for hematologic malignancies. Biol Blood Marrow Transplant. 2010;16:1037–1069. doi: 10.1016/j.bbmt.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiden PL, Flournoy N, Thomas ED, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. 1979;300:1068–1073. doi: 10.1056/NEJM197905103001902. [DOI] [PubMed] [Google Scholar]
- 7.Horowitz MM, Gale RP, Sondel PM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–562. [PubMed] [Google Scholar]
- 8.Blazar BR, Korngold R, Vallera DA. Recent advances in graft-versus-host disease (GVHD) prevention. Immunol Rev. 1997;157:79–109. doi: 10.1111/j.1600-065x.1997.tb00976.x. [DOI] [PubMed] [Google Scholar]
- 9.Korngold R. Biology of graft-vs.-host disease. Am J Pedtr Hematol Oncol. 1993;15:18–27. doi: 10.1097/00043426-199302000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Korngold R, Sprent J. Graft-versus-host disease in experimental allogeneic bone marrow transplantation. Proc Soc Exp Biol Med. 1991;197:12–18. doi: 10.3181/00379727-197-43217a. [DOI] [PubMed] [Google Scholar]
- 11.Borrello IM, Sotomayor EM. Cancer vaccines for hematologic malignancies. Cancer Control. 2002;9:138–151. doi: 10.1177/107327480200900206. [DOI] [PubMed] [Google Scholar]
- 12.Oostvogels R, Minnema MC, van Elk M, et al. Towards effective and safe immunotherapy after allogeneic stem cell transplantation: identification of hematopoietic-specific minor histocompatibility antigen UTA2-1. Leukemia. 2013;27:642–649. doi: 10.1038/leu.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spaapen R, Mutis T. Targeting haematopoietic-specific minor histocompatibility antigens to distinguish graft-versus-tumour effects from graft-versus-host disease. Best Pract Res Clin Haematol. 2008;21:543–557. doi: 10.1016/j.beha.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Parmiani G, De Filippo A, Novellino L, Castelli C. Unique human tumor antigens: immunobiology and use in clinical trials. J Immunol. 2007;178:1975–1979. doi: 10.4049/jimmunol.178.4.1975. [DOI] [PubMed] [Google Scholar]
- 15.Kessler JH, Melief CJ. Identification of T-cell epitopes for cancer immunotherapy. Leukemia. 2007;21:1859–1874. doi: 10.1038/sj.leu.2404787. [DOI] [PubMed] [Google Scholar]
- 16.Bocchia M, Defina M, Aprile L, Sicuranza A. Peptide vaccines for hematological malignancies: a missed promise? Int J Immunol. 2014;99:107–116. doi: 10.1007/s12185-013-1497-3. [DOI] [PubMed] [Google Scholar]
- 17.van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 18.Traversari C, van der Bruggen P, Luescher IF, et al. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J Exp Med. 1992;176:1453–1457. doi: 10.1084/jem.176.5.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costa FF, Le Blanc K, Brodin B. Concise review: cancer/testis antigens, stem cells, and cancer. Stem Cells. 2007;25:707–711. doi: 10.1634/stemcells.2006-0469. [DOI] [PubMed] [Google Scholar]
- 20.Kalejs M, Erenpreisa J. Cancer/testis antigens and gametogenesis: a review and “brain-storming” session. Cancer Cell Int. 2005;5:4. doi: 10.1186/1475-2867-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kronig H, Hofer K, Conrad H, et al. Allorestricted T lymphocytes with a high avidity T-cell receptor towards NY-ESO-1 have potent anti-tumor activity. Int J Cancer. 2009;125:649–655. doi: 10.1002/ijc.24414. [DOI] [PubMed] [Google Scholar]
- 22.Boel P, Wildmann C, Sensi ML, et al. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity. 1995;2:167–175. doi: 10.1016/s1074-7613(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 23.de Carvalho F, Vettore AL, Colleoni GW. Cancer/testis antigen MAGE-C1/CT7: new target for multiple myeloma therapy. Clin Devel Immunol. 2012;2012:257695. doi: 10.1155/2012/257695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson LD, Jr, Cook DR, Yamamoto TN, et al. Identification of MAGE-C1 (CT-7) epitopes for T-cell therapy of multiple myeloma. Cancer Immunol Immunother. 