

Abstract
Post injury epilepsy (PIE) is a common complication following brain insults, including ischemic and traumatic brain injuries. At present there are no means to identify the patients at-risk to develop PIE or to prevent its development. Seizures can occur months or years after the insult, do not respond to anti-seizure medications in over third of the patients, and are often associated with significant neuropsychiatric morbidities. We have previously established the critical role of blood-brain barrier dysfunction in PIE, demonstrating that exposure of brain tissue to extravasated serum albumin induces activation of inflammatory transforming growth factor beta (TGF-β) signaling in astrocytes and eventually seizures. However, the link between the acute astrocytic inflammatory responses and reorganization of neural networks that underlie recurrent spontaneous seizures, remains unknown. Here we demonstrate in-vitro and in-vivo that activation of the astrocytic ALK5/TGF-β-pathway induces excitatory, but not inhibitory, synaptogenesis that precedes the appearance of seizures. Moreover, we show that treatment with SJN2511, a specific ALK5/TGF-β inhibitor, prevents synaptogenesis and epilepsy. Our findings point to astrocyte-mediated synaptogenesis as a key epileptogenic process, and highlight manipulation of the TGF-β-pathway as a potential strategy for the prevention of PIE.
Keywords: Epilepsy, Albumin, TGF-β, Synaptogenesis, Blood-Brain barrier (BBB), Seizures, Astrocytes, Post-insult epilepsy (PIE), Post-traumatic epilepsy (PTE), ALK5
Introduction
Traumatic, ischemic and infectious brain injuries often initiate a cascade of epileptogenic events, ultimately leading to post injury epilepsy (PIE) after a latent period of months to years. Accumulating evidence suggests a key role of post-injury dysfunction of the blood-brain barrier (BBB) in the development of PIE: BBB dysfunction is a well-documented finding in patients following brain trauma (occuring within hours of the event and often persisting for days to weeks, for review (Abbott and Friedman, 2012; Cunningham et al., 2005; Rosenberg, 2012; Shlosberg et al., 2010)) and is more frequent in post-traumatic patients with epilepsy (Raabe et al., 2012; Schmitz et al., 2013; Tomkins et al., 2008). In experimental animals, BBB dysfunction was associated with increased propensity for symptomatic seizures and the development of epilepsy (Friedman et al., 2009; Frigerio et al., 2012; Marchi et al., 2007; Seiffert et al., 2004; van Vliet et al., 2007).
Previous studies have linked the brain’s immune response with seizures and epilepsy, as activation of the pro-inflammatory IL-1 receptor/Toll-like receptor (IL1R/TLR) system was shown to promote seizure onset and recurrence in mice and rats (for review see (Devinsky et al., 2013; Marchi et al., 2013; Vezzani et al., 2011a, 2011b)). Recent findings have highlighted the involvement of a specific inflammatory pathway in PIE, identifying the epileptogenic role of transforming growth factor beta (TGF-β) signaling in animal models of BBB dysfunction. Serum albumin was shown to enter BBB-disrupted brain tissue and bind to TGF-β receptors in astrocytes (Ivens et al., 2007), inducing inflammatory signaling (Cacheaux et al., 2009) and SMAD-2/3 phosphorylation (Bar-Klein et al., 2014), thereby modifying astrocytic function (Braganza et al., 2012; David et al., 2009; Seiffert et al., 2004). TGF-β signaling was also associated with immediate changes in extracellular potassium and glutamate, and a lower threshold for neuronal activation in slices (David et al., 2009; Lapilover et al., 2012). However, the immediate and short-lived nature of these changes (Ivens et al., 2007; Lapilover et al., 2012) suggests the involvement of additional modifications, underlying permanent network changes that sustain chronic recurrent seizures. While axonal sprouting and synaptogenesis were shown to contribute to seizures and persistent network alterations (Babb et al., 1991; Bragin et al., 2000; Marco and DeFelipe, 1997), the detailed changes and mechanisms underlying network reorganization in PIE are not well understood.
Glial cells are known to play a key role in the formation and elimination of both excitatory and inhibitory synapses in developing and mature brain (Chung and Barres, 2012; Clarke and Barres, 2013; Elmariah et al., 2005; Eroglu and Barres, 2010). Specifically, astrocytes were shown to regulate synaptogenesis through secretion of thrombospondins (TSPs)(Christopherson et al., 2005; Crawford et al., 2012; Eroglu et al., 2009; Liauw et al., 2008), Hevin (Kucukdereli et al., 2011) and Glypicans 4 and 6 (Allen et al., 2012). Moreover, synapse formation was also associated with activation of TGF-β signaling in Schwann cells (Feng and Ko, 2008; Packard et al., 2003), and more recently in astrocytes via induction of Smad3 (Yu et al., 2014), secretion of D-serine (Diniz et al., 2012) and CaM kinase II signaling (Diniz et al., 2014). Since dysfunction of astroglia has been shown following brain injury, in models of acquired epilepsy (Crunelli et al., 2014; Devinsky et al., 2013; Friedman et al., 1996; Marchi et al., 2013) and in the BBB-compromised brain, here we set out to study the role of albumin-induced TGF-β pathway activation in synaptogenesis and chronic seizures. We further challenged the hypothesis that inhibition of the TGF-β ALK5 receptor is sufficient to prevent the development of epilepsy. Through a combination of in vitro and in vivo models of inflammation post BBB dysfunction, our study sheds light on the epileptogenic cascade following brain injury, and highlights novel targets for PIE prevention in at-risk patients.
Materials and Methods
All animal procedures were approved by the UC Berkeley Animal Care and Use Committee or the animal care and use ethical committees at the Ben-Gurion University of the Negev, Beer-Sheva.
IntraCerebroVentricular (ICV) Pump implantation
Two-month old male FVB-N mice (Harlan, Israel) were anesthetized with isoflurane (0.8–1.2%) and placed in a stereotaxic frame. A 0.7 mm diameter hole was drilled through the skull over the somatosensory cortex (0.5 mm caudal, 1 mm lateral to bregma) and anterior to the hippocampus. Micro-osmotic pumps (ALZET, Cupertino, CA) were filled with 200 μL of either 0.4 mM bovine serum albumin (BSA; Sigma) solution or 100 ng/mL TGF-β1 (Peprotech, Rocky Hill, NJ) in artificial cerebrospinal fluid (aCSF) as previously described (Seiffert et al., 2004), and placed subcutaneously between the shoulder blades. In a subset of animals, 10% of the BSA was replaced with fluorescein isothiocyanate (FITC) conjugated BSA (Sigma) or Alexa Fluor 488 conjugated BSA (Life Technologies) (2.68gr/l). Other animals were infused with BSA and 300 μM SJN2511 (Tocris, Bristol, UK) to block TGF-β signaling. Sham controls were implanted with pumps containing aCSF with FITC conjugated 70 kD dextran (Sigma). Pumps delivered their contents into the right lateral cerebral ventricle via a brain infusion kit (ALZET, 0008851). Pumps were extracted under isoflurane anesthesia 7 days after implantation.
