

Abstract
The CLN2 form of neuronal ceroid lipofuscinosis is an autosomal recessively inherited lysosomal storage disease that is characterized by progressive vision loss culminating in blindness, cognitive and motor decline, neurodegeneration, and premature death. CLN2 disease results from mutations in the gene that encodes the soluble lysosomal enzyme tripeptidyl peptidase-1. A null mutation in the TPP1 gene encoding this enzyme causes a CLN2-like disease in Dachshunds. Dachshunds that are homozygous for this mutation serve as a model for human CLN2 disease, exhibiting clinical signs and neuropathology similar to those of children with this disorder. Affected dogs reach end-stage terminal disease status at 10–11 months of age. In addition to retinal changes typical of CLN2 disease, a retinopathy consisting of multifocal, bullous retinal detachment lesions was identified in 65% of (TPP1−/−) dogs in an established research colony. These lesions did not occur in littermates that were heterozygous or homozygous for the normal TPP1 allele. Retinal changes and the functional effects of this multifocal retinopathy were examined objectively over time using ophthalmic examinations, fundus photography, electroretinography (ERG), quantitative pupillary light response (PLR) recording, fluorescein angiography, optical coherence tomography (OCT) and histopathology. The retinopathy consisted of progressive multifocal serous retinal detachments. The severity of the disease-related retinal thinning was no more serious in most detached areas than in adjacent areas of the retina that remained in close apposition to the retinal pigment epithelium. The retinopathy observed in these dogs was somewhat similar to canine multifocal retinopathy (CMR), a disease caused by a mutation of the bestrophin gene BEST1. ERG a-wave amplitudes were relatively preserved in the Dachshunds with CLN2 disease, whether or not they developed the multifocal retinopathy. The retinopathy also had minimal effects on the PLR. Histological evaluation indicated that the CLN2 disease-related retinal degeneration was not exacerbated in areas where the retina was detached except where the detached areas were very large. DNA sequence analysis ruled out a mutation in the BEST1 exons or splice junctions as a cause for the retinopathy. Perfect concordance between the TPP1 mutation and the retinopathy in the large number of dogs examined indicates that the retinopathy most likely occurs as a direct result of the TPP1 mutation. Therefore, inhibition of the development and progression of these lesions can be used as an indicator of the efficacy of therapeutic interventions currently under investigation for the treatment of CLN2 disease in the Dachshund model. In addition, these findings suggest that TPP1 mutations may underlie multifocal retinopathies of unknown cause in animals and humans.
Keywords: dog, canine, neuronal ceroid lipofuscinosis, bullous retinopathy, retinal detachment, TPP1, CLN2, BEST1
1. Introduction
The CLN2 form of neuronal ceroid lipofuscinosis (NCL) is an autosomal recessive lysosomal storage disease characterized by progressive vision loss culminating in blindness, declines in cognitive and motor function, degeneration of the brain and retina, seizures and premature death (Mole et al., 2011). In human patients, 14 genetically distinct forms of NCL have been identified (Warrier et al., 2013). Histologically the NCLs are characterized by accumulation of intracellular autofluorescent storage bodies in nervous tissues, including the retina, as well as in other tissues (Haltia, 2006). NCLs have been reported in numerous dog breeds, and of the canine NCLs, eight causative mutations have been identified in the canine orthologs of genes that contain NCL-causing mutations in human patients (Awano et al., 2006a; Awano et al., 2006b; Farias et al., 2011; Guo et al., 2014; Guo et al., 2015; Katz et al., 2011; Katz et al., 2005a; Melville et al., 2005; Sanders et al., 2010). A form of NCL that results from a null mutation in the TPP1 gene encoding the lysosomal storage enzyme tripeptidyl-peptidase-1 (TPP1) was discovered in miniature longhaired Dachshunds (Awano et al., 2006a). TPP1 is orthologous to the human CLN2 gene, mutations in which cause CLN2 disease, a late-infantile onset form of NCL. CLN2 disease is usually characterized by early onset of neurological signs (2–4 years of age) progressive vision loss and death typically by the teenage years (Mole et al., 2011).
Dachshunds that are homozygous for the TPP1 null mutation (TPP1−/−) develop clinical signs similar to those in children with CLN2 disease. Affected Dachshunds reach end-stage disease status at 10–11 months of age (Katz et al., 2014). These TPP−/− dogs have normal visual behavior and retinal function at 3 months, show clear vision impairment and ERG deficits at 7 months, and suffer continued visual and neurologic decline through 10 months of age (Katz et al., 2008; Katz et al., 2014; Whiting et al., 2013a). Histological examination of the retina in affected dogs revealed inner nuclear layer cell loss, thinning of the inner plexiform layer, reduced photoreceptor cell density and the accumulation of autofluorescent storage material in ganglion cells and adjacent to the outer limiting membrane (Katz et al., 2008). The TPP1−/− dog has been adopted as a model for human CLN2 disease and has been bred extensively for laboratory studies since the initial retinal investigative study (Awano et al., 2006a; Katz et al., 2008; Katz et al., 2014; Sanders et al., 2011; Vuillemenot et al., 2011; Whiting et al., 2013a). Therapeutic intervention studies with this dog model served as the basis for a human clinical trial of an enzyme replacement therapy treatment for the CLN2 disease that is currently under way. During routine retinal examination of purpose bred TPP1−/− dogs, a retinopathy consisting of multifocal bullous retinal detachment lesions was identified. This study was conducted to document the objective retinal changes and the functional effects of the multifocal retinopathy in TPP1−/− dogs utilizing fundus photography, electroretinography, quantitative pupillary light response (PLR) recording, fluorescein angiography, optical coherence tomography (OCT) and histopathology.
2. Materials and Methods
2.1. Animals
Purpose-bred miniature longhaired Dachshunds housed in a research facility at the University of Missouri were used in this study. The colony was established by mating two Dachshunds, both heterozygous for a one nucleotide deletion in TPP1 (c.325delC) that causes a shift in the reading frame and a premature stop codon (Awano et al., 2006a). Tissues from affected dogs do not have detectable levels of TPP1 enzyme activity (Awano et al., 2006a). Most breeding consisted of carrier to carrier crosses but also consisted of normal-carrier and affected-carrier crosses. Homozygous normal dogs (TPP1+/+) (n=26), heterozygous carriers (TPP1+/−) (n=7) and affected mutants (n=35) were included in the study. Dogs received routine husbandry care, were maintained on a 12:12 daily light cycle and were socialized daily. All studies were performed in compliance with the EU Directive 2010/63/EU for animal experiments and were approved by the University of Missouri Animal Care and Use Committee. Seventeen dogs were concurrently enrolled in an ongoing gene therapy study (12 affected, 2 carriers and 3 normal dogs) (unpublished). Twenty six dogs were being used in enzyme replacement therapy studies (12 affected and 14 normal dogs) (Katz et al., 2014). Twenty five dogs received no treatment (11 affected, 5 carrier and 9 normal dogs). All affected dogs were followed until they reached end-stage disease at which time they were euthanized. Although untreated dogs reached end- stage between 10 and 11 months of age, dogs receiving either enzyme replacement therapy (Katz et al., 2014) or gene therapy (unpublished) survived up to 19 months of age.
2.2. Clinical examinations and in vivo retinal imaging
All dogs received a complete ophthalmic examination at 3 months of age prior to inclusion in the study. Animals were excluded from the study if they exhibited any retinal abnormalities that would compromise vision prior to the onset of retinal or neurological disease due to CLN2 disease. Ophthalmic examination was performed monthly on TPP1 −/− dogs and every other month for TPP1+/+ and TPP1 +/− dogs. Examination consisted of vision-mediated behavior assessment, slit lamp biomicroscopy and indirect ophthalmoscopy. Slit lamp biomicroscopy was performed prior to dilation (SL14; Kowa Col Ltd., Tokyo, Japan). Pupils were dilated with 1% tropicamide (Alcon, Fort Worth, TX) prior to performing indirect ophthalmoscopy with an indirect wireless headset (12,500; Welch Allyn Inc., Skaneateles Falls, NY, USA) and a handheld lens (30 diopter clear lens, Volk Optical Inc., Mentor, OH). Fundus photographs were taken following examination (NM-100; Nidek Co. Ltd., Freemont, CA) and archived electronically. Dogs were evaluated for retinopathy severity during each examination and a grading scheme was established as follows: Grade 1 indicating that less than 15% of the retina was affected by lesions; Grade 2 indicating that greater than 15% but less than 30% of the retina was affected by lesions; and Grade 3 indicating that 30% or greater of the retina was affected by lesions.