2011;60:985–997. doi: 10.1007/s00262-011-1009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernandez J, Garcia-Pons F, Lone YC, et al. Identification of a human telomerase reverse transcriptase peptide of low affinity for HLA A2.1 that induces cytotoxic T lymphocytes and mediates lysis of tumor cells. Proc Natl Acad Sci U S A. 2002;99:12275–12280. doi: 10.1073/pnas.182418399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedrichs B, Siegel S, Andersen MH, et al. Survivin-derived peptide epitopes and their role for induction of antitumor immunity in hematological malignancies. Leuk Lymphoma. 2006;47:978–985. doi: 10.1080/10428190500464062. [DOI] [PubMed] [Google Scholar]
- 27.Andersen MH, thor SP. Survivin–a universal tumor antigen. Histol Histopathol. 2002;17:669–675. doi: 10.14670/HH-17.669. [DOI] [PubMed] [Google Scholar]
- 28.Knights AJ, Weinzierl AO, Flad T, et al. A novel MHC-associated proteinase 3 peptide isolated from primary chronic myeloid leukaemia cells further supports the significance of this antigen for the immunotherapy of myeloid leukaemias. Leukemia. 2006;20:1067–1072. doi: 10.1038/sj.leu.2404234. [DOI] [PubMed] [Google Scholar]
- 29.Fujiwara H, El Ouriaghli F, Grube M, et al. Identification and in vitro expansion of CD4+ and CD8+ T cells specific for human neutrophil elastase. Blood. 2004;103:3076–3083. doi: 10.1182/blood-2003-07-2424. [DOI] [PubMed] [Google Scholar]
- 30.Oka Y, Elisseeva OA, Tsuboi A, et al. Human cytotoxic T-lymphocyte responses specific for peptides of the wild-type Wilms’ tumor gene (WT1) product. Immunogenetics. 2000;51:99–107. doi: 10.1007/s002510050018. [DOI] [PubMed] [Google Scholar]
- 31.Oka Y, Udaka K, Tsuboi A, et al. Cancer immunotherapy targeting Wilms’ tumor gene WT1 product. J Immunol. 2000;164:1873–1880. doi: 10.4049/jimmunol.164.4.1873. [DOI] [PubMed] [Google Scholar]
- 32.Apostolopoulos V, McKenzie IF. Cellular mucins: targets for immunotherapy. Crit Rev Immunol. 1994;14:293–309. doi: 10.1615/critrevimmunol.v14.i3-4.40. [DOI] [PubMed] [Google Scholar]
- 33.Matsushita M, Yamazaki R, Ikeda H, Kawakami Y. Preferentially expressed antigen of melanoma (PRAME) in the development of diagnostic and therapeutic methods for hematological malignancies. Leuk Lymphoma. 2003;44:439–444. doi: 10.1080/1042819021000035725. [DOI] [PubMed] [Google Scholar]
- 34.Arai J, Yasukawa M, Ohminami H, et al. Identification of human telomerase reverse transcriptase-derived peptides that induce HLA-A24-restricted antileukemia cytotoxic T lymphocytes. Blood. 2001;97:2903–2907. doi: 10.1182/blood.v97.9.2903. [DOI] [PubMed] [Google Scholar]
- 35.Rezvani K, Yong AS, Tawab A, et al. Ex vivo characterization of polyclonal memory CD8+ T-cell responses to PRAME-specific peptides in patients with acute lymphoblastic leukemia and acute and chronic myeloid leukemia. Blood. 2009;113:2245–2255. doi: 10.1182/blood-2008-03-144071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kessler JH, Beekman NJ, Bres-Vloemans SA, et al. Efficient identification of novel HLA-A(*)0201-presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis. J Exp Med. 2001;193:73–88. doi: 10.1084/jem.193.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greiner J, Schmitt M, Li L, et al. Expression of tumor-associated antigens in acute myeloid leukemia: Implications for specific immunotherapeutic approaches. Blood. 2006;108:4109–4117. doi: 10.1182/blood-2006-01-023127. [DOI] [PubMed] [Google Scholar]
- 38.Greiner J, Ringhoffer M, Simikopinko O, et al. Simultaneous expression of different immunogenic antigens in acute myeloid leukemia. Exp Hematol. 2000;28:1413–1422. doi: 10.1016/s0301-472x(00)00550-6. [DOI] [PubMed] [Google Scholar]