Immunohistochemistry (IHC)
Brain sections were stained according to commonly accepted protocols (Ippolito and Eroglu, 2010). Mice were cardio-perfused with phosphate-buffered saline (PBS) and brains were removed and fixed in 4% PFA in PBS overnight at 4 °C. The brains were then transferred to PBS containing 30% sucrose for 24 hours and then embedded in a 2:1 mixture of 20% sucrose in PBS:Tissue-Tek OCT (VWR, Radnor, PA). Once frozen, 12 μm sections were collected using a freezing microtome. Slices were dried at 37 °C, washed three times in PBS and blocked with 10% normal goat serum in PBS for 1 hour. Glial fibrillary acidic protein (GFAP) stains were performed using a monoclonal mouse anti-GFAP (Sigma). Excitatory synapses were labeled with primary antibodies for PSD-95 (Millipore, 1:200) and synapsin1 (Synaptic Systems, Goettingen, DE; 1:200). Alexa Fluor 555 and Alexa Fluor 488 conjugated secondary antibodies (Abcam, Cambridge, MA; 1:400) in 1% normal donkey serum in PBS were applied for 2 hours at room temperature. The nuclear stain (ToPro3, Life Technologies; 1:1000) was added prior to mounting coverslips. Each treated slice was stained together with a matched control.
Western Blot
Extracted brain tissue was immediately frozen on dry ice and homogenized in 20 mM Tris-HCl (pH 7.4), and 1 mM EDTA, 5 mM EGTA, 1 mM Na-vanadate, 2 ug/ul aprotinin, 1 ug/ul pepstatin, 2 ug/ul leupeptin (30 mg tissue/150 ul homogenization buffer). Equal amounts of protein (30 μg) were separated by SDS-PAGE (11%) and transferred to nitrocellulose membranes (Bio-Rad Laboratories). Membranes were rinsed with PBS containing 0.1% Tween 20 and blocked with 5% nonfat milk in 1× PBS overnight at 4 °C. The membranes were incubated with mouse monoclonal antibody against GFAP (1:20000; Chemicon Int Inc.) for 1 h at room temperature. After washes in PBS–1% Tween (PBS-T), membranes were incubated with peroxidase-conjugated anti-mouse secondary antibody (1:10,000; Sigma-Aldrich) in PBS with 3% milk for 1 h. Membranes were then washed repeatedly in PBS-T and incubated with ECL. Immunoreactive bands were visualized and series of ECL exposures were performed to ensure that non-saturated bands were used for quantification. Western blot data acquisition and analysis were accomplished by measuring the pixel density and area of Immunoreactive bands from the ECL films using ImageJ. Values were normalized to β-actin.
Telemetric electrocorticography and seizure detection
ECoG activity was monitored telemetrically (Data Science International, St. Paul, MN) for up to a month after pump implantation. During pump implantation, a subset of animals was implanted with a small transmitter (TA10EAF20), placed subcutaneously on the dorsal side. Two 0.7 mm diameter holes were drilled through the skull over the cortex (0.5 mm anterior, 2.5 mm lateral to lambda) for screws serving as electrodes. The epicranial side of each screw was connected to a transmitter lead and secured with bone cement. Signals were transmitted to a receiver placed below the animal’s cage and saved on a personal computer at a sampling rate of 1 KHz. For the unbiased detection of seizures, ECoG data was analyzed using an in-house algorithm, based on feature extraction and artificial neural network (ANN) classification (Bar-Klein et al., 2014). Seizures were defined an electrographic events lasting a minimum of 5 seconds.
Ex-vivo GFAP and Synaptic quantification
Brain slices were imaged using z-stack confocal microscopy (Olympus IX50) and a 40x (GFAP) or 63x (synaptic) objective with 1μm z-slices. Images were analyzed automatically using Matlab (Mathworks, Natick, MA). For GFAP quantification, positive voxels were considered as those with intensities higher than the averaged intensity of control images (mean minus standard deviation). For synaptic quantification we adapted from (Eroglu et al., 2009; Ippolito and Eroglu, 2010), [http://fohs.bgu.ac.il/neurophysio/]. In brief, maximum intensity projections (MIPs) were generated from groups of 3 consecutive sections (1μm each). The puncta size was set at 4–15 voxels and synapses were defined as at least 50% co-localization between pre- and post-synaptic staining.
Primary cell culture
Brains from embryonic day 19 Sprague Dawley rats (Charles River, Hollister, CA) were removed and the temporal lobe was microdissected. Cells were dissociated with a papain solution (10 units/mL) for 20 minutes at 37 °C, after which they were pelleted by centrifugation, and the papain solution was replaced with HBSS (Life Technologies, Grand Island, NY). A single cell suspension was achieved by mechanical trituration and the cells were counted using a hemocytometer. The cells were plated at a density of 12.5×104 cells per mL in growth medium (MEM with Earle’s salts, 2.5% B27 supplement (Life Technologies), 5% FBS (Life Technologies), 20 mM glucose and 5 mM L-glutamine) into glass-bottomed chamber slides (Thermo Fisher Scientific, Portsmouth, NH). After a 4 hour incubation, the cell culture medium was replaced with Neurobasal medium (Life Technologies) supplemented with 2% B27 and 0.5 mM GlutaMAX (Life Technologies). The cells were maintained in 5% CO2 at 37 °C. For pure neuronal cultures, cytosine arabinofuranoside (AraC; 10 μM; Sigma, St. Louis, MO) was added at 3 days in vitro (DIV) to the cultures to inhibit mitotically dividing cells. Cells were treated for 72 hours with 0.2 mM BSA, 10 ng/mL TGF-β1 or Neurobasal media. For TGF-β blocking experiments, cells were pre-incubated for 1 hour in 30 μM SJN 2511.
Immunocytochemistry (ICC)
For ICC in vitro preparations, cells were fixed with 4% PFA for 15 min at room temperature and washed in PBS. Cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature. Primary antibodies were incubated overnight at 4 °C in 1% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) in PBS. Excitatory synapses were labeled with primary antibodies for PSD-95 (UC Davis, Neuromab, Davis, CA; 1:800) and synapsin1 (Sigma; 1:800). Inhibitory synapses were labeled with primary antibodies for V-Gat (Millipore, Billerica, MA; 1:200) and Gephyrin (BD Bioscience, San Jose, CA; 1:100). Cy3 and Alexa Fluor 488 conjugated secondary antibodies (Jackson ImmunoResearch; 1:500) in 1% normal donkey serum in PBS were applied for 2 hours at room temperature. The nuclear stain (DAPI; 1:20,000) was added prior to mounting coverslips in DABCO. Fluorescent images were obtained with an inverted fluorescence microscope using the Metamorph imaging system.