All retinal examinations and grading of lesion status were done by the same veterinary ophthalmologist (JWP) in order to maintain consistency. The accuracy of the grading system was confirmed by quantitative measurements of the areas of the fundus occupied by the lesions in cSLO images from the majority of the samples. A masked investigator manually traced the outlines of the lesions in the cSLO images and Image J software was used to measure the total area of the lesions and the total area of the retina from which the lesions were analyzed.
In vivo imaging was performed using a combined confocal scanning laser ophthalmoscope/spectral-domain optical coherence tomography (cSLO/sdOCT) instrument (Spectralis HRA/OCT, Heidelberg, Germany). Dogs were dilated with 1% tropicamide (Alcon, Fort Worth, TX) and placed under general anesthesia for cSLO/sdOCT imaging and fluorescein angiography. The cSLO imaging was performed using the 55° lens. The sdOCT scans were done with the 30° lens in groups of parallel linear scans of 40–50 sequential B-scans each separated by 200 μm in the superior-temporal retinal quadrant OS. Fluorescein angiography was performed on one affected dog without retinopathy lesions and on 3 affected dogs with retinopathy lesions (n=1 for Grade 1, n=1 for Grade 2 and n=1 for Grade 3). For fluorescein angiography all dogs received maropitant citrate (1.0 mg/kg SQ) and diphenhydramine (2.0 mg/kg SQ) 20 minutes prior to the procedure. Standard cSLO fundus images were obtained prior to the administration of 10% sodium fluorescein (20 mg/kg IV) and real-time cSLO images were obtained throughout all phases of fluorescein passage.
2.3. Electroretinography
Bilateral electroretinogram (ERG) evaluations were performed as previously described (Whiting et al., 2013a) on a selection of untreated TPP1−/− dogs at either 8 months or 10 months of age. An ERG was obtained from eleven 8 month-old TPP1−/− dogs [Grade 0 (n=6), Grade 1 (n=3), Grade 2 (n=1), Grade 3 (n=1)] and from eight 10 month-old TPP1−/− dogs [Grade 0 (n=3), Grade 1 (n=3), Grade 2 (n=1), Grade 3 (n=1)]. ERG evaluation was performed within one week after the PLR recording session. Dogs were prepared for ERG recording in ambient room light. Prior to recording, both pupils were dilated with 1% tropicamide, and dogs were deeply sedated with intramuscular administration of dexmedetomidine (30 – 40 μg/kg).
ERGs were bilaterally elicited and simultaneously recorded with a portable unit (HMsERG model 2000; RetVet Corp., Columbia, MO). The right and left mini-Ganzfeld domes were positioned approximately 2 cm from the corresponding eye. Each ERG session consisted of scotopic and photopic recordings in accordance with the Dog Diagnostic Protocol, recommended by the European College of Veterinary Ophthalmology, primarily for evaluation of rod and cone function (Ekesten et al., 2013). During a 20 minute period of dark adaptation, scotopic low-intensity rod responses were elicited at a stimulus intensity of 10.2 log photons/cm2/s (0.01 cd.s/m2) with 4 minutes of dark adaptation between recordings. Following this, scotopic responses were elicited using flashes of 12.65 and 13.2 log photons/cm2/s (3 cd.s/m2 and 10 cd.s/m2) to evaluate mixed rod and cone function. The eyes were then exposed to diffuse white light at a luminance of 13.65 log photons/cm2/s (30 cd/m2) for 10 minutes, immediately after which responses to single 12.65 log photons/cm2/s (3 cd.s/m2) flash stimuli were recorded. This was immediately followed by evaluation of responses to 30-Hz photopic flicker stimuli at the same light intensity. ERG waveforms in all recordings were evaluated, and the amplitudes and implicit times for the a- and b-waves were measured as previously described (Whiting et al., 2013a).
2.4. Pupillary light response recordings
PLR recordings were performed at 8 months and 10 months in a selection of untreated TPP1−/− dogs. The PLR recording was performed at 8 months in eleven TPP1−/− dogs [Grade 0 (n=6), Grade 1 (n=3), Grade 2 (n=1), Grade 3 (n=1)] and at 10 months in eight TPP1−/− dogs [Grade 0 (n=3), Grade 1 (n=3), Grade 2 (n=1), Grade 3 (n=1)]. All recordings were done during the light period of the daily 12:12 light-dark cycle. The detailed methods for obtaining the PLRs have been described previously (Whiting et al., 2013b). Dogs were kept in dim light (0.9 lux) for at least 1 hour, including preparation time, and in complete darkness for 10 minutes prior to recording. After 30 minutes of dim light adaptation, dogs were pre-medicated with dexmedetomidine (20–25 μg/kg IM) prior to induction of anesthesia with propofol [IV to effect, 1.49 ± 0.59 mg/kg (mean ± SD); PropoFlo 28, Abbott Laboratories, Abbott Park, IL]. Dogs were intubated with a cuffed endotracheal tube and anesthesia maintained with isoflurane (1.5% vaporizer setting; Terrell, Piramal Healthcare, Boise, ID) in oxygen. The cornea of the right eye was anesthetized with proparacaine and a lid speculum was inserted to ensure that the nictitating membrane and eyelids did not interfere with light exposure or visualization of the pupil. In addition, a small stay suture was placed in the bulbar conjunctiva on the central axis approximately 5 mm superior to the limbus to facilitate globe manipulation to maintain centration of the pupil on the optical axis of the recording apparatus. The eye was regularly lubricated with saline eye wash solution throughout the procedure.
Recordings were performed with a custom apparatus (Whiting et al., 2013b) which provided timed delivery of a visible light stimulus from a mounted high-power broad spectrum LED (MCWHL2; Thorlabs Inc., Newton, NJ) and concurrently recorded pupil images at 30 frames per second using an infrared-sensitive camera (PC164CEX-2; Supercircuits Inc., Austin, TX) under continuous infrared illumination (880 nm LED) for visualization of the eye. The direct PLR of the right eye was evaluated using a standardized protocol of 100 millisecond flashes of broad spectrum white light at each of 10 intensities between 8 and 15 log photons/cm2/s (0.1 to 1400 lux).
Pupil images were analyzed using the batch processing feature in Photoshop (Adobe Systems Inc; San Jose, CA). A list of image frame number and corresponding pupil area was exported to a spreadsheet and used to calculate desired parameters. Area measurements were converted from pixels to mm2 based on the known size of the lid speculum present in each pupil image (Whiting et al., 2013b).
For the studies described here, baseline pupil area was the average pupil area, in a dark- adapted dog, over a 1-second period before the light stimulus. Baseline pupil diameter was calculated from area measurements assuming a round pupil. PLR constriction amplitude was defined as the difference between baseline pupil area and minimum pupil area attained following the light stimulus. These values were then converted to a percentage of baseline pupil size.
2.5. Histological analysis
Globes of TPP1 −/− animals were enucleated shortly after euthanasia, and the corneas were removed immediately. The samples were then placed either in a fixative consisting of 2.0% glutaraldehyde, 1.2% paraformaldehyde, 0.12 M Na-cacodylate, 0.5 mM CaCl2, pH 7.4 (EM fix) or in 0.1% glutaraldehyde, 3.5% paraformaldehyde, 0.13 M sodium cacodylate, and 0.13 mM CaCl2; pH 7.40 (FM fix). Within an hour of enucleation, the iris, lens and vitreous were removed from each globe and the remainder of the eye was then incubated in the fixative with gentle agitation for at least 24 hours. Samples fixed in FM fix were transferred to 0.17 M sodium cacodylate, pH 7.4 after 24 hours in the fixative. After initial fixation in EM fix, a region of the eyecup adjacent to the optic nerve head along the equator of the eye was dissected from each sample and processed for light and electron microscopy as described previously (Katz et al., 2005b). Sections of the samples were cut at a thickness of 0.5 μm, mounted on glass slides, and stained with toluidine blue. Digital photographs of the sections were obtained using conventional transmitted light microscopy. Samples fixed in FM fix were processed for cryostat sectioning and 8 μm thick cryostat sections were examined with fluorescence microscopy as described previously (Katz et al., 2008).