- 39.Lacey SF, La Rosa C, Kaltcheva T, et al. Characterization of immunologic properties of a second HLA-A2 epitope from a granule protease in CML patients and HLA-A2 transgenic mice. Blood. 2011;118:2159–2169. doi: 10.1182/blood-2011-04-349951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molldrem J, Dermime S, Parker K, et al. Targeted T-cell therapy for human leukemia: cytotoxic T lymphocytes specific for a peptide derived from proteinase 3 preferentially lyse human myeloid leukemia cells. Blood. 1996;88:2450–2457. [PubMed] [Google Scholar]
- 41.Anguille S, Van Tendeloo VF, Berneman ZN. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia. 2012;26:2186–2196. doi: 10.1038/leu.2012.145. [DOI] [PubMed] [Google Scholar]
- 42.Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a National Cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Driessche A, Berneman ZN, Van Tendeloo VF. Active specific immunotherapy targeting the Wilms’ tumor protein 1 (WT1) for patients with hematological malignancies and solid tumors: lessons from early clinical trials. Oncologist. 2012;17:250–259. doi: 10.1634/theoncologist.2011-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maslak PG, Dao T, Krug LM, et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood. 2010;116:171–179. doi: 10.1182/blood-2009-10-250993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brossart P, Schneider A, Dill P, et al. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 2001;61:6846–6850. [PubMed] [Google Scholar]
- 46.Burton J, Mishina D, Cardillo T, et al. Epithelial mucin-1 (MUC1) expression and MA5 anti-MUC1 monoclonal antibody targeting in multiple myeloma. Clin Cancer Res. 1999;5:3065s–3072s. [PubMed] [Google Scholar]
- 47.Brossart P, Heinrich KS, Stuhler G, et al. Identification of HLA-A2-restricted T-cell epitopes derived from the MUC1 tumor antigen for broadly applicable vaccine therapies. Blood. 1999;93:4309–4317. [PubMed] [Google Scholar]
- 48.Xing PX, Apostolopoulos V, Trapani J, et al. Peptide epitopes in breast cancer mucins. Adv Exp Med Biol. 1994;353:9–16. doi: 10.1007/978-1-4615-2443-4_2. [DOI] [PubMed] [Google Scholar]
- 49.Andrulis M, Ellert E, Mandel U, et al. Expression of Mucin-1 in multiple myeloma and its precursors: correlation with glycosylation and subcellular localization. Histopathology. 2014;64:799–806. doi: 10.1111/his.12330. [DOI] [PubMed] [Google Scholar]
- 50.Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother. 2005;54:187–207. doi: 10.1007/s00262-004-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat Rev Cancer. 2004;4:371–380. doi: 10.1038/nrc1365. [DOI] [PubMed] [Google Scholar]
- 52.Sherwood RA, Brent L, Rayfield LS. Presentation of alloantigens by host cells. Eur J Immunol. 1986;16:569–574. doi: 10.1002/eji.1830160519. [DOI] [PubMed] [Google Scholar]
- 53.Korngold R, Sprent J. Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T cells from marrow. J Exp Med. 1978;148:1687–1698. doi: 10.1084/jem.148.6.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Bueger M, Bakker A, Van Rood JJ, et al. Tissue distribution of human minor histocompatibility antigens. Ubiquitous versus restricted tissue distribution indicates heterogeneity among human cytotoxic T lymphocyte-defined non-MHC antigens. J Immunol. 1992;149:1788–1794. [PubMed] [Google Scholar]
- 55.Warren EH, Greenberg PD, Riddell SR. Cytotoxic T-lymphocyte-defined human minor histocompatibility antigens with a restricted tissue distribution. Blood. 1998;91:2197–2207. [PubMed] [Google Scholar]
- 56.Marijt WA, Heemskerk MH, Kloosterboer FM, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci U S A. 2003;100:2742–2747. doi: 10.1073/pnas.0530192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mommaas B, Kamp J, Drijfhout JW, et al. Identification of a novel HLA-B60-restricted T cell epitope of the minor histocompatibility antigen HA-1 locus. J Immunol. 2002;169:3131–3136. doi: 10.4049/jimmunol.169.6.3131. [DOI] [PubMed] [Google Scholar]
- 58.den Haan JM, Meadows LM, Wang W, et al. The minor histocompatibility antigen HA-1: a diallelic gene with a single amino acid polymorphism. Science. 1998;279:1054–1057. doi: 10.1126/science.279.5353.1054. [DOI] [PubMed] [Google Scholar]