In-vitro Synaptic quantification
Cell culture images were collected using epifluorescence microscopy (Zeiss Axio Observer Z1), and analyzed using the Metamorph Software automatic cell scoring application. Excitatory synapses were defined as co-localization between Synapsin1 (presynaptic) and PSD-95 (postsynaptic) markers, while inhibitory synapses were defined as co-localization between V-Gat (presynaptic) and Gephyrin (postsynaptic) markers.
Real-time RT-PCR
Total RNA was isolated using the TRIzol reagent (Life Technologies) in accordance with the manufacturer’s protocol. mRNA expression levels were determined by quantitative reverse transcriptase-PCR using real-time kinetic analysis with an iQ5 detection system (Bio-Rad, Hercules, CA). Real-time PCR data were analyzed using the PCR Miner program (Leonoudakis et al., 2008), and RPLP (ribosomal protein L16) RNA levels were used as internal controls for variations in sample preparation. Primer sequences were as follows: GFAP (+): AGA AAA CCG CAT CAC CAT TC; GFAP (−): TCC TTA ATG ACC TCG CCA TC. PSD-95 (+) TGC GAA GCA ACC CCA AGA GG, (−) ATG AAG CAC ATC CCC GAA GCG; VGlut1 (+) CCA CAC ACA GCA CAG TTC AGC C, (−) TCC TCG ACA CTG CCG TTT AGG
Statistics
All statistical comparisons were performed using GraphPad Prism (GraphPad Software, San Diego, CA). Single variable comparisons between groups were performed using either one-way ANOVA with Dunnett’s posthoc test or nonparametric Kruskal-Wallis test with Dunn’s multiple comparisons test. Two variable comparisons were performed using two-way ANOVA with post-hoc of repeated t-tests, corrected using the Sidak-Bonferroni method. P-values smaller than 0.05 were considered significant.
Results
ICV albumin induces epileptogenesis in vivo
To simulate BBB dysfunction in naïve animals and study the role of serum albumin in epileptogenesis, mice were infused with albumin (0.4 mM in ACSF, 1 uL/hour) through an ICV mini-osmotic pump for 7 days. ECoG data was acquired for up to 32 days (32 days: n=4; 14 days: n=7; 10 days: n=2), and analyzed using an automated seizure detection algorithm (see Methods and (Bar-Klein et al., 2014)). Albumin treatment did not induce status epilepticus (SE) nor seizures during the first 72 hours after pump implantation, in contrast to the pilocarpine model of epilepsy, in which mice develop SE shortly after drug injection (Fig. 1B). After a latent period of 4.9 ± 1.3 days, ten of the thirteen albumin-infused mice presented recurring spontaneous seizures (1.16 ± 0.16 seizures per day, lasting 47.2 ± 12.7 sec). Notably, spontaneous seizures continued throughout the duration of ECoG monitoring, well after pump removal on day 7 (Fig. 1C,D). These results indicate that, similar to human PIE, exposure to albumin leads to self-sustained recurrent seizures, preceded by a latent period with no seizure activity.
Figure 1. ICV albumin induces latent appearance of spontaneous recurrent seizures.

(A) The experimental paradigm in vivo. (B) Continuous ECoG recordings show that ICV albumin does not induce acute seizures or status epilepticus (common in other models of acquired epilepsy, e.g pilocarpine), yet results in latent appearance of spontaneous seizures. (C) Representative seizure recorded 72 h after pump implantation. (D) Comparison of number of seizures per day (averaged over 3 day periods) shows higher incidence of seizures in the albumin-infused mice (black line, n=13), compared to controls (red line, n=10). Notably, seizures appeared 3 days after initiating albumin infusion, and lasted well after the extraction of the pumps on day 7. Error bars indicate SEM.
ICV albumin induces activation of hippocampal astrocytes
To study the pathological changes that precede the appearance of seizures, a cohort of mice was infused with albumin for 72 hours. Imaging of fluorescein-conjugated albumin revealed prominent albumin accumulation in the hippocampi and cortical regions (Fig. 2A). Western blot analysis confirmed hippocampal accumulation of albumin, comparable to the levels observed 24 hours following SE in the pilocarpine model of epilepsy (ctrl, 0.58 ± 0.2 μM; 72h-alb, 3.2 ± 0.2 μM; n=4, p<0.05; ctrl, 0.31 ± 0.06; 24h post-SE 0.82 ± 0.2 μM; n=7, p<0.05). Consistent with findings following cortical exposure to albumin (Bar-Klein et al., 2014; Braganza et al., 2012; Frigerio et al., 2012; Ivens et al., 2007), fluorescent albumin was evident in astroglial elements (Fig. 2B). GFAP staining 72h after infusion of fluorescein-conjugated albumin suggest astrocytic activation in the hippocampus, the somatosensory cortex, corpus callosum and adjacent neocortical regions. Quantification of GFAP mRNA and protein (through immunohistochemistry, ctrl=4 alb=5 p<0.0001; real time PCR, ctrl=3 alb=3 p=0.0004; and western blot, ctrl=4 alb=6 p<0.01) confirmed astrocytic activation in albumin-exposed hippocampi (Fig. 2D,E). Hematoxylin eosin staining 1 month after ICV albumin did not reveal any gross abnormalities (Fig. 2F), in contrast to SE-induced models of epilepsy.
Figure 2. ICV Albumin induces activation of astrocytes.