2.6. BEST1 sequencing
DNA samples from a control dog and from a TPP1 −/− dog with multifocal retinopathy were PCR-amplified with primer pairs flanking each of the 11 BEST1 coding exons. Table 1 lists the PCR primer sequences and the expected sizes of the resulting amplicons. The PCR amplifications were performed using a GoTaq® enzyme and buffer kit (Promega, Madison, WS). Amplifications were performed with a theromocycler using 40 cycles of 95 °C for 30 seconds, 60 °C for 30 seconds, followed by an additional 30 seconds at 72 °C. The resulting amplicons were evaluated with a microcapillary electrophoresis system (eGene, Qiagen Inc., Alameda, CA). The amplicons were purified with a spin column (QIAquick PCR Purification Kit, Qiagen Inc., Alameda, CA) and sequenced in both directions with an automated DNA sequencer (Model 3730xl, Applied Biosystems, Foster City, CA). DNA sequence analysis software (Sequencher®4.10.1, Gene Codes Corporation, Ann Arbor, MI 48108) was used to compare the nucleotide sequences from individual dogs to each other and to build 3.1 of the NCBI canine reference sequence.
Table 1.
PCR primer sequences and expected sizes of resulting amplicons for BEST1 sequencing.
| Target | Forward Primer / Reverse Primer | Amplicon Size (bp) |
|---|---|---|
| exon 1 | TCAGTGTACCTCCCTGTCTGT/ TGGCTATTTGTTCCAAGACCC | 282 |
| exon 2 | CAAAGAGGCAGCCACACTCCC/ CAGCCCTGAACTGCCGAGA | 282 |
| exon 3 | CGCGCTCCGGTCAAGGGAGATG/ GGTCCTTCGCCCGTCTGAAGCC | 387 |
| exon 4 | ATCCCGGCCACGCTGCCAA/ TCTACCCAGAGCACAACCGTCCC | 292 |
| exon 5 | GTGGTCAGAACCCCAACCCTC/ AAGTGAGCCCTTCCCATCACA | 201 |
| exon 6 | TCACAGAGGGCCAAAGTCAC/ TCAGCCATGACCCTTCTAAAGC | 336 |
| exon 7 | AACAGAAGTTTAAGGGCAAGG/ CACAGGACAGGCTCCGTCTC | 188 |
| exon 8 | CTTGGGCTGACTTTTCTGTTGGA/ CTTTCCTAAGTCCTTGCCCTTG | 264 |
| exon 9 (5′) | GTCCTGCTTTGGTCCTTTGTCC/ GCAGCAGTCTCAAAAGCACCC | 310 |
| exon 9 (center) | GCCAGCCCAAGAAACTCGG/ ACTCGACAGTTTTCCTCTTCACG | 342 |
| exon 9 (3′) | AGACCAGTTCTGAAGCACTCC/ TGGGTGTAAATCCAGCATCCTC | 386 |
| exon 10 | TCCAAGGTCCCCAAGTATGTCT/ TTTGACCACAGAAGTGACTGC | 294 |
| exon 11 | AACCTTTGCCCTAACTCCC/ TCCAGTGGTTGATTCCAGC | 338 |
2.7. Statistical analyses
All statistical tests were performed using SigmaPlot (Systat Software Inc., San Jose, CA). Data were subjected to the Shapiro-Wilk test to confirm normal distribution. One-way repeated measures analysis of variance was used to test for differences in total retinal thickness between lesion grades. For 8 month and 10 month ERG and PLR data, one way analysis of variance was used to test for differences due to lesion grade amongst age-matched CLN2 affected dogs. Follow-up pairwise comparisons were performed with the Holm-Sidak correction (α=0.05) to control family-wise error rate.
3. Results
3.1. Ophthalmic exams
Initial examination in all dogs revealed normal adnexal and ocular structures. Both TPP1+/+ and TPP1+/− dogs had normal ophthalmic examination findings throughout the study period. The TPP1−/− dogs in this study showed ophthalmic visual behavior and retinal changes consistent with previous reports (Katz et al., 2008; Katz et al., 2005b; Whiting et al., 2013a). In addition, the majority of TPP1−/− dogs developed a retinopathy consisting of progressive multifocal retinal detachment lesions. Twenty-three of thirty-five (65%) of theTPP1−/− dogs developed lesions between the ages of 5 and 12 months. The average age at onset was 7.5 months. Other than retinal changes, no other significant ophthalmic abnormalities were detected in the TPP1−/− dogs throughout the study period. Among the twenty-three dogs that developed lesions, twenty-one had Grade 1 lesions when first observed; the initial lesions for the other two dogs were Grade 2. Nine of the Grade 1 progressed to Grade 2 when the dogs were between 5 and 14 months old. The lesions in three of the dogs progressed to Grade 3 between 6 and 9 months old. The median time for progression from Grade 1 to Grade 2 was 82 days with a range of 23–230 days (n=9; note that two dogs first presented at Grade 2 and therefore did not progress from Grade 1 to Grade 2). Estimated median time for progression from Grade 2 to Grade 3 was 65 days with a range of 23–92 days (n=3). Representative cSLO fundus images for Grades 1, 2 and 3 are shown in Figure 1. In the early stages of the TPP1 retinopathy, the first focal detachments most often appear temporally. As the disease progresses, the appearance of additional detachments is not biased toward any specific area of the retina. This can be seen in Figure 1. Three affected dogs exhibited apparent resorption of serous fluid from under some of their bullous lesions between the ages of 7 and 18 months with an average age of 12.3 months as the retinal lesions appeared to flatten out over time. One dog with Grade 1 lesions developed a retinal hole in the center of a large retinal bullous lesion. As stated earlier, some of the dogs in this study received intraventricular infusions of recombinant TPP1 protein. This treatment had no effect on the development of the focal retinal detachments among the TPP1−/− dogs (Table 2).
Figure 1.
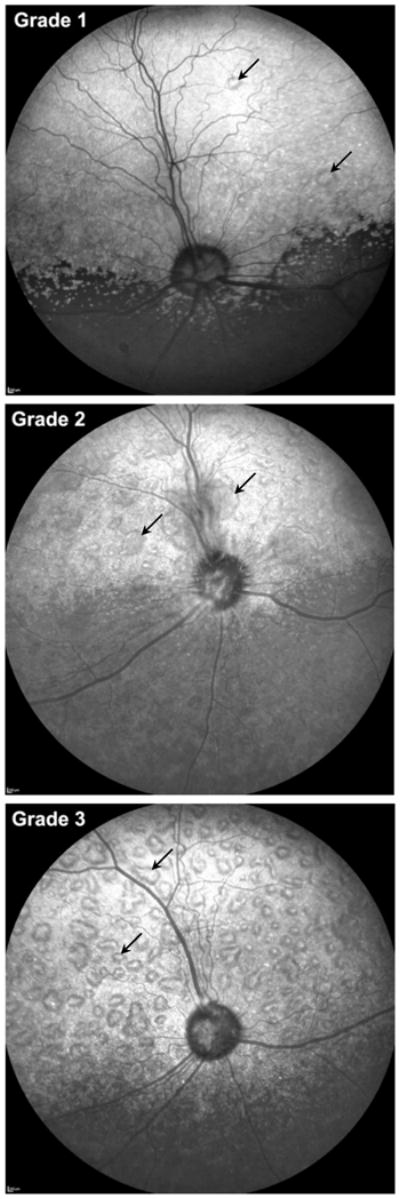
Representative combined scanning laser ophthalmoscopic (cSLO) fundus images of retinas exhibiting grades 1, 2 and 3 mutlifocal lesions. All images are from the left eye.
Table 2.
