

Abstract
Differential Sensing (DS) methods traditionally use spatially arrayed receptors and optical signals to create score plots from multivariate data that classify individual analytes or complex mixtures. Herein, a new approach is described, in which nucleic acid sequences and sequence counts are used as the multivariate data without the necessity of a spatial array. To demonstrate this approach to DS, previously selected aptamers, identified from the literature, were used as semi-specific receptors, Next-Gen DNA sequencing was used to generate data, and cell line differentiation was the test-bed application. Analysis of a Principal Component Analysis (PCA) loading plot revealed cross-reactivity between the aptamers. The technique generates high-dimensionality score plots, and should be applicable to any mixture of complex and subtly different analytes for which nucleic acid-based receptors exist.
Keywords: Next Generation Sequencing, Principal Component Analysis, Aptamer, Differential Sensing, quantitative Real Time PCR
Differential Sensing (DS) uses a suite of cross-reactive receptors that generate a unique pattern of interaction via chemometric routines to differentiate complex mixtures.[1] DS is a powerful approach when the analytes are not fully characterized or even known. There are currently many kinds of receptors used in DS routines, including biomolecules, synthetic receptors,[2] solid composites,[1,3] and nanoparticles.[4] Optical spectroscopy is the most common experimental technique to analyze the suite of solution-based receptor responses.[5] While spectroscopy is simple and routine, because the observed bands are broad, the absorbance/emission values often co-vary, and therefore there is little differential reactivity for creating high dimensionality chemometric patterns.[6] Further, with the exception of the use of suspension arrays,[7] DS methods most commonly use spatially separated receptors.[4,8,9] An approach that does not involve the use of optical signals or spatial arrays could dramatically improve and simplify DS routines.
In the study described herein, we advance the field of DS by showing that, rather than using optical values, the abundance of captured nucleic acids measured by Next Generation Sequencing (NGS) can be used as input data for chemometric protocols.[10] To demonstrate this approach, we used nucleic acid-based receptors, aptamers, that were targeted against antigens on cell surfaces in order to classify cell types (Table S1). In contrast to protein receptors (such as antibodies) the sequences of nucleic acid aptamers can be easily interrogated using NGS. Moreover, while aptamers can be highly selective receptors,[11] the selectivities of most aptamers have not been fully vetted, especially against complex targets such as cells. Indeed, we have previously found that aptamers selected against single targets are often found to be cross-reactive (i.e. semi-specific) in the context of cells.[12] We therefore viewed aptamers as a convenient source of semi-specific receptors for DS, where relaxed selectivity is actually an advantage.[13] Further, we anticipated that, unlike optical bands, individual aptamer sequences would have low co-variance, ultimately resulting in improved chemometric classification.
Forty-six previously selected aptamers, grouped into three sets, were recreated for use in this study: aptamers selected to bind specific cell types (C1–C17), aptamers selected to bind molecular targets found on a cell surface (T1–T27), and aptamers not expected to bind cells (N1–N2) (Table S1). It was also important that the aptamers not undergo degradation by nucleases, and thus only 2’-fluoropyrimidines modified aptamers were chosen for use in this study.[14] It was not the purpose of these experiments to validate the purported selectivities of the aptamers against cells, since this information is largely unknown.
Among the C1–C17 aptamers, the individual members had been reported to bind gliomas (U87MG, U251), prostate cancer (PC3), non-small cell lung carcinoma (NSCLC H358), small cell lung carcinoma (SNLC H562), rat adrenal medulla cells that express the human RETC634Y mutant receptor (PC12/MEN2A), and HeLa cells. Aptamers T1–T27 had been selected against cell surface targets, including extracellular matrix proteins (TN-c, PAI-1), surface receptors (EGFR, EGFRviii, OX40, VCAM-1, NTS-1, PfEMP-1, αvβ3), non-receptor proteins (PSMA, CD4, 4-1BB), and the carbohydrate Sialyl Lewis X. Finally, aptamers N1 and N2 were included as negative controls because they were not expected to bind cells. Because molecular targets are common between cell lines, it is unsurprising that many of the chosen aptamers show broad specificities. For example, aptamer C2 was originally selected against U87MG cells, but is known to bind to the LN-18, LN-229, U87MGΔVIII, and TB10 cell lines.[15]
The aptamers were modified with common primer extensions at their 5’- and 3’-ends in order to facilitate co-amplification within mixtures. The primer sequences were chosen so that they did not lead to significant pairing with any of the chosen aptamer sequences. Nonetheless, because it was possible that the extension sequences would lead to misfolding or otherwise impair the functionality of a chosen aptamer, oligonucleotide complements to the primer sequences were added to hybridize and block the primer regions.