(A) 72 hour infusion of fluorescein-conjugated albumin (10%; Alb-488, green) into the lateral ventricle results in florescence accumulation in the hippocampus (DG, CA1,3), the somatosensory cortex (S1), corpus callosum, entorhinal cortex (EC) and adjacent neocortical regions, as evident here in coronal slices 72h after pump implantation (right,~ bregma −1.58mm; left,~bregma −2.92mm). (B) Best representative images of albumin (Alb-488) co-localization with neurons (NeuN, somatosensory cortex), microglia (IBA1, hippocampal hilus) and astrocytes (GFAP, hippocampal hilus). White arrowheads mark co-localization of cells with fluorescein-conjugated albumin. (C) GFAP immunohistochemical staining of a coronal slice, 72h after infusion of fluorescein-conjugated albumin. GFAP and albumin florescent intensity maps (right panels) show high levels of fluorescence in the hippocampus, the somatosensory cortex, corpus callosum and adjacent neocortical regions. (D) Immunohistochemical (IHC) staining of cell nuclei (ToPro3, green) and GFAP positive cells (red) demonstrates increased levels of GFAP following ICV albumin treatment in the hilus of the hippocampus. (E) Quantitative analyses of IHC staining (IHC, ctrl=4 alb=5) of the hippocampus reveals a significant increase in GFAP expression, further confirmed by real time RT-PCR (mRNA, ctrl=3 alb=3), and western blots (western, ctrl=4 alb=6). (F) Hematoxylin eosin (HE) hippocampal staining shows no gross abnormalities one month after albumin infusion, compared to controls (ctrl=3 alb=3). Error bars indicate SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Albumin induces excitatory synaptogenesis
Astrocytes are known to regulate the generation and elimination of both excitatory and inhibitory synapses (Chung and Barres, 2012; Clarke and Barres, 2013; Elmariah et al., 2005; Eroglu and Barres, 2010). Thus, the prominent albumin-induced astrocytic activation observed during the pre-seizure period suggested a possible role in synaptic network reorganization. To study the synaptogenic effect of albumin, we quantified excitatory and inhibitory synaptic puncta in primary mixed neuronal and glial cells of temporal origin exposed to 0.2 mM serum albumin (50% of normal serum concentration, Alb=17; no treatment (NT)=12) for 72 h. Quantification of excitatory presynaptic (synapsin1) and postsynaptic (PSD-95) markers along dendritic lengths revealed a significant increase in excitatory presynaptic (p<0.0001), postsynaptic (p=0.0017), and co-localized (p=0.0010) puncta (Fig. 3A). Since inhibitory synapses occur both on dendrites and on the axon initial segment (Nusser et al., 1995), inhibitory presynaptic (V-Gat) and postsynaptic (Gephyrin) markers were quantified along dendritic lengths and per somatic area. No significant changes in the number of inhibitory synapses were observed (Fig. 3B, per soma: Alb=17; NT=15; p=0.2569; and per dendrite Alb=19; NT=18. P=0.0854), indicating that the synaptogenic effect of albumin is specific to excitatory synapses. These observations suggest a mechanism whereby vascular pathology, BBB breakdown and the extravasation of albumin, shift the balance of the neural network in favor of increased excitability.
Figure 3. Albumin induces excitatory, but not inhibitory, synaptogenesis in temporal cultures.

(A) Staining for excitatory presynaptic (Syn1; green) and postsynaptic (PSD-95, red) proteins, revealed a significant albumin-induced increase in presynaptic, postsynaptic and co-localized counts along dendritic lengths (Alb=17; no treatment (NT)=12). (B) Staining for inhibitory presynaptic (V-Gat, green) and postsynaptic (Gephyrin, red) proteins showed no significant albumin-induced differences in synaptic counts, quantified along dendritic lengths and the somatic area (per soma: Alb=17; NT=15; p=0.2569; and per dendrite Alb=19; NT=18. P=0.0854). Error bars indicate SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Albumin-induced excitatory synaptogenesis is mediated by TGF-β/ALK5 signaling
We have previously shown that albumin initiates TGF-β signaling in astrocytes (Cacheaux et al., 2009; Ivens et al., 2007) via the ALK5 receptor (Bar-Klein et al., 2014). To test the potential involvement of TGF-β signaling in albumin-induced excitatory synaptogenesis, we treated cultures with TGF-β1 (10 ng/mL) for 72 h and repeated the synaptic quantification assay. Similar to the effect of albumin, exposure to TGF-β1 increased the number of excitatory presynaptic (n=18, p < 0.0001), postsynaptic (n=18, p < 0.0001) and co-localized (n=18, p < 0.0001) puncta (Fig. 4A). A subset of cultures was co-incubated with the selective TGF-β type I receptor ALK5 inhibitor SJN2511 (SJN; 30 μM, applied 1 h prior to TGF-β1). SJN prevented both albumin- and TGF-β1-induced increase in excitatory presynaptic (Alb=17 Alb+SJN=21, p < 0.0001; TGFβ=18 TGFβ+SJN=20, p < 0.0001), postsynaptic (Alb+SJN, p < 0.0001; TGFβ+SJN, p < 0.0001) and co-localized puncta (Alb+SJN, p=0.00012; TGFβ+SJN, p < 0.0001), to a level comparable with the untreated control group (Fig. 4A). These results demonstrate that activation of the ALK5/TGF-β pathway is critical for albumin–induced excitatory synaptogenesis. Moreover, ALK5/TGF-β1 inhibition in control cultures was sufficient per se to reduce synaptic counts (p=0.0102, Fig. 4A), suggesting that baseline release of TGF-β1 promotes synaptogenesis in cultures.
Figure 4. Albumin-induced excitatory synaptogenesis is mediated by astrocyte-driven TGF-β\ALK5 signaling.
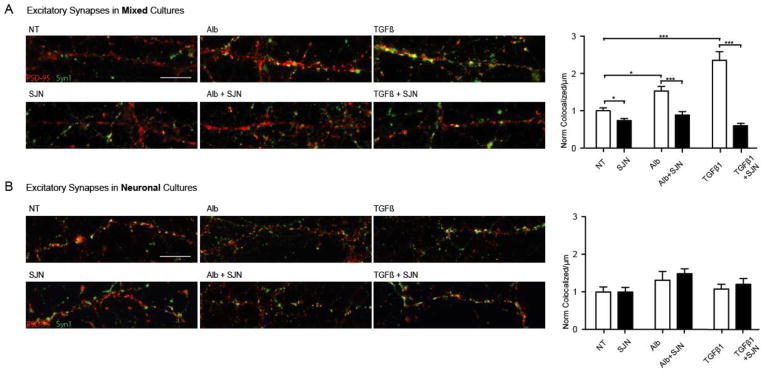
(A) In mixed cultures of astrocytes and neurons, 72 hour TGF-β1 incubation (n=18) induced a synaptogenic effect similar to that of albumin (n=17), with both treatments resulting in significantly higher synaptic counts compared to untreated controls (no treatment (NT)=17). Both albumin- and TGF-β1- induced synaptogenesis was significantly blocked by the selective TGF-β type I receptor ALK5 inhibitor (SJN2511, SJN) (NT=15;Alb+SJN=21;TGFβ+SJN=20). (B) No significant increase in synaptic counts was observed in enriched neuronal cultures (P>0.6; Alb=13; TGFβ=14; NT=15), and pre-incubation with SJN had no effect on synaptic counts in the absence of astrocytes (P>0.9; SJN=15; SJN+Alb=13; SJN+TGFβ1=13). Error bars indicate SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Albumin\TGF-β-induced excitatory synaptogenesis is astrocyte dependent
We have previously demonstrated that exposure to albumin induces activation of the TGF-β pathway and secretion of TGF-β1 in astrocytes, and not in neurons (Bar-Klein et al., 2014). In order to test the role of astrocytes in albumin-induced synaptogenesis, we created neuronal enriched cultures by eliminating astrocytes with the mitotic inhibitor AraC. Notably, in the absence of a significant astrocytic population, exposure to albumin did not induce any significant changes in the number of excitatory or inhibitory synaptic puncta (NT=15; Alb=13; Fig. 4B). Similarly, exposure to TGF-β1 or the ALK5 blocker SJN did not induce any significant changes in the number of excitatory or inhibitory synaptic puncta in the absence of astrocytes (TGF-β1=14; SJN=15; SJN&Alb=13; SJN&TGF-β1=13; Fig. 4B). These results demonstrate that astrocytes are necessary for albumin- and TGF-β1-induced synaptogenesis.