Relationship between age and CSF infusion of recombinant TPP1 on focal retinal detachment development
| Number of Dogs and Ages of Lesion Onset | |||
|---|---|---|---|
| Total | Untreated | Treated | |
| Number that developed lesions | 23 of 35 dogs | 8 of 11 dogs | 15 of 24 dogs |
| Age range of lesion onset (months) | 5 m to 12 m | 5 m to 10 m | 5 m to 12 m |
| Number and age range of progression from Grade 1 to Grade 2 | 9 dogs; 5 m to 14 m | 4 dogs; 5 m to 11 m | 5 dogs; 5 m to 14 m |
| Number and age range of progression from Grade 2 to Grade 3 | 3 dogs; 6 m to 9 m | 2 dogs; 8 m to 9 m | 1 dog; 6 m |
3.2. In vivo retinal imaging
Studies using sdOCT definitively confirmed that multifocal retinal lesions were serous retinal detachments (Figure 2). Lesions with apparent fluid resorption were also observed using sdOCT, which confirmed flattening of retinopathy lesions and thinning of the overlying retina in areas where lesions had been previously observed on ophthalmic examination (Figure 3). The sdOCT scans were performed directly on the localized flattened lesions once they were observed, prior sdOCT scans of the affected retinal area were not obtained. However, presence of typical raised lesions in the area had been previously documented on ophthalmic examination reports. Three total retinal thickness measurements were taken in normal appearing retinal regions adjacent to lesions from each patient at each grade via sdOCT archived scans. Lesions were defined as small [<0.5 disc diameter (DD)], large (>1 DD) and resorptive/flattened for retinal thickness measurements. Three retinal thickness measurements were taken for small and large lesions from dogs of each grade and for resorptive/flattened lesions via sdOCT archived scans. Results of retinal thickness measurements are summarized in Figure 4. Retinal thickness was not significantly affected in small or large lesions from Grades 1 and 2 or in small lesions from Grade 3. Retinal depth of large, Grade 3 lesions was significantly thinner than normal retinal depth without lesions present (p=0.012). Resorptive/flattened retinal lesions had significantly reduced retinal thickness compared to all other categories (p<0.005). Fundus autofluorescence imaging with the Spectralis instrument did not reveal any differences in the appearance of retinas with the lesions compared to retinas lacking the lesions from either normal dogs or dogs with CLN2 that did not develop the lesions.
Figure 2.
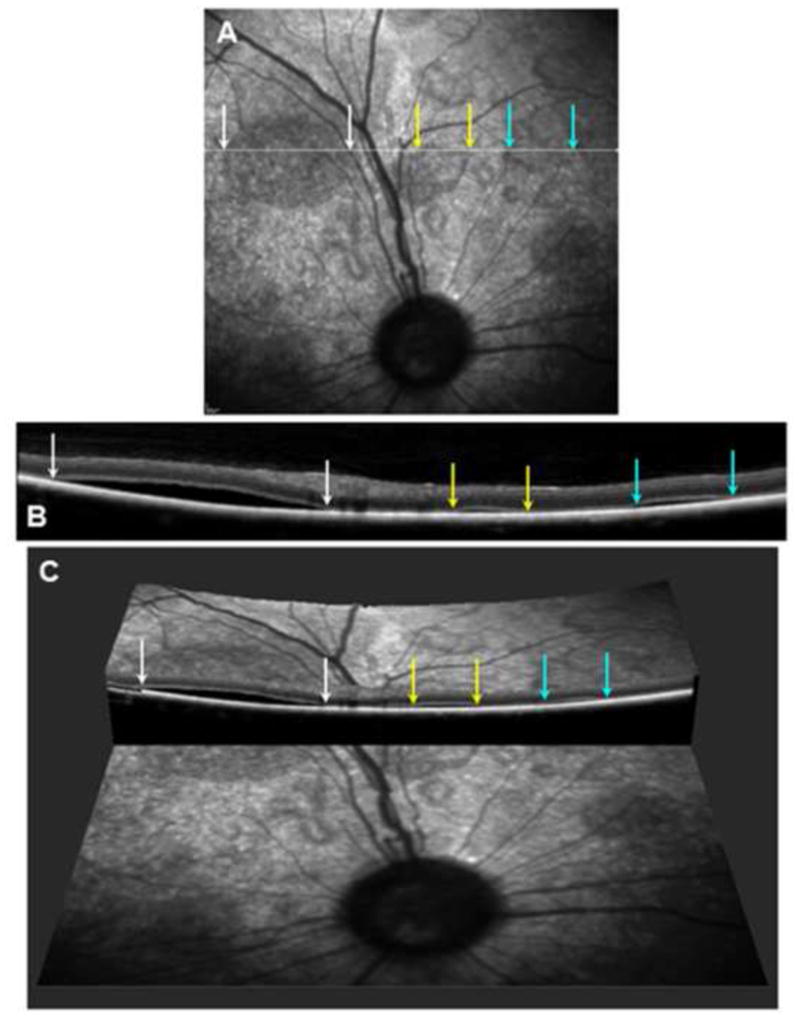
(A) Scanning laser ophthalmoscopic image of the fundus from a dog exhibiting multiple localized serous detachments. A scan line for the OCT image shown in (B) is shown in white. The edges of 3 of these lesions traversed by the scan line are indicated by colored arrows. (B) An OCT cross-section image of the retina from along the scan line shown in (A) with the edges of the same 3 lesions indicated by the same colored arrows. (C) A 3-dimensional reconstruction of the same area of the retina made by combining multiple OCT images showing the same 3 serous detachments.
Figure 3.
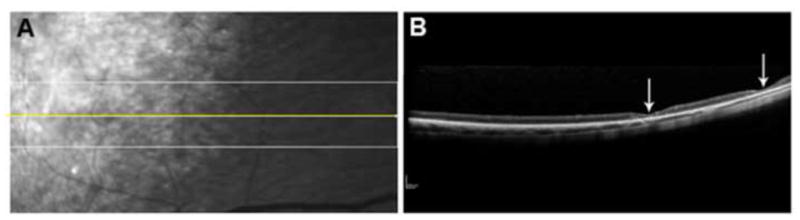
(A) An SLO image of the fundus of a 12.5 month old dog at the tapetal/nontapetal boundary. Yellow line shows the OCT scan line used to generate the image in (B). (B) sdOCT scan of resorptive/flattened lesions demonstrating evidence of focal thinning and degeneration of the overlying retina (arrows). These areas of thinning are not apparent in the SLO image.
Figure 4.
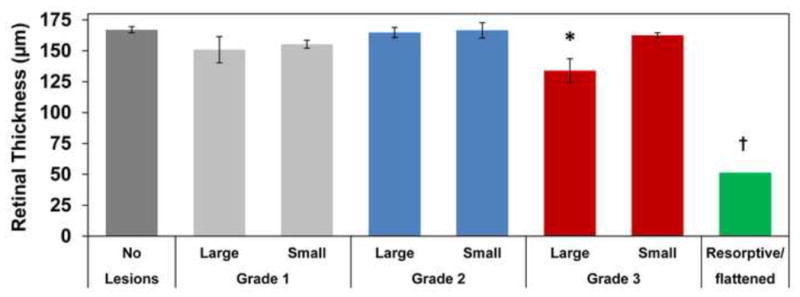
Retinal thickness in NCL-affected dogs. Lesions were defined as small [<0.5 disc diameter (DD)], large (>1 DD) and resorptive/flattened for retinal thickness measurements. Relative to areas where the retina remained attached to the RPE, retinal thickness was not affected except in large Grade 3 lesions. Retinal thickness in large, Grade 3 lesions was significantly thinner than retina without lesions (*p=0.012). Resorptive/flattened lesion retinal thickness was significantly reduced from all other categories (†, p<0.005).
Fluorescein angiography using cSLO real-time images revealed normal fluorescein passage throughout all phases. No obvious filling defects or changes were noted in any of the 3 dogs imaged (Figure 5).
Figure 5.