- 59.Dolstra H, de Rijke B, Fredrix H, et al. Bi-directional allelic recognition of the human minor histocompatibility antigen HB-1 by cytotoxic T lymphocytes. Eur J Immunol. 2002;32:2748–2758. doi: 10.1002/1521-4141(2002010)32:10<2748::AID-IMMU2748>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 60.Brickner AG, Evans AM, Mito JK, et al. The PANE1 gene encodes a novel human minor histocompatibility antigen that is selectively expressed in B-lymphoid cells and B-CLL. Blood. 2006;107:3779–3786. doi: 10.1182/blood-2005-08-3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spierings E, Kim YH, Hendriks M, et al. Multicenter analyses demonstrate significant clinical effects of minor histocompatibility antigens on GvHD and GvL after HLA-matched related and unrelated hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19:1244–1253. doi: 10.1016/j.bbmt.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 62.van Bergen CA, Kester MG, Jedema I, et al. Multiple myeloma-reactive T cells recognize an activation-induced minor histocompatibility antigen encoded by the ATP-dependent interferon-responsive (ADIR) gene. Blood. 2007;109:4089–4096. doi: 10.1182/blood-2006-08-043935. [DOI] [PubMed] [Google Scholar]
- 63.de Rijke B, van Horssen-Zoetbrood A, Beekman JM, et al. A frameshift polymorphism in P2X5 elicits an allogeneic cytotoxic T lymphocyte response associated with remission of chronic myeloid leukemia. J Clin Invest. 2005;115:3506–3516. doi: 10.1172/JCI24832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng X, Hui KM, Younes HM, Brickner AG. Targeting minor histocompatibility antigens in graft versus tumor or graft versus leukemia responses. Trends Immunol. 2008;29:624–632. doi: 10.1016/j.it.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hobo W, Broen K, van der Velden WJ, et al. Association of disparities in known minor histocompatibility antigens with relapse-free survival and graft-versus-host disease after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19:274–282. doi: 10.1016/j.bbmt.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goulmy E, Schipper R, Pool J, et al. Mismatches of minor histocompatibility antigens between HLA-identical donors and recipients and the development of graft-versus-host disease after bone marrow transplantation. N Engl J Med. 1996;334:281–285. doi: 10.1056/NEJM199602013340501. [DOI] [PubMed] [Google Scholar]
- 67.Lin MT, Gooley T, Hansen JA, et al. Absence of statistically significant correlation between disparity for the minor histocompatibility antigen-HA-1 and outcome after allogeneic hematopoietic cell transplantation. Blood. 2001;98:3172–3173. doi: 10.1182/blood.v98.10.3172. [DOI] [PubMed] [Google Scholar]
- 68.Fanning SL, Zilberberg J, Stein J, et al. Unraveling graft-versus-host disease and graft-versus-leukemia responses using TCR Vbeta spectratype analysis in a murine bone marrow transplantation model. J Immunol. 2013;190:447–457. doi: 10.4049/jimmunol.1201641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolpert EZ, Grufman P, Sandberg JK, et al. Immunodominance in the CTL response against minor histocompatibility antigens: interference between responding T cells, rather than with presentation of epitopes. J Immunol. 1998;161:4499–4505. [PubMed] [Google Scholar]
- 70.Wolpert E, Franksson L, Karre K. Dominant and cryptic antigens in the MHC class I restricted T cell response across a complex minor histocompatibility barrier: analysis and mapping by elution of cellular peptides. Int Immunol. 1995;7:919–928. doi: 10.1093/intimm/7.6.919. [DOI] [PubMed] [Google Scholar]
- 71.Sekimata M, Griem P, Egawa K, et al. Isolation of human minor histocompatibility peptides. Int Immunol. 1992;4:301–304. doi: 10.1093/intimm/4.2.301. [DOI] [PubMed] [Google Scholar]
- 72.de Bueger M, Verreck F, Blokland E, et al. Isolation of an HLA-A2.1 extracted human minor histocompatibility peptide. Eur J Immunol. 1993;23:614–618. doi: 10.1002/eji.1830230305. [DOI] [PubMed] [Google Scholar]