ICV albumin and TGF-β induce anatomically specific excitatory synaptogenesis in vivo
To verify the in vitro evidence of albumin-induced synaptogenesis in-vivo, mice were ICV infused for 72 hours with either: aCSF (ctrl=4), albumin (Alb =4), TGF-β1 (TGFβ=3) or albumin & SJN2511 (Alb+SJN=3). Both albumin and TGF-β1 significantly increased excitatory synaptic counts in the entorhinal cortex (EC), motor cortical areas (CTX), and the CA1 area of the hippocampus (p<0.0001, compared to ctrl, Fig. 5B), with no significant effect in the DG or CA3 (p>0.2, compared to ctrl, Fig. 5B). Notably, co-infusion with SJN prevented synaptogenesis in all three brain areas (p<0.0001, compared to albumin, Fig. 5B). In accordance with the immunohistochemistry, mRNA levels revealed that exposure to albumin (n=8) significantly increased excitatory synaptic proteins PSD95 (p<0.0001) and VGlut (p=0.0025) in the neocortex, but not in the homogenates from the entire hippocampus. Thus, similar to the in vitro findings, albumin induces excitatory synaptogenesis through TGF-β signaling, and this process can be prevented in-vivo with SJN2511.
Figure 5. Albumin exposure in-vivo is associated with region-specific, TGF-β1 mediated synaptogenesis.

(A) Representative z-stacked images of pre-synaptic (Syn1, green) and post-synaptic (PSD-95) markers 72 h after pump implantation. (B) Synaptic quantification demonstrates that TGF-β1 (n=3 × 6 slices) and albumin (n=4 × 6 slices) induce similar synaptogenic effects, blocked by SJN2511. (C) RT-PCR demonstrates a significant increase in cortical expression of post-synaptic (PSD95) and pre-synaptic (Vglut) protein mRNA in albumin treated mice (n=8), compared to controls (n=12). Error bars indicate SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
SJN2511 prevents albumin-induced seizures in-vivo
Exposure to TGF-β1 was previously reported to induce increased hyperexcitability in evoked potentials recoded in vitro (Cacheaux et al., 2009). As both albumin and TGF-β1 have similar synaptogenic effects, we set out to test whether albumin-induced seizures (Fig. 1) could be replicated by infusion of TGF-β1, and prevented by ALK5 inhibition. Thus, an additional cohort of mice was implanted with epidural ECoG electrodes and ICV pumps loaded with either TGF-β1 (TGFβ =5) or albumin & SJN2511 (Alb+SJN =8). ECoG data was acquired over 14 days and analyzed using the automated seizure detection algorithm (see Methods and (Bar-Klein et al., 2014)). While 77% of albumin treated mice developed epilepsy (p=0.0004, compared to controls), TGF-β1 induced spontaneous recurring seizures in 100% of infused animals (p=0.0003, compared to controls, Fig. 6A). Notably, similar to albumin, TGF-β1-induced seizures appeared after a latent period of several days (5.8 ± 1.76 days, p=0.8039 compared to albumin), having a comparable seizure duration (39.3 ± 13.4 sec) and frequency (2.1857 ± 0.428 per day). Most importantly, SJN2511 prevented the development of epilepsy in all but one albumin-treated mice (p=0.0075, compared to controls, Fig. 6B), demonstrating that blocking albumin activation of ALK5 signaling is sufficient to significantly reduce the incidence of epilepsy in this model.
Figure 6. Albumin-induced seizures are mediated by TGF-β and can be prevented with SJN2511.

(A) ICV TGF-β induces spontaneous epileptic seizures, comparable to those observed following ICV albumin. However, while albumin induced epilepsy in 77% of mice (n=13), all TGF-β-infused mice developed seizures (n=5). Moreover, co-infusion with SJN2511 (n=8) prevented seizures in all but one animal. (B) Albumin and TGF-β-induced seizures persisted throughout the monitoring period (and well after pump extraction on day 7). The one epileptic animal in the SJN group suffered two seizures on day 9.
Discussion
Here we demonstrate an albumin-induced and TGF-β-mediated synaptogenic mechanism critical in the epileptogenic process. We show both in vitro and in vivo that albumin induces excitatory synapse formation, and that this effect is astrocyte and ALK5 dependent.
TGF-β signaling in astrocytes following exposure to albumin was previously shown to promote several functional changes that may be associated with increased neuronal excitability, including induction of pro-ictogenic inflammatory cytokines (Frigerio et al., 2012), down-regulation of gap junction proteins (connexin 30 and 43 (Braganza et al., 2012) and astrocytic potassium, glutamate and water channels (Cacheaux et al., 2009; Ivens et al., 2007). This transcriptional response was then associated with a frequency dependent increase in the concentration of extracellular potassium ([K+]o) and hyperexcitability of neighboring neurons (David et al., 2009; Lapilover et al., 2012). However, the reduced capacity of [K+]o buffering was found to be immediate and short-lived, returning to pre-albumin levels within 4 days (Braganza et al., 2012). In contrast, the tendency of the network for seizures post exposure to albumin was found to be delayed and long-lasting (Bar-Klein et al., 2014; Seiffert et al., 2004), resembling the delayed appearance of epilepsy in patients following insults to the brain. Re-organization of the neural network has been suggested to underlie chronic seizures, in light of evidence of axonal sprouting and synapse formation in status epilepticus and traumatic injury models of epilepsy (Jin et al., 2006; Liauw et al., 2008; Scheff et al., 2005). Our findings demonstrate a mechanism linking brain injury to synaptogenesis and epilepsy, through BBB dysfunction and albumin–induced, astrocytic TGF-β\ALK5- mediated selective formation of excitatory synapses.