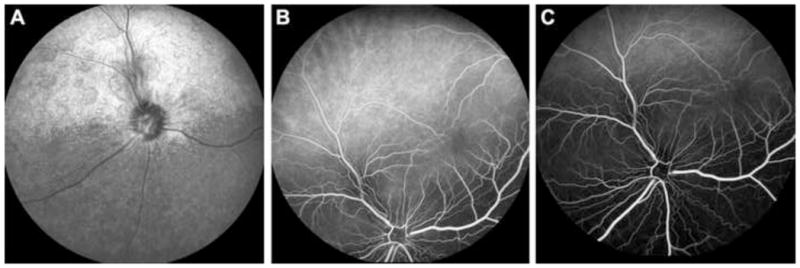
(A) cSLO fundus image of a dog with Grade 2 retinopathy lesions. (B) cSLO fluorescein angiogram of the same dog detailing transition between choroidal and arterial phases with no obvious leakage of fluorescein dye around lesions. (C) cSLO fluorescein angiogram of the same dog detailing late venous phase with no obvious leakage of fluorescein dye around lesions.
3.3. Retinal function tests (ERG and PLR)
The presence of concurrent retinal degenerative changes and multifocal retinal detachments in this study population represented a potential confounding factor for ERG analysis. Previous reports have shown significant reduction in the ERG b-wave amplitudes of TPP1−/− dogs by 6 months of age with relative preservation of a-wave amplitude (Katz et al., 2005b; Whiting et al., 2013a). In this study a-wave amplitude was selected and examined to compare retinal function in TPP1−/− dogs of various lesion statuses (no lesions and Grade 1–3 lesions), since the b-wave amplitude was already significantly decreased due to the CLN2 disease. No statistically significant differences in a-wave amplitudes were observed between age-matched groups of different lesion statuses (Figure 6). Due to the limited number of dogs available, the statistical power of the comparison was too low to rule out the possibility that the severity of the lesions did affect the ERG a-wave.
Figure 6.
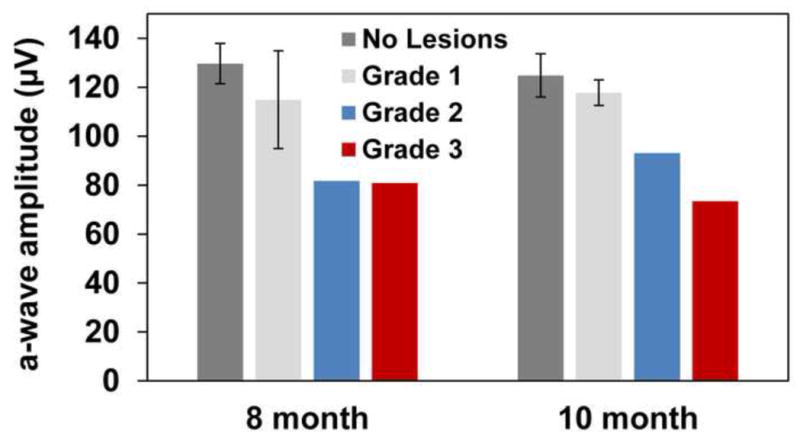
ERG a-wave amplitude by age and retinal lesion status. Measures are from a dark-adapted dog, in response to a stimulus of 13.2 log photons/cm2/s (10cd.s/m2). No statistically significant differences exist amongst the groups. However, due to the limited number of dogs available the statistical power of the comparison was too low to rule out the possibility that the severity of the lesions did affect the ERG a-wave.
The PLR constriction amplitude was not significantly different in TPP1 −/− dogs without lesions compared to those with Grade 1 or 2 lesions at 8 and 10 months of age or in 10 month old dogs with Grade 3 lesions at any stimulus intensity tested. However, for 8 month old dogs with Grade 3 lesions, PLR constriction amplitude was significantly reduced from that of other 8 month old affected dogs in response to a stimulus of 10 log photons/cm2/s (p=0.011) but not at other stimulus intensities examined (Figure 7).
Figure 7.
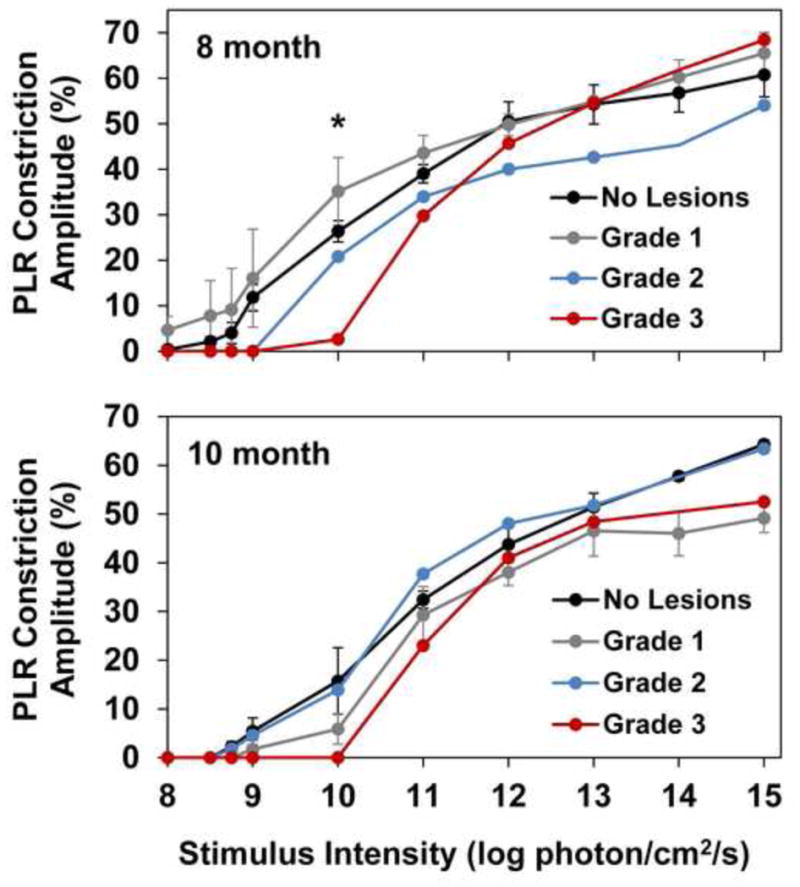
For 8 month old dogs with grade 3 lesions, PLR constriction amplitude is significantly reduced from that of other 8 month old affected dogs in response to a stimulus of 10 log photons/cm2/s (*p=0.011).
3.4. Histopathology
Light microscopic examination of the retinas confirmed the observation from OCT imaging that for most detachment lesions there were no apparent differences in morphology between the areas of the retina that had lifted away from the underlying retinal pigment epithelium (RPE) and adjacent areas of the retina where the normal apposition of the retina to the RPE was retained (Figure 8). In all of the affected dogs, the photoreceptor outer segments were reduced in length compared to those of normal retinas, and the inner nuclear and inner plexiform layers were reduced in thickness.
Figure 8.
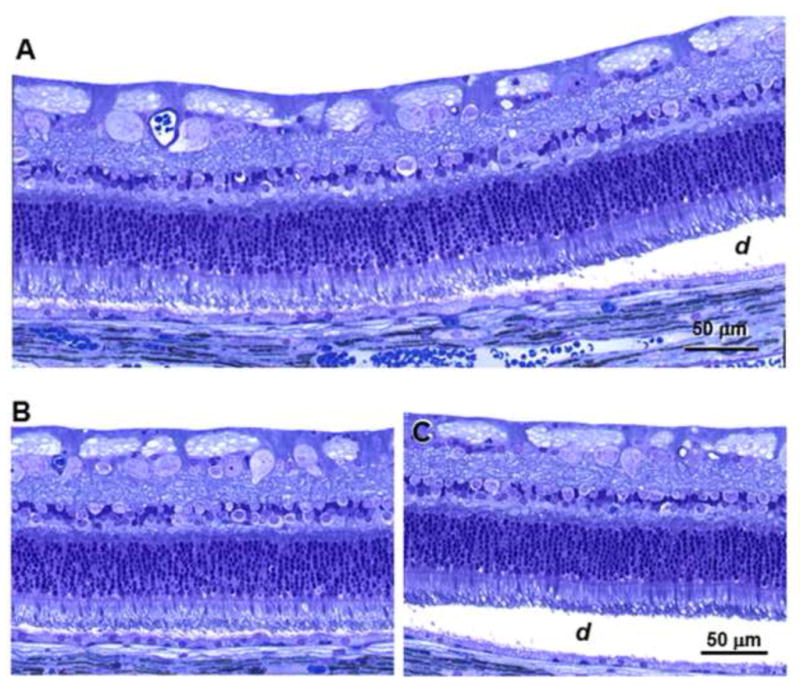
Light micrographs of an area of a retina near a grade 2 lesion. (A) The boundary between an area where the retina remains in close apposition to the RPE and the edge of the detached region (d). (B) An area from the same retina distant from the region of detachment. (C) An area of the retina within the region of detachment. Bar in (C) indicates the magnification in both panels (B) and (C).