While the semi-specificity of affinity reagents can be an advantage for chemometrics, we wished to validate that any differences in sequence representation observed were indeed due to differential binding. Therefore, the aptamer panel was exposed to two cell lines: A431, an epidermoid carcinoma cell line that expresses high EGFR,[16] and MDA-MB-435, a breast carcinoma that has four-fold lower expression of EGFR.[17] The response of four of the aptamers (T5, T15, C19, C16) was quantified using qRT-PCR (Figure S1). Briefly, cells were grown to confluency in 6-well plates and five panel concentrations were tested: 0, 0.01, 0.1, 1, and 2 pmol of each aptamer, with four experimental replicates per concentration (SI experimental). Cells were incubated with the panels for 30 minutes at RT, washed with DPBS, and lysed. Bound aptamer was quantified using qRT-PCR with primers specific to that aptamer and its abundance (Ct) divided by that in the naïve panel (amplified aptamers without exposure to the cells) was calculated to give fold change. T5 is an anti-EGFR aptamer that has been shown to have differential binding to these lines using FACS,[18] and this aptamer showed enrichment with A431 cells and depletion with MDA-MB-435 cells across all concentrations tested (Figure S1A). The other three aptamers also showed differential responses to the two cell lines, although they were not selected against epitopes present on either of those cells (Figure S1B-D).
This experiment was repeated using NGS, and the response of all of the aptamers in the panel was quantified. Captured aptamers were reverse-transcribed and PCR-amplified using a common primer for all of the aptamers. The abundance of each aptamer was expressed as the proportion of reads for each sequence over the total reads (fold change). The NGS results for T5 confirmed the qRT-PCR results and validated the use of NGS for representation of differential aptamer binding (Figure S2). Each aptamer was then used at a concentration of 1 pmol in further experiments.
Four different cell lines were selected for chemometric analysis using NGS as the readout. In addition to A431 and MDA-MB-435, we tested U87MGΔVIII, a glioma, and HEK, a line derived from non-cancerous human embryonic kidney cells. Figure 1 shows the fold change of each of the aptamers as ratios of relative abundance of bound aptamer compared to abundance in the naïve panel. Individual members of the aptamer pool exhibited a range of specificities, as predicted. The ubiquitous internalizer C15 [19] showed a positive fold change with all cell lines. Similarly, T27 showed a positive fold change with three of the four cell lines, including U87MGVIII. Large positive fold changes in binding were seen for aptamers T1 (selected against PAI-1),[20] T27 (selected against EGFRvIII),[21] and T11 (selected against PSMA)[22] when the panel was exposed to MDA-MB-435. In general, different sequences had very different and reproducible fold changes across the cell types, resulting in a unique pattern for each cell type.
Figure 1.
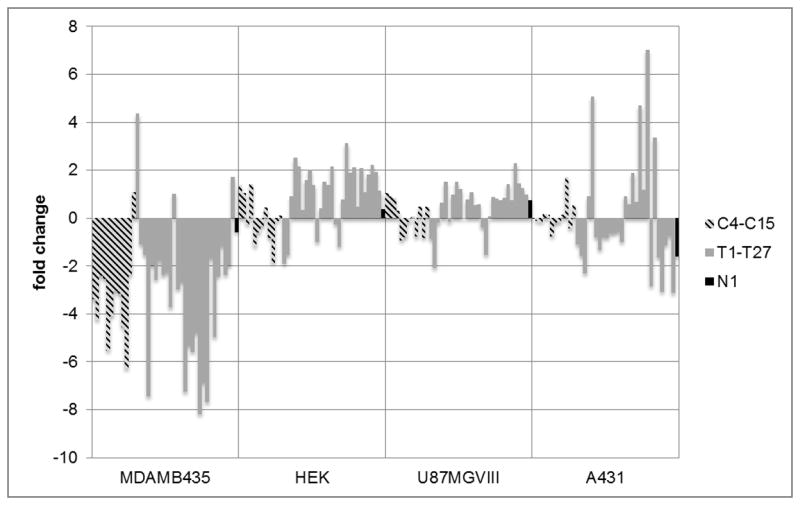
Fold change (defined as log2(Ct lysate/Ct panel) for each aptamer with the cell lines. Each cell line has a unique pattern of response to the sequenced aptamer panel. Both cell binding (C4-C15) and epitope targeting (T1–T27) aptamers are able to bind to cell lines with varying specificites. Some of the aptamers only bind to a particular cell line while many of the aptamers bind to multiple cell lines in variable amounts. The contribution of each aptamer to classification of cell type is clarified using PCA.