BBB dysfunction has been well documented following epileptogenic insults and in epileptic tissue (Aronica et al., 2008; Kaya et al., 2012; Marcon et al., 2009; Raabe et al., 2012; Shlosberg et al., 2010; Tomkins et al., 2001). Animal studies indicate that BBB dysfunction promotes seizure generation (Marchi et al., 2007; van Vliet et al., 2007) and is associated with epileptogenesis (Bar-Klein et al., 2014; Seiffert et al., 2004). Furthermore, the delayed effect of chemical BBB opening by bile salts (Ivens et al., 2007; Seiffert et al., 2004; Tomkins et al., 2007) was reproduced through direct cortical exposure to the most common serum protein, albumin, suggesting albumin as a key initiator of the epileptogenic process following BBB injury (Bar-Klein et al., 2014; Seiffert et al., 2004). Here we focused on the mechanisms underlying albumin-induced network reorganization, establishing a minimally invasive mouse model of BBB dysfunction and epilepsy, through ICV infusion of albumin.
To date, many existing models of acquired epilepsy are initiated by status epilepticus, temporal lobe sclerosis and massive cell loss and dispersion. However status epilepticus is a rare event in human patients and 30–40% of patients with temporal lobe epilepsy do not have sclerosis (De Lanerolle et al., 2012). Here we present a non-status epilepticus model of chronic epilepsy, leading to delayed appearance of recurrent seizures in mice. Furthermore, although we did not rule out endfolium sclerosis or pyramidal interneuron loss, we found no apparent evidence of gross abnormalities for up to 32 days, thus allowing the study of epileptogenic mechanisms independent of sclerosis. Moreover, direct infusion of albumin mimics the pathological accumulation of albumin observed in human epileptic tissue (Raabe et al., 2012; Schmitz et al., 2013; van Vliet et al., 2007) and in animal models of status epilepticus (Ndode-Ekane et al., 2010; Pont et al., 1995; Ruth, 1984; Saija et al., 1992; van Vliet et al., 2014, 2007; Zucker et al., 1983). However, while albumin is sufficient to induce the development of epilepsy in a naïve brain, the interplay of BBB dysfunction and albumin-induced network alterations with other epileptogenic insults (e.g. neuronal sprouting, sclerosis, channelopathies), in the context of an epileptic brain remains to be studied (Friedman and Heinemann, 2012).
We further demonstrate that albumin and TGF-β1 have similar epileptogenic effects that are prevented by specific inhibition of the TGF-β\ALK5 pathway. Our findings validate the role of TGF-β in PIE, supporting the epileptogenic role of the ALK5 SMAD 2,3 signaling (Bar-Klein et al., 2014). While TGF-β1 infusion induced seizures in all mice, albumin induced seizures in only 77%. Interestingly, albumin also induced a significant yet variable increase in GFAP, encouraging further studies to explore the cause of this variability and whether it may be correlated to albumin accumulation and/or seizure onset/frequency. Blocking the TGF-β pathway eliminated the occurrence of spontaneous seizures in all but one mouse, which presented a total of two seizures (Fig. 6D,E). These results suggest either an incomplete block or the involvement of additional mechanisms in albumin-induced epilepsy.
Our in-vitro results showed that both albumin and TGF-β1 induce the formation of excitatory synapses, with no significant effect on inhibitory synaptic counts in astrocyte/neuronal mixed temporal cell cultures. Moreover, the observed excitatory synaptogenesis was found to be astrocyte dependent, as it was absent in astrocyte depleted cultures. These results are consistent with the absence of TGF-β mediated synaptogenesis in astrocyte-free cultures in retinal ganglion cells (Christopherson et al., 2005), and the plethora of reports of astrocyte-driven excitatory synapse formation, through secretion of thrombospondins (TSPs)(Cekanaviciute et al., 2014; Christopherson et al., 2005; Liauw et al., 2008), hevin (Kucukdereli et al., 2011) and glypicans 4 and 6 (Allen et al., 2012). Cell cultures are derived from temporal lobe embryonic tissue representing hippocampal and entorhinal cortex. To enable a detailed analysis of regional albumin-induced changes in synaptogenesis we next analyzed excitatory synapses in in-vivo exposure of albumin exposure, and found that both albumin and TGF-β1 increase excitatory synaptic counts in the EC, CTX, and the CA1, with no significant effect in the DG or CA3. Interestingly, recent reports have associated TGF-β signaling with both excitatory and inhibitory synaptogenesis in mixed and in pure neuronal cortical cultures (Diniz et al., 2014, 2012; Yu et al., 2014). Together with these studies, our findings suggest that TGF-β may effect different cellular targets and regulate various processes in a region and\or dose dependent manner.
Further studies are required to understand the detailed characteristics of TGF-β signaling and the mechanisms of its synaptogenic effect. NMDA post-synaptic currents are likely to be part of this mechanism, since: [1] TGF-β mediated synaptogenesis is associated with secretion of the NMDA co-agonist D-serine (Diniz et al., 2012); [2] post-albumin accumulation of [K+]o predicts over-activation of NMDA channels (David et al., 2009); and [3] albumin-induced up-regulation of IL1β or HMGB1 in astrocytes may enhance Ca influx through NMDA receptor channels (Balosso et al., 2008; Frigerio et al., 2012; Maroso et al., 2010; Viviani et al., 2003).
Using the new model of BBB dysfunction in mice, we reveal a synaptogenic cascade leading to spontaneous recurrent seizures, and demonstrate a method for preventing this outcome. While it was suggested that brain injuries elicit inflammatory signaling and synaptogenesis as part of a compensatory repair mechanism (Falo et al., 2008; Li et al., 2013; Lin et al., 2003; Ruscher et al., 2011; Scheff et al., 2005), here we suggest that this repair response may in and of itself lead to enhanced pathological connectivity and tendency for spontaneous seizures. Furthermore, we show that serum albumin can act as a potent pro-synaptogenic signaling molecule, suggesting that regulation of BBB integrity may allow controlled synaptogenesis in injured tissue. Future research is needed to understand the therapeutic role of post-injury synaptogenesis and the regulating mechanisms, required for the maintenance of a normal excitatory\inhibitory balance. While the neuronal pathways activated following albumin-mediated inflammatory response remain to be identified, they may offer an opportunity to further manipulate and refine post-injury synapse formation.
Highlights.
Injection of albumin into the ventricles of mice results in gliosis and seizures
TGF-β1 and albumin have similar epileptogenic effects
Albumin and TGF-β1 induce similar excitatory synaptogenesis in vitro and in vivo
The synaptogenic effect is astrocyte-dependent
Inhibition of TGF-β/ALK5 pathway prevents excitatory synaptogenesis and epilepsy
Acknowledgments
The research leading to these results has received funding from the European Union’s Seventh Framework Program (FP7/2007-2013) under grant agreement no.602102 (EPITARGET, AF), the Israel Science Foundation (713/11, AF), the National Institute of Health (RO1/NINDS NS066005, DK, AF) and the German Science Foundation (DFG-SFB TR3, AF).