Both of these changes were similar in the detached and non-detached areas of the retina for the lesions examined. As reported previously, the Dachshund CLN2 disease is accompanied by an accumulation of autofluorescent storage material in the retina, primarily in the ganglion cells and along the outer limiting membrane (Katz et al., 2008). The RPE also contains autofluorescent storage material, but this appears to be normal lipofuscin and its presence is not disease-related (Katz et al. 2008). In dogs that exhibited the focal retinal detachments, there was no apparent difference in the amount or distribution of this autofluorescent material between regions of the retina that remained in normal apposition to the RPE and those regions in which the retina had separated from the RPE (Figure 9). In addition, the amount of autofluorescent pigment in the RPE did not vary to an appreciable extent between areas where the retina remained attached to the RPE and areas where the retina was separated from the RPE.
Figure 9.
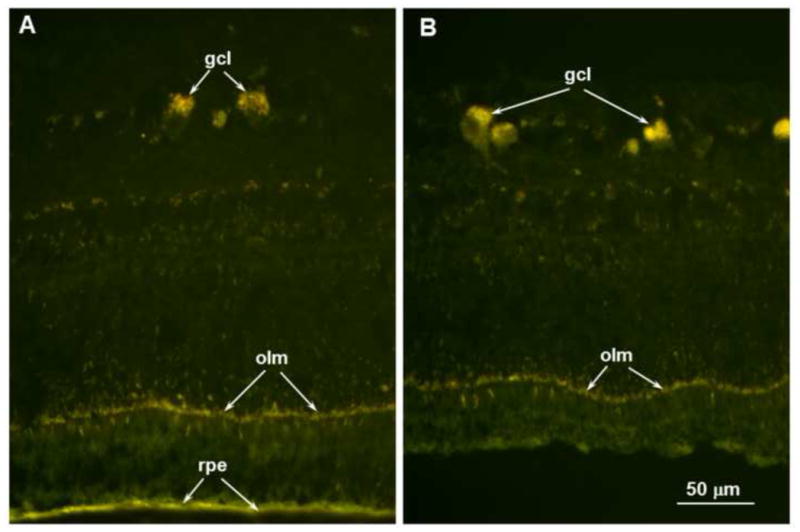
Fluorescence micrographs of an unstained cryostat section of the retina from a region where the retina remained attached to the RPE (A) and a region within one of the localized retina detachments (B). In both areas the disease-related autofluorescent material was present primarily in the ganglion cells (gcl) and outer limiting membrane (olm). The retinal pigment epithelium (rpe) also contained autofluorescent material with similar spectral properties, but this likely consisted primarily of normal age pigment since similar RPE autofluorescence is present in genetically normal dogs. Scale bar in (B) indicates magnification for both micrographs.
3.5. Best1 sequencing
Resequencing of all 11 BEST1 exons and flanking segments of the introns failed to reveal a plausible causal mutation. There were no sequence variants relative to the canine reference sequence that would predict changes in the amino sequence of the BEST1 protein. Relative to build 3.1 of the NCBI canine reference sequence, two sequence variants were found in the BEST1 coding regions that did not predict changes in amino acid coding: c. 66G>A and c. 309C>A.
4. Discussion
In this study we have documented for the first time the characteristics of progressive, multifocal serous retinal detachments in the TPP1−/− miniature longhaired Dachshund in relation to disease status. The retinopathy has some phenotypic similarities to that of canine multifocal retinopathy (CMR), although the degeneration of the inner retinal layers seen in the CLN2 disease is not a feature in reported cases of CMR. All CMRs currently identified are caused by mutations in BEST1 (Guziewicz et al., 2011; Guziewicz et al., 2007; Hoffmann et al., 2012; Zangerl et al., 2010). Mutations in human BEST1 cause several retinal disorders known as bestrophinopathies and CMR affected dogs serve as an animal model for the study of human bestrophinopathies. Human disorders characterized by retinal lesions similar to those seen in the bestrophinopathies have also been associated with mutations in IMPG1 and PRPH2 (Manes et al., 2013; Meunier et al, 2011), but to date no mutations in these genes have been associated with CMR. CMR has been documented in many canine breeds including the Great Pyrenees (CMR1) (Grahn and Sandmeyer, 2006), Coton de Tulear (CMR2) (Grahn and Sandmeyer, 2006) and the Lapponian Herder (CMR3) (Zangerl et al., 2010). Most CMRs develop in the first year of life, and the retinopathy in this study appeared in TPP1 −/− dogs between the ages of 5 and 12 months and at an average age of 7.5 months. In most types of CMR the retinopathy progresses over time and then becomes static. The retinopathy reported here was progressive throughout the course of observation, but because of the terminal nature of the CLN2 disease the course of lesion development could followed for a much shorter period than in dogs with CMR in which affected dogs can live a normal life span. Three TPP1−/− dogs with the retinopathy developed gradual loss of serous fluid from beneath some of the bullae based on ophthalmic examination findings. Significant decreases in the thickness of the retina overlying these flattened bullous lesions were observed via sdOCT. The phenomenon of serous fluid loss from within the bullae and subsequent development of adjacent retinal thinning has also been noted in dogs with CMR (Grahn and Sandmeyer, 2006; Grahn et al., 2006). The resorptive/flattened lesions appeared in dogs with an average age of 12.3 months, which is a considerably advanced age for TPP1−/− dogs. All three of these dogs received TPP1 enzyme replacement therapy, which extended their survival time beyond the 10 to 11 months that untreated TPP1−/− dogs survive. This observation suggests that if it were possible to achieve an even greater lifespan extension in the CLN2-diseased dogs, the prevalence and the progression of the retinopathy would be increased, with increased development of flattened bullae and retinal thinning. However, unlike in CMR, the retinal thinning in TPP1−/− dogs with extended survival times due to TPP1 administration to the CSF occurred across the entire retina and not just in the areas of the bullous detachments (unpublished). Fluorescein leakage from bullous lesions has not been observed in CMR (Grahn and Sandmeyer, 2006; Grahn et al., 2006) and was also not noted in the TPP1−/− dogs affected with the retinopathy in this study.
The development of these multifocal serous retinal detachments was clearly associated with the TPP1 mutation and not related to any genetic variants in the coding regions of BEST1. Not only was the coding sequence of BEST1 normal in a TPP1−/− dog with the retinal lesions, but these lesions only appeared in the dogs that were homozygous for the TPP1 null mutation. Had the lesions been the direct result of a BEST1 mutation, equal frequencies of the retinal lesions would be expected in the TPP1−/−, TPP1+/−, and TPP1+/+ dogs. Although we did not investigate the 3′UTR containing regulatory sequences of BEST1 in this study, if any mutations in this region were the direct cause of the multifocal retinal detachments, we would expect the frequency of these lesions to be similar in dogs with all three possible TPP1 genotypes. Since this was clearly not the case, we can rule out the mutations in this region of BEST1 as being the cause of the retinopathy. However, we cannot rule out the possibility that certain sequence variants in the 3′UTR region of BEST1 contributed to the development of the retinal lesions in the TPP1 −/− dogs. Although only 7 heterozygous carriers were followed formally as part of this study, we have maintained over 30 TPP1+/− dogs in our breeding colony up to ages of greater than 8 years and none of these dogs ever developed the multifocal retinopathy described in this study. Thus, clearly the retinopathy is restricted to TPP1−/− dogs.