We then employed Principal Component Analysis (PCA) in order to make it easier to interpret the contributions of each aptamer to overall classification. PCA reduces the dimensionality in a data set by transforming multi-dimensional variable space to new, orthogonal axes that describe decreasing extents of variance. Such analysis aids in interpretation of multivariate data, such as that presented in Figure 1. Five PCA axes were found to carry variance greater than the noise in the data, with the first three axes accounting for nearly 86% of the variance (Figure 2), with 5 and 4% on the next two axes. The fact that several axes of the score plot carry significant variance demonstrates a high degree of cross-reactivity between the aptamers.[23]
Figure 2.

PCA score plot of 4 different cell lines across the first three components derived from responses to the aptamers at 1 pmol. Scores for each cell line are a result of a combination of responses to each aptamer in the pool. Repeats of each cell line are grouped and the four cell lines are classified based solely on the variance in the fold change of the aptamer pool.
PCA score plots show the response of each cell line to the aptamer panel in the new, transformed variable space. Each axis in a score plot is a principal component (PC) that is derived from linear combinations of the original variables. In our experiments, the original variables are the relative abundances of sequence counts for each aptamer and the PCs are combinations of these variables that capture the variance in the data. The score of a cell type replicate on a PC is derived by multiplying the data shown in Figure 1 for each aptamer by the contribution of each aptamer to the variance described by that PC.
The score plot in Figure 2 for the first three principal components shows four distinct groupings of replicates corresponding to each of the four cell lines with the same stock mixture of aptamers. The grouping in the score plot confirms the high reproducibility of our method because the operator does not bias PCA. The clustering of replicates within the same cell type and the separation of different cell types arise from the intrinsic variance within the data.
PCA loading plots illustrate the contribution of each of the original variables to each of the derived PCs. The loading plot in Figure 3 shows that several aptamers have large loadings along the major PC axes, and thus contribute nearly equally to discrimination between cell lines. Further, the loading vectors for individual aptamers often point between cell groups, meaning that many aptamers bind and contribute to differentiating more than one cell line, further emphasizing the semi-specific nature of these reagents.
Figure 3.
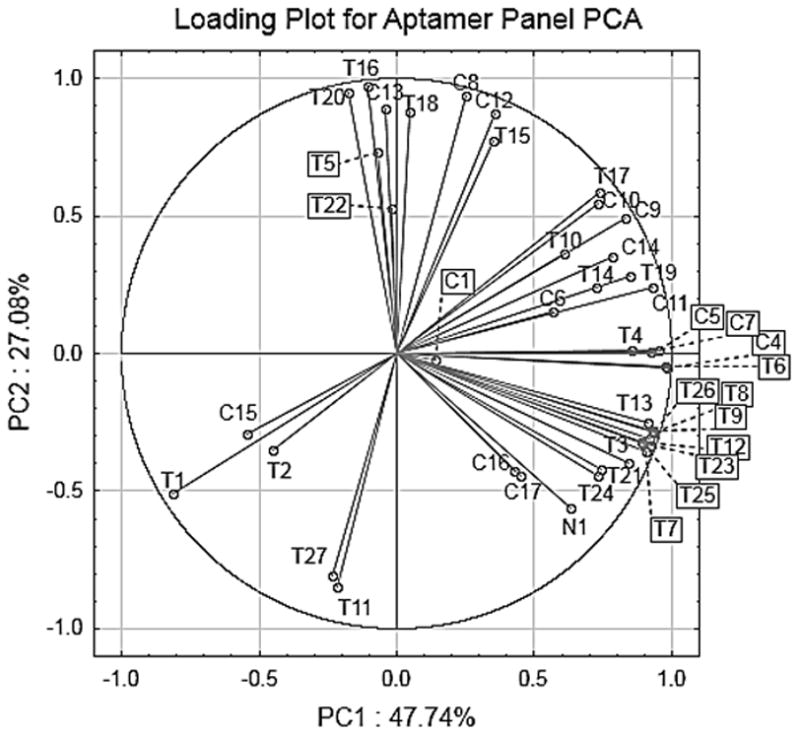
The loading plot of the aptamer panel on the first two principal components for the PCA of the four cell lines tested. Many aptamers contribute to each differentiation axis. Combinations of responses to each aptamer result variable scores for each cell line and thus cell line differentiation.