- 73.Kawakami Y, Fujita T, Matsuzaki Y, et al. Identification of human tumor antigens and its implications for diagnosis and treatment of cancer. Cancer Sci. 2004;95:784–791. doi: 10.1111/j.1349-7006.2004.tb02182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Warren EH, Otterud BE, Linterman RW, et al. Feasibility of using genetic linkage analysis to identify the genes encoding T cell-defined minor histocompatibility antigens. Tissue Antigens. 2002;59:293–303. doi: 10.1034/j.1399-0039.2002.590407.x. [DOI] [PubMed] [Google Scholar]
- 75.Spaapen RM, Lokhorst HM, van den Oudenalder K, et al. Toward targeting B cell cancers with CD4+ CTLs: identification of a CD19-encoded minor histocompatibility antigen using a novel genome-wide analysis. J Exp Med. 2008;205:2863–2872. doi: 10.1084/jem.20080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spaapen RM, de Kort RA, van den Oudenalder K, et al. Rapid identification of clinical relevant minor histocompatibility antigens via genome-wide zygosity-genotype correlation analysis. Clin Cancer Res. 2009;15:7137–7143. doi: 10.1158/1078-0432.CCR-09-1914. [DOI] [PubMed] [Google Scholar]
- 77.Van Bergen CA, Rutten CE, Van Der Meijden ED, et al. High-throughput characterization of 10 new minor histocompatibility antigens by whole genome association scanning. Cancer Res. 2010;70:9073–9083. doi: 10.1158/0008-5472.CAN-10-1832. [DOI] [PubMed] [Google Scholar]
- 78.Griffioen M, Honders MW, van der Meijden ED, et al. Identification of 4 novel HLA-B*40:01 restricted minor histocompatibility antigens and their potential as targets for graft-versus-leukemia reactivity. Haematologica. 2012;97:1196–1204. doi: 10.3324/haematol.2011.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Siewert K, Malotka J, Kawakami N, et al. Unbiased identification of target antigens of CD8+ T cells with combinatorial libraries coding for short peptides. Nat Med. 2012;18:824–828. doi: 10.1038/nm.2720. [DOI] [PubMed] [Google Scholar]
- 80.Hombrink P, Hassan C, Kester MG, et al. Discovery of T cell epitopes implementing HLA-peptidomics into a reverse immunology approach. J Immunol. 2013;190:3869–3877. doi: 10.4049/jimmunol.1202351. [DOI] [PubMed] [Google Scholar]
- 81.Maecker B, von B-B, Anderson KS, et al. Linking genomics to immunotherapy by reverse immunology–’immunomics’ in the new millennium. Curr Mol Med. 2001;1:609–619. doi: 10.2174/1566524013363447. [DOI] [PubMed] [Google Scholar]
- 82.Schuler MM, Nastke MD, Stevanovikc S. SYFPEITHI: database for searching and T-cell epitope prediction. Methods Mol Biol. 2007;409:75–93. doi: 10.1007/978-1-60327-118-9_5. [DOI] [PubMed] [Google Scholar]
- 83.Schuler MM, Donnes P, Nastke MD, et al. SNEP: SNP-derived epitope prediction program for minor H antigens. Immunogenetics. 2005;57:816–820. doi: 10.1007/s00251-005-0054-5. [DOI] [PubMed] [Google Scholar]
- 84.Shen WJ, Zhang S, Wong HS. An effective and effecient peptide binding prediction approach for a broad set of HLA-DR molecules based on ordered weighted averaging of binding pocket profiles. Proteome Sci. 2013;11:S15. doi: 10.1186/1477-5956-11-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang L, Chen Y, Wong HS, et al. TEPITOPEpan: extending TEPITOPE for peptide binding prediction covering over 700 HLA-DR molecules. PLoS One. 2012;7:e30483. doi: 10.1371/journal.pone.0030483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Popovic J, Li LP, Kloetzel PM, et al. The only proposed T-cell epitope derived from the TEL-AML1 translocation is not naturally processed. Blood. 2011;118:946–954. doi: 10.1182/blood-2010-12-325035. [DOI] [PubMed] [Google Scholar]
- 87.Hombrink P, Hadrup SR, Bakker A, et al. High-throughput identification of potential minor histocompatibility antigens by MHC tetramer-based screening: feasibility and limitations. PLoS One. 2011;6:e22523. doi: 10.1371/journal.pone.0022523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larsen MV, Lundegaard C, Lamberth K, et al. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol. 2005;35:2295–2303. doi: 10.1002/eji.200425811. [DOI] [PubMed] [Google Scholar]