The ERG findings indicated that the presence of the multifocal retinal lesions did not cause a significant further decrease in overall retinal function, compared to the deficits found in CLN2 without retinal lesions. This correlates with the finding that the degree of disease-related retinal pathology was similar in detached and non-detached regions within the same eyes. In the Dachshund CLN2 disease, ERG b-wave amplitudes are significantly diminished by 5 months of age with progressive decline through end-stage disease (Whiting et al., 2013b). The ERG a-wave is relatively well preserved in TPP1−/− dogs in the early stages of the disease, however a-wave amplitude is significantly decreased at 10 months of age when compared to normal (TPP1+/+) dogs (Whiting et al., 2013b). We selected a-wave amplitude measurement as a way to compare effects of the retinopathy due to the relative preservation of this portion of the waveform in similarly aged CLN2 disease-affected dogs. TPP1−/− dogs with retinopathy in this study did not have significantly different a-wave amplitudes compared to TPP1−/− dogs without the retinopathy. This finding is consistent with the histological data that showed the degree of retinal degeneration was similar in regions of retinal detachment and in adjacent regions of the same retina where the retina remained normally apposed to the RPE (Figure 8). Long-standing retinal detachment has been shown to result in retinal degeneration due to the necessity for the RPE to be in close apposition to the photoreceptor outer segments in order for the RPE to perform functions essential to photoreceptor survival and function (Murakami et al., 2013). It is not apparent why, for the most part, degeneration of the neural retina was not associated with the areas of retinal detachment. It is possible that most of the individual localized detachments were in the acute phase and had not been present for a long enough duration to result in photoreceptor damage. The three dogs with resorptive/flattened lesions did show evidence of retinal thinning and degeneration on the sdOCT studies and were of comparatively advanced age. It is also possible that the sizes of most detached areas were small enough that RPE support functions were maintained within these areas.
The ERG is widely used to assess retinal function in animals and people but has limited sensitivity (Brown, 1968). A PLR can be stimulated with lower intensity light stimuli than can the ERG (Whiting et al., 2013a). Quantitative evaluation of the PLR can be combined with ERG data to evaluate the function of the entire complex network of neuronal circuitry involved in modulating pupil size including retina (Fotiou et al., 2000; Park et al., 2011). As the CLN2 disease progresses, almost complete loss of detectable ERG responses develop but PLR, particularly to bright stimuli, is relatively preserved. Therefore, PLR is much more sensitive than ERG in detecting residual retinal function in animal models of retinal degenerative disease. The presence of PLR with profoundly depressed ERGs correlates with preservation of visually- mediated behavior even late in CLN2 disease (Whiting et al., 2013a). Due to the preservation of the PLR in late-stages of this disorder in the dog model, PLR measurement was used as an additional quantitative measurement to evaluate the effect of the retinopathy on retinal function. Generally there was no significant effect of retinopathy lesion presence or grade on PLR at most stimulus intensities. Again, this is consistent with the preservation of retinal morphological integrity over most of the areas of detachment. However, in 8 month old dogs with Grade 3 retinopathy (highest recorded grade), a significant decrease in PLR constriction amplitude was noted at the low stimulus intensity of 10 log photons/cm2/s. This indicates that the presence of a very large number of the retinopathy lesions may have a small effect on overall retinal function. However, the effect of the retinopathy is likely to be minimal in these dogs in terms of actual clinical visual function based on the PLR data presented here.
The association of these lesions with CLN2 disease indicates that their presence and numbers can be useful markers in assessing the efficacy of potential treatments in slowing the progression of disease-related retinal abnormalities. Delivery of recombinant TPP1 protein to the brain via periodic infusion into the cerebrospinal fluid (CSF) was shown to significantly delay the progression of neurological disease signs and brain atrophy in the Dachshund CLN2 disease model (Katz et al., 2014). In these studies, disease-related decline in retinal function was not inhibited, most likely because TPP1 protein administered to the CSF did not reach the retina. In the current study, some of the dogs received the CSF TPP1 infusions. As with the ERG, these infusions had no effect on the age at which the focal retinal detachments first appeared or on the rate at which they increased in number and size. Research is currently under way to determine whether chronic delivery of the TPP1 protein to the retina from the vitreous will be effective in inhibiting the effects of the disease on the retina. If this approach to treatment is effective, it should not only preserve retinal function as assessed with the ERG and PLR, but should inhibit the development of the multifocal retinal detachments.
It is noteworthy that the disease-related retinal detachments observed in the TPP1−/− dogs have not been observed in human subjects with the CLN2 disease (Hainsworth et al., 2009; Weleber, 1998). The basis for this species difference is not apparent. It most likely reflects an underlying difference in retinal physiology between people and dogs.
The similarity of the multifocal retinal lesions in the TPP1−/− dogs to those observed in dogs with BEST1 mutations suggested the possibility that an allelic variant in BEST1 was involved in the Dachshunds. However, resequencing of all 11 BEST1 exons from one of the TPP1−/− dogs affected with the retinopathy did not reveal a plausible causal mutation. In addition, perfect concordance between homozygosity for the TPP1 null mutation and the retinopathy in the large number of dogs examined strongly suggests that the TPP1 mutation is responsible for the lesions. The association of the TPP1 null mutation with the development of these lesions suggests that other more mild TPP1 mutations may be responsible for retinopathies characterized by multifocal retinal detachments occurring in animals and humans that are not associated with BEST1 mutations.
Highlights.
CLN2 neuronal ceroid lipofuscinosis results in progressive retinal degeneration.
Dogs with this disease develop multifocal retinal detachments.
Retinal degenerative changes are not exacerbated in the detached areas.
The retina appears to tolerate localized detachments without adverse effects.
Acknowledgments
This study was supported by grants EY018815 and EY023968 from the U.S. National Institutes of Health, and by grants from the Batten Disease Support and Research Association and the University of Missouri Research Board. These sponsors were not involved in the study design, the collection, analysis and interpretation of data, the writing of this report or in the decision to submit the article for publication.