To determine which aptamers contribute to each PC axis and to cell line differentiation, the loading plot in Figure 3 can be examined in conjunction with the score plot in Figure 2. C4 had the highest correlation value for PC1 at 0.98; this aptamer was selected using PC3 cells, high-metastatic potential prostate cancer cells that are PSMA negative.[24] This aptamer also contributes highly to classification of HEK and to a lesser extent to that of U87MGΔVIII, illustrating that the aptamer may be capable of binding targets for which it was not selected. The anti-PSMA aptamer T11 showed a strong negative correlation on PC2 (−0.86) and three cell lines score negatively on this axis. Aptamer T5 was positively associated with PC2 and, as anticipated, contributes to the location of A431 since this line over-expresses EGFR.[16] Similarly T16, an aptamer selected for cell surface CD4, has an even higher correlation (0.97) along PC2 and also contributes to the positive score of A431. MDA-MB-435, U87MGVIII, and HEK, all have positive fold changes for T11 and all score negatively on PC2. This illustrates again the semi-specific nature of the aptamers, as one aptamer can bind many cell lines. Finally, the only aptamer that showed strong correlation along PC3 was C6 (value of −0.80). This aptamer was selected against H526 cells, a small cell lung carcinoma line, but shows a positive fold change with only U87MGVIII, which contributes to the classification and negative score of this glioma line.
The success of the method relies on the fact that several aptamers combine together to differentiate the cells, without foreknowledge of how each aptamer will behave. While it is possible that the differences revealed by the aptamers reflect an underlying set of specific interactions (the response of T5, for example), this is neither required nor necessarily desired in the context of DS. Thus, we find that although the aptamers used in this study were originally selected to specifically bind target cells or epitopes present on cell surfaces, they behave as semi-specific binding reagents capable of differential responses to each of the cellular analytes. In fact, it may be possible to achieve similar classification results using virtually any set of divergent sequences, potentially even a series of randomly generated sequences.
Irrespective of the cross-reactivity of aptamers revealed by this study, the most important advance described herein is the use of NGS as input data for chemometric analysis. This represents a simplified experimental technique relative to spatial separated arrays and optical spectroscopy. This is because NGS occurs in one single rapid process, thereby interrogating all sequences and their copy numbers in a single event. Further, because each aptamer acts independently, there is reduced covariance compared to multiple optical signals from the same receptor, and thus chemometric score plots that carry greater variance along each axis. Of course, the limitation of this method is that the receptors must be nucleic acid based.
In summary, the DS method we have introduced exploits advances in nucleotide sequencing that allows the use of aptamer identity and relative abundance as chemometric input, avoiding the need to spatially array the receptors. This was done without the need to perform experiments to assess the affinity of each aptamer for the chosen analytes. The high information density inherently carried in sequence and sequence count makes this kind of data excellent for generating PCA plots with high levels of variance on numerous axes, potentially allowing a variety of complex mixtures of otherwise subtly different compounds to be readily discriminated.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from both the NIH (R01-GM065515) and the Welch Foundation (F.1151).
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Dr. Eric Anslyn, Email: anslyn@austin.utexas.edu.
Dr. Andrew Ellington, Email: andy.ellington@mail.utexas.edu.