Glossary
- PIE
Post injury epilepsy
- TGF-β
Transforming growth factor beta
- BBB
blood-brain barrier
- IL1R/TLR
Pro-inflammatory IL-1 receptor/Toll-like receptor
- TSPs
Thrombospondins
- ICV
IntraCerebroVentricular
- aCSF
artificial cerebrospinal fluid
- FITC
fluorescein isothiocyanate
- IHC
Immunohistochemistry
- PBS
phosphate-buffered saline
- GFAP
Glial fibrilic acidic protein
- ANN
Artificial neural network
- DIV
Days in vitro
- ICC
Immunocytochemistry
- SE
Status epilepticus
- SJN
SJN2511
- Alb
Albumin
- EC
Enthorinal cortex
- CTX
Motor cortical areas
Footnotes
Competing interests:
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott NJ, Friedman A. Overview and introduction: the blood-brain barrier in health and disease. Epilepsia. 2012;53(Suppl 6):1–6. doi: 10.1111/j.1528-1167.2012.03696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486:410–4. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Boer K, Becker A, Redeker S, Spliet WG, van Rijen PC, Wittink F, Breit T, Wadman WJ, Lopes da Silva FH, Troost D, Gorter JA. Gene expression profile analysis of epilepsy-associated gangliogliomas. Neuroscience. 2008;151:272–292. doi: 10.1016/j.neuroscience.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Babb TL, Kupfer WR, Pretorius JK, Crandall PH, Levesque MF. Synaptic reorganization by mossy fibers in human epileptic fascia dentata. Neuroscience. 1991;42:351–363. doi: 10.1016/0306-4522(91)90380-7. [DOI] [PubMed] [Google Scholar]
- Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, Vezzani A. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain. 2008;131:3256–65. doi: 10.1093/brain/awn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Klein G, Cacheaux LP, Kamintsky L, Prager O, Weissberg I, Schoknecht K, Cheng P, Kim SY, Wood L, Heinemann U, Kaufer D, Friedman A. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann Neurol. 2014;75:n/a–n/a. doi: 10.1002/ana.24147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braganza O, Bedner P, Hüttmann K, von Staden E, Friedman A, Seifert G, Steinhäuser C, Huttmann K, Steinhauser C. Albumin is taken up by hippocampal NG2 cells and astrocytes and decreases gap junction coupling. Epilepsia. 2012;53:1898–1906. doi: 10.1111/j.1528-1167.2012.03665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Engel J. Chronic epileptogenesis requires development of a network of pathologically interconnected neuron clusters: a hypothesis. Epilepsia. 2000;41(Suppl 6):S144–52. doi: 10.1111/j.1528-1157.2000.tb01573.x. [DOI] [PubMed] [Google Scholar]
- Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, Heinemann U, Friedman A, Kaufer D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014 doi: 10.1002/glia.22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–33. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Chung WS, Barres BA. The role of glial cells in synapse elimination. Curr Opin Neurobiol. 2012;22:438–45. doi: 10.1016/j.conb.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14:311–21. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Jiang X, Taylor A, Mennerick S. Astrocyte-derived thrombospondins mediate the development of hippocampal presynaptic plasticity in vitro. J Neurosci. 2012;32:13100–10. doi: 10.1523/JNEUROSCI.2604-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Carmignoto G, Steinhäuser C. Astrocytic Targets Provide New Avenues for the Therapeutic Treatment of Epilepsy. Neuroscientist. 2014 doi: 10.1177/1073858414523320. 1073858414523320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham AS, Salvador R, Coles JP, Chatfield DA, Bradley PG, Johnston AJ, Steiner LA, Fryer TD, Aigbirhio FI, Smielewski P, Williams GB, Carpenter TA, Gillard JH, Pickard JD, Menon DK. Physiological thresholds for irreversible tissue damage in contusional regions following traumatic brain injury. Brain. 2005;128:1931–42. doi: 10.1093/brain/awh536. [DOI] [PubMed] [Google Scholar]
- David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009;29:10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36:174–84. doi: 10.1016/j.tins.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Diniz LP, Almeida JC, Tortelli V, Vargas Lopes C, Setti-Perdigão P, Stipursky J, Kahn SA, Romão LF, de Miranda J, Alves-Leon SV, de Souza JM, Castro NG, Panizzutti R, Gomes FCA. Astrocyte-induced synaptogenesis is mediated by transforming growth factor β signaling through modulation of D-serine levels in cerebral cortex neurons. J Biol Chem. 2012;287:41432–45. doi: 10.1074/jbc.M112.380824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz LP, Tortelli V, Garcia MN, Araújo APB, Melo HM, Seixas da Silva GS, De Felice FG, Alves-Leon SV, de Souza JM, Romão LF, Castro NG, Gomes FCA. Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia. 2014:n/a–n/a. doi: 10.1002/glia.22713. [DOI] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–50. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–92. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–31. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falo MC, Reeves TM, Phillips LL. Agrin expression during synaptogenesis induced by traumatic brain injury. J Neurotrauma. 2008;25:769–83. doi: 10.1089/neu.2008.0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Ko CP. Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-beta1. J Neurosci. 2008;28:9599–609. doi: 10.1523/JNEUROSCI.2589-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Heinemann U. Role of Blood-Brain Barrier Dysfunction in Epileptogenesis. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. Oxford University Press; Bethesda (MD): 2012. [PubMed] [Google Scholar]
- Friedman A, Kaufer D, Heinemann U. Blood-brain barrier breakdown-inducing astrocytic transformation: Novel targets for the prevention of epilepsy. Epilepsy Res. 2009;85:142–9. doi: 10.1016/j.eplepsyres.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Kaufer D, Shemer J, Hendler I, Soreq H, Tur-Kaspa I. Pyridostigmine brain penetration under stress enhances neuronal excitability and induces early immediate transcriptional response. Nat Med. 1996;2:1382–1385. doi: 10.1038/nm1296-1382. [DOI] [PubMed] [Google Scholar]