- 89.Feldhahn M, Donnes P, Schubert B, et al. miHA-Match: computational detection of tissue-specific minor histocompatibility antigens. J Immunol Methods. 2012;386:94–100. doi: 10.1016/j.jim.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 90.Nielsen M, Lundegaard C, Blicher T, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PloS One. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ivanov R, Hol S, Aarts TI, et al. T cell receptor-transgenic primary T cells as a tool for discovery of leukaemia-associated antigens. Clin Exp Immunol. 2006;143:78–84. doi: 10.1111/j.1365-2249.2005.02967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Willemsen RA, Debets R, Chames P, Bolhuis RL. Genetic engineering of T cell specificity for immunotherapy of cancer. Hum Immunol. 2003;64:56–68. doi: 10.1016/s0198-8859(02)00730-9. [DOI] [PubMed] [Google Scholar]
- 94.Friedman TM, Goldgirsh K, Berger SA, et al. Overlap between in vitro donor antihost and in vivo posttransplantation TCR Vbeta use: a new paradigm for designer allogeneic blood and marrow transplantation. Blood. 2008;112:3517–3525. doi: 10.1182/blood-2008-03-145391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jones SC, Friedman TM, Murphy GF, Korngold R. Specific donor Vbeta-associated CD4 T-cell responses correlate with severe acute graft-versus-host disease directed to multiple minor histocompatibility antigens. Biol Blood Marrow Transplant. 2004;10:91–105. doi: 10.1016/j.bbmt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 96.Margolis DA, Casper JT, Segura AD, et al. Infiltrating T cells during liver graft-versus-host disease show a restricted T-cell repertoire. Biol Blood Marrow Transplant. 2000;6:408–415. doi: 10.1016/s1083-8791(00)70017-6. [DOI] [PubMed] [Google Scholar]
- 97.Liu C, He M, Rooney B, et al. Longitudinal analysis of T-cell receptor variable beta chain repertoire in patients with acute graft-versus-host disease after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2006;12:335–345. doi: 10.1016/j.bbmt.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 98.Patterson AE, Korngold R. Infusion of select leukemia-reactive TCR Vbeta+ T cells provides graft-versus-leukemia responses with minimization of graft-versus-host disease following murine hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2001;7:187–196. doi: 10.1053/bbmt.2001.v7.pm11349805. [DOI] [PubMed] [Google Scholar]
- 99.Friedman TM, Jones SC, Statton D, et al. Evolution of responding CD4+ and CD8+ T-cell repertoires during the development of graft-versus-host disease directed to minor histocompatibility antigens. Biol Blood Marrow Transplant. 2004;10:224–235. doi: 10.1016/j.bbmt.2003.12.303. [DOI] [PubMed] [Google Scholar]
- 100.Gerlinger M, Quezada SA, Peggs KS, et al. Ultra-deep T cell receptor sequencing reveals the complexity and intratumour heterogeneity of T cell clones in renal cell carcinomas. J Pathol. 2013;231:424–432. doi: 10.1002/path.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Emerson RO, Sherwood AM, Rieder MJ, et al. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J Pathol. 2013;231:433–440. doi: 10.1002/path.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sherwood AM, Emerson RO, Scherer D, et al. Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer Immunol Immunother. 2013;62:1453–1461. doi: 10.1007/s00262-013-1446-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011;29:550–557. doi: 10.1016/j.tibtech.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rezvani K. Posttransplantation vaccination: concepts today and on the horizon. Hematology Am Soc Hematol Educ Program. 2011;2011:299–304. doi: 10.1182/asheducation-2011.1.299. [DOI] [PubMed] [Google Scholar]