The authors thank Dr. Kristina Narfstrom for her support and guidance with many aspects of this work. Drs. Christine Sibigtroth and Melissa Carpentier assisted with anesthesia. We also thank Dr. Gayle C. Johnson for her assistance with necropsies. Ms. Karen Clifford provided assistance with the preparation of images and figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Awano T, Katz ML, O’Brien DP, Sohar I, Lobel P, Coates JR, Khan S, Johnson GC, Giger U, Johnson GS. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006a;89:254–260. doi: 10.1016/j.ymgme.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Awano T, Katz ML, O’Brien DP, Taylor JF, Evans J, Khan S, Sohar I, Lobel P, Johnson GS. A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006b;87:341–348. doi: 10.1016/j.ymgme.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Brown KT. The electroretinogram: its components and their origins. Vision Res. 1968;8:633–677. doi: 10.1016/0042-6989(68)90041-2. [DOI] [PubMed] [Google Scholar]
- Ekesten B, Komaromy AM, Ofri R, Petersen-Jones SM, Narfstrom K. Guidelines for clinical electroretinography in the dog: 2012 update. Doc Ophthalmol. 2013;127:79–87. doi: 10.1007/s10633-013-9388-8. [DOI] [PubMed] [Google Scholar]
- Farias FH, Zeng R, Johnson GS, Wininger FA, Taylor JF, Schnabel RD, McKay SD, Sanders DN, Lohi H, Seppala EH, Wade CM, Lindblad-Toh K, O’Brien DP, Katz ML. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis. 2011;42:468–474. doi: 10.1016/j.nbd.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Fotiou F, Fountoulakis KN, Goulas A, Alexopoulos L, Palikaras A. Automated standardized pupillometry with optical method for purposes of clinical practice and research. Clin Physiol. 2000;20:336–347. doi: 10.1046/j.1365-2281.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- Grahn BH, Sandmeyer LS. Multifocal retinopathy of Great Pyrenees dogs. Can Vet J. 2006;47:491–492. [PMC free article] [PubMed] [Google Scholar]
- Grahn BH, Sandmeyer LS, Breaux CB. Multifocal bullous retinopathy of coton de Tulear dogs. Can Vet J. 2006;47:929–930. [PMC free article] [PubMed] [Google Scholar]
- Guo J, Johnson GS, Brown HA, Provencher ML, da Costa RC, Mhlanga-Mutangadura T, Taylor JF, Schnabel RD, O’Brien DP, Katz ML. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian Shepherd ancestry. Mol Genet Metab. 2014;112:302–309. doi: 10.1016/j.ymgme.2014.05.014. [DOI] [PubMed] [Google Scholar]
- Guo J, O’Brien DP, Mhlanga-Mutangadura T, Olby NJ, Taylor JF, Schnabel RD, Katz ML, Johnson GS. A rare homozygous MFSD8 single-base-pair deletion and frameshift in the whole genome sequence of a Chinese Crested dog with neuronal ceroid BMC Vet. Res BMC Veterinary Research. 2015;10:960. doi: 10.1186/s12917-014-0181-z. electronic resource. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Slavik J, Lindauer SJ, Aguirre GD, Zangerl B. Molecular consequences of BEST1 gene mutations in canine multifocal retinopathy predict functional implications for human bestrophinopathies. Invest Ophthalmol Vis Sci. 2011;52:4497–4505. doi: 10.1167/iovs.10-6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Zangerl B, Lindauer SJ, Mullins RF, Sandmeyer LS, Grahn BH, Stone EM, Acland GM, Aguirre GD. Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Invest Ophthalmol Vis Sci. 2007;48:1959–1967. doi: 10.1167/iovs.06-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainsworth DP, Liu GT, Hamm CW, Katz ML. Funduscopic and angiographic appearance in the neuronal ceroid lipofuscinoses. Retina. 2009;29:657–668. doi: 10.1097/IAE.0b013e31819b0542. [DOI] [PubMed] [Google Scholar]
- Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochim Biophys Acta. 2006;1762:850–856. doi: 10.1016/j.bbadis.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Hoffmann I, Guziewicz KE, Zangerl B, Aguirre GD, Mardin CY. Canine multifocal retinopathy in the Australian Shepherd: a case report. Vet Ophthalmol. 2012;15(Suppl 2):134–138. doi: 10.1111/j.1463-5224.2012.01005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ML, Coates JR, Cooper JJ, O’Brien DP, Jeong M, Narfstrom K. Retinal pathology in a canine model of late infantile neuronal ceroid lipofuscinosis. Invest Ophthalmol Vis Sci. 2008;49:2686–2695. doi: 10.1167/iovs.08-1712. [DOI] [PubMed] [Google Scholar]
- Katz ML, Coates JR, Sibigtroth CM, Taylor JD, Carpentier M, Young WM, Wininger FA, Kennedy D, Vuillemenot BR, O’Neill CA. Enzyme Replacement Therapy Attenuates Disease Progression in a Canine Model of Late Infantile Neuronal Ceroid Lipofuscinosis (CLN2 Disease) J Neurosci Res. 2014;92:1591–1598. doi: 10.1002/jnr.23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ML, Farias FH, Sanders DN, Zeng R, Khan S, Johnson GS, O’Brien DP. A missense mutation in canine CLN6 in an Australian shepherd with neuronal ceroid lipofuscinosis. J Biomed Biotech. 2011;2011:198042. doi: 10.1155/2011/198042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ML, Khan S, Awano T, Shahid SA, Siakotos AN, Johnson GS. A mutation in the CLN8 gene in English Setter dogs with neuronal ceroid-lipofuscinosis. Biochem Biophys Res Commun. 2005a;327:541–547. doi: 10.1016/j.bbrc.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Katz ML, Narfstrom K, Johnson GS, O’Brien DP. Assessment of retinal function and characterization of lysosomal storage body accumulation in the retinas and brains of Tibetan Terriers with ceroid-lipofuscinosis. Am J Vet Res. 2005b;66:67–76. doi: 10.2460/ajvr.2005.66.67. [DOI] [PubMed] [Google Scholar]
- Manes G, Meunier I, Avila-Fernandez A, Banfi S, Le Meur G, Zanlonghi X, Corton M, Simonelli F, Brabet P, Labesse G, Audo I, Mohand-Said S, Zeitz C, Sahel JA, Weber M, Dollfus H, Dhaenens CM, Allorge D, De Baere E, Koenekoop RK, Kohl S, Cremers FP, Hollyfield JG, Senechal A, Hebrard M, Bocquet B, Ayuso Garcia C, Hamel CP. Mutations in IMPG1 cause vitelliform macular dystrophies. Am J Human Genet. 2013;93:571–578. doi: 10.1016/j.ajhg.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville SA, Wilson CL, Chiang CS, Studdert VP, Lingaas F, Wilton AN. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border collie dogs. Genomics. 2005;86:287–294. doi: 10.1016/j.ygeno.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Meunier I, Senechal A, Dhaenens CM, Arndt C, Puech B, Defoort-Dhellemmes S, Manes G, Chazalette D, Mazoir E, Bocquet B, Hamel CP. Systematic screening of BEST1 and PRPH2 in juvenile and adult vitelliform macular dystrophies: a rationale for molecular analysis. Ophthalmology. 2011;118:1130–1136. doi: 10.1016/j.ophtha.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Mole SE, Williams RE, Goebel HH. The Neuronal Ceroid Lipofuscinoses (Batten Disease) 2. Oxford University Press; Oxford: 2011. [Google Scholar]
- Murakami Y, Notomi S, Hisatomi T, Nakazawa T, Ishibashi T, Miller JW, Vavvas DG. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog Retin Eye Res. 2013;37:114–140. doi: 10.1016/j.preteyeres.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JC, Moura AL, Raza AS, Rhee DW, Kardon RH, Hood DC. Toward a clinical protocol for assessing rod, cone, and melanopsin contributions to the human pupil response. Invest Ophthalmol Vis Sci. 2011;52:6624–6635. doi: 10.1167/iovs.11-7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DN, Farias FH, Johnson GS, Chiang V, Cook JR, O’Brien DP, Hofmann SL, Lu JY, Katz ML. A mutation in canine PPT1 causes early onset neuronal ceroid lipofuscinosis in a Dachshund. Mol Genet Metab. 2010;100:349–356. doi: 10.1016/j.ymgme.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DN, Kanazono S, Wininger FA, Whiting RE, Flournoy CA, Coates JR, Castaner LJ, O’Brien DP, Katz ML. A reversal learning task detects cognitive deficits in a Dachshund model of late-infantile neuronal ceroid lipofuscinosis. Genes, Brain, & Behavior. 2011;10:798–804. doi: 10.1111/j.1601-183X.2011.00718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemenot BR, Katz ML, Coates JR, Kennedy D, Tiger P, Kanazono S, Lobel P, Sohar I, Xu S, Cahayag R, Keve S, Koren E, Bunting S, Tsuruda LS, O’Neill CA. Intrathecal tripeptidyl-peptidase 1 reduces lysosomal storage in a canine model of late infantile neuronal ceroid lipofuscinosis. Mol Genet Metab. 2011;104:325–337. doi: 10.1016/j.ymgme.2011.06.018. [DOI] [PubMed] [Google Scholar]
- Warrier V, Vieira M, Mole SE. Genetic basis and phenotypic correlations of the neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2013;1832:1827–1830. doi: 10.1016/j.bbadis.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Weleber RG. The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye. 1998;12:580–590. doi: 10.1038/eye.1998.148. [DOI] [PubMed] [Google Scholar]
- Whiting REH, Narfstrom K, Yao G, Pearce JW, Coates JR, Castaner LJ, Katz ML. Pupillary light reflex deficits in a canine model of late infantile neuronal ceroid lipofuscinosis. Exp Eye Res. 2013a;116:402–410. doi: 10.1016/j.exer.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting REH, Yao G, Narfstrom K, Pearce JW, Coates JR, Dodam JR, Castaner LJ, Katz ML. Quantitative assessment of the canine pupillary light reflex. Invest Ophthalmol Vis Sci. 2013b;54:5432–5440. doi: 10.1167/iovs.13-12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangerl B, Wickstrom K, Slavik J, Lindauer SJ, Ahonen S, Schelling C, Lohi H, Guziewicz KE, Aguirre GD. Assessment of canine BEST1 variations identifies new mutations and establishes an independent bestrophinopathy model (cmr3) Mol Vis. 2010;16:2791–2804. [PMC free article] [PubMed] [Google Scholar]