- Frigerio F, Frasca A, Weissberg I, Parrella S, Friedman A, Vezzani A, Noé FM, Noe’ FM. Long-lasting pro-ictogenic effects induced in vivo by rat brain exposure to serum albumin in the absence of concomitant pathology. Epilepsia. 2012;53:1887–97. doi: 10.1111/j.1528-1167.2012.03666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C. Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp. 2010 doi: 10.3791/2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer v pyramidal neurons of chronically injured epileptogenic neocortex in rats. J Neurosci. 2006;26:4891–900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya M, Becker AJ, Gürses C. Blood-brain barrier, epileptogenesis, and treatment strategies in cortical dysplasia. Epilepsia. 2012;53(Suppl 6):31–6. doi: 10.1111/j.1528-1167.2012.03700.x. [DOI] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A. 2011;108:E440–9. doi: 10.1073/pnas.1104977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapilover EG, Lippmann K, Salar S, Maslarova A, Dreier JP, Heinemann U, Friedman A. Peri-infarct blood-brain barrier dysfunction facilitates induction of spreading depolarization associated with epileptiform discharges. Neurobiol Dis. 2012;48:495–506. doi: 10.1016/j.nbd.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008;28:2119–30. doi: 10.1523/JNEUROSCI.5159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LY, Li JL, Zhang HM, Yang WM, Wang K, Fang Y, Wang Y. TGFβ1 treatment reduces hippocampal damage, spontaneous recurrent seizures, and learning memory deficits in pilocarpine-treated rats. J Mol Neurosci. 2013;50:109–23. doi: 10.1007/s12031-012-9879-1. [DOI] [PubMed] [Google Scholar]
- Liauw J, Hoang S, Choi MM, Eroglu C, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- Lin T-n, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY. Differential Regulation of Thrombospondin-1 and Thrombospondin-2 After Focal Cerebral Ischemia/Reperfusion. Stroke. 2003;34:177–186. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, Hallene K, Diglaw T, Franic L, Najm I, Janigro D. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia. 2007;48:732–42. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Granata T, Janigro D. Inflammatory pathways of seizure disorders. Trends Neurosci. 2013;37:55–65. doi: 10.1016/j.tins.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco P, DeFelipe J. Altered synaptic circuitry in the human temporal neocortex removed from epileptic patients. Exp Brain Res. 1997;114:1–10. doi: 10.1007/pl00005608. [DOI] [PubMed] [Google Scholar]
- Marcon J, Gagliardi B, Balosso S, Maroso M, Noé F, Morin M, Lerner-Natoli M, Vezzani A, Ravizza T. Age-dependent vascular changes induced by status epilepticus in rat forebrain: implications for epileptogenesis. Neurobiol Dis. 2009;34:121–32. doi: 10.1016/j.nbd.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–9. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- Ndode-Ekane XE, Hayward N, Gröhn O, Pitkänen A. Vascular changes in epilepsy: functional consequences and association with network plasticity in pilocarpine-induced experimental epilepsy. Neuroscience. 2010;166:312–32. doi: 10.1016/j.neuroscience.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Roberts JD, Baude A, Richards JG, Sieghart W, Somogyi P. Immunocytochemical localization of the alpha 1 and beta 2/3 subunits of the GABAA receptor in relation to specific GABAergic synapses in the dentate gyrus. Eur J Neurosci. 1995;7:630–46. doi: 10.1111/j.1460-9568.1995.tb00667.x. [DOI] [PubMed] [Google Scholar]
- Packard M, Mathew D, Budnik V. Wnts and TGF beta in synaptogenesis: old friends signalling at new places. Nat Rev Neurosci. 2003;4:113–20. doi: 10.1038/nrn1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont F, Collet A, Lallement G. Early and transient increase of rat hippocampal blood-brain barrier permeability to amino acids during kainic acid-induced seizures. Neurosci Lett. 1995;184:52–4. doi: 10.1016/0304-3940(94)11166-g. [DOI] [PubMed] [Google Scholar]
- Raabe A, Schmitz AK, Pernhorst K, Grote A, von der Brelie C, Urbach H, Friedman A, Becker AJ, Elger CE, Niehusmann P. Cliniconeuropathologic correlations show astroglial albumin storage as a common factor in epileptogenic vascular lesions. Epilepsia. 2012;53:539–48. doi: 10.1111/j.1528-1167.2012.03405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA. Neurological diseases in relation to the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1139–51. doi: 10.1038/jcbfm.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscher K, Shamloo M, Rickhag M, Ladunga I, Soriano L, Gisselsson L, Toresson H, Ruslim-Litrus L, Oksenberg D, Urfer R, Johansson BB, Nikolich K, Wieloch T. The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke. Brain. 2011;134:732–746. doi: 10.1093/brain/awq367. [DOI] [PubMed] [Google Scholar]
- Ruth RE. Increased cerebrovascular permeability to protein during systemic kainic acid seizures. Epilepsia. 1984;25:259–68. doi: 10.1111/j.1528-1157.1984.tb04185.x. [DOI] [PubMed] [Google Scholar]
- Saija A, Princi P, Pisani A, Santoro G, De Pasquale R, Massi M, Costa G. Blood-brain barrier dysfunctions following systemic injection of kainic acid in the rat. Life Sci. 1992;51:467–77. doi: 10.1016/0024-3205(92)90023-i. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719–32. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- Schmitz AK, Grote A, Raabe A, Urbach H, Friedman A, von Lehe M, Becker AJ, Niehusmann P. Albumin storage in neoplastic astroglial elements of gangliogliomas. Seizure. 2013;22:144–50. doi: 10.1016/j.seizure.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins O, Friedman O, Ivens S, Reiffurth C, Major S, Dreier JP, Heinemann U, Friedman A. Blood-brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol Dis. 2007;25:367–377. doi: 10.1016/j.nbd.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Tomkins O, Kaufer D, Korn A, Shelef I, Golan H, Reichenthal E, Soreq H, Friedman A. Frequent blood-brain barrier disruption in the human cerebral cortex. Cell Mol Neurobiol. 2001;21:675–691. doi: 10.1023/A:1015147920283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins O, Shelef I, Kaizerman I, Eliushin A, Afawi Z, Misk A, Gidon M, Cohen A, Zumsteg D, Friedman A. Blood-brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry. 2008;79:774–7. doi: 10.1136/jnnp.2007.126425. [DOI] [PubMed] [Google Scholar]
- Van Vliet EA, da Costa Araujo S, Redeker S, van Schaik R, Aronica E, Gorter JA, da Costa Araújo S. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130:521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- Van Vliet EA, Otte WM, Gorter JA, Dijkhuizen RM, Wadman WJ. Longitudinal assessment of blood-brain barrier leakage during epileptogenesis in rats. A quantitative MRI study. Neurobiol Dis. 2014;63:74–84. doi: 10.1016/j.nbd.2013.11.019. [DOI] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011a;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun. 2011b;25:1281–9. doi: 10.1016/j.bbi.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C-Y, Gui W, He H-Y, Wang X-S, Zuo J, Huang L, Zhou N, Wang K, Wang Y. Neuronal and Astroglial TGFβ-Smad3 Signaling Pathways Differentially Regulate Dendrite Growth and Synaptogenesis. Neuromolecular Med. 2014 doi: 10.1007/s12017-014-8293-y. [DOI] [PubMed] [Google Scholar]
- Zucker DK, Wooten GF, Lothman EW. Blood-brain barrier changes with kainic acid-induced limbic seizures. Exp Neurol. 1983;79:422–33. doi: 10.1016/0014-4886(83)90223-6. [DOI] [PubMed] [Google Scholar]