References
- 1.Albert K, Lewis N, Schauer C, Sotzing G, Stitzel S, Vaid D, Walt T. Chem Rev. 2000;100:2595–2626. doi: 10.1021/cr980102w. [DOI] [PubMed] [Google Scholar]
- 2.Anslyn E. J Org Chem. 2007;72:687–699. doi: 10.1021/jo0617971. [DOI] [PubMed] [Google Scholar]; Umali A, LeBoeuf S, Newberry R, Kim S, Tran L, Rome W, Tian T, Taing D, Hong J, Kwan M. Chem Sci. 2011;2:439–445. [Google Scholar]
- 3.Zhang C, Suslick K. J Amer Chem Soc. 2005;127:11548–11549. doi: 10.1021/ja052606z. [DOI] [PubMed] [Google Scholar]
- 4.a) Bajaj A, Miranda O, Kim I, Phillips R, Jerry D, Bunz U, Rotello V. Proc Natl Acad Sci USA. 2009;106:10912–10916. doi: 10.1073/pnas.0900975106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bajaj A, Rana S, Miranda O, Yawe J, Jerry D, Bunz U, Rotello V. Chem Sci. 2010;1:134–138. [Google Scholar]; c) Rana S, Singla A, Bajaj A, Elci S, Miranda O, Mout R, Yan B, Jirik F, Rotello V. ACS Nano. 2012;6:8233–8240. doi: 10.1021/nn302917e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu Q, Yeh Y, Rana S, Jiang Y, Guo L, Rotello V. Cancer Lett. 2013;334:196–201. doi: 10.1016/j.canlet.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) El-Boubbou K, Zhu D, Vasileiou C, Borhan B, Prosperi D, Li W, Huang X. J Am Chem Soc. 2010;132:4490–4499. doi: 10.1021/ja100455c. [DOI] [PubMed] [Google Scholar]
- 5.Adams M, Joyce L, Anslyn E. Uses of Differential Sensing and Arrays in Chemical Analysis. In: Gale PA, Steed JW, editors. Supramolecular Chemistry: From Molecules to Nanomaterials. Vol. 2. John Wiley & Sons, Ltd; West Sussex: 2012. pp. 709–732. [Google Scholar]
- 6.It is very common in DS routines to obtain multiples absorbance values for each individual host. Inherently, these often co-vary and therefore do not contribute to differentiation along the factor axes. .
- 7.Nolan J, Sklar L. Trends Biotechnol. 2002;20:9–12. doi: 10.1016/s0167-7799(01)01844-3. [DOI] [PubMed] [Google Scholar]
- 8.a) Kong H, Liu D, Zhang S, Zhang X. Anal Chem. 2011;83:1867–1870. doi: 10.1021/ac200076c. [DOI] [PubMed] [Google Scholar]; b) Liu Y, Liu Y, Zhang S, Wang S, Zhang S, Zhang X. Anal Chem. 2013;85:6571–6574. doi: 10.1021/ac4014594. [DOI] [PubMed] [Google Scholar]
- 9.Bajaj A, Miranda O, Phillips R, Kim I-B, Jerry D, Bunz U, Rotello V. J Am Chem Soc. 2010;132:1018–1022. doi: 10.1021/ja9061272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.NGS for concentration of proteins, not classification. Turner D, Tuytten R, Janssen K, Lammertyn J, Wuyts J, Pollet J, Eyckerman S, Brown C, Kas K. Anal Chem. 2010;83:666–670. doi: 10.1021/ac102666n.
- 11.a) Jayasena S. Clin Chem. 1999;45:1628–1650. [PubMed] [Google Scholar]; b) Nimjee S, Rusconi C, Sullenger B. Annu Rev Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]; c) Proske D, Blank M, Buhmann R, Resch A. Appl Microbial Biotechnol. 2005;69:367–374. doi: 10.1007/s00253-005-0193-5. [DOI] [PubMed] [Google Scholar]; d) Xiao Z, Shangguan D, Cao Z, Fang X, Tan W. Chem Eur J. 2008;14:1769–1775. doi: 10.1002/chem.200701330. [DOI] [PubMed] [Google Scholar]; e) Keefe A, Pai S, Ellington A. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li N, Ebright J, Stovall G, Chen X, Nguyen H, Singh A, Syrett A, Ellington A. J Proteome Res. 2009;8:2438–2448. doi: 10.1021/pr801048z. [DOI] [PubMed] [Google Scholar]
- 13.Stewart S, Syrett A, Pothukuchy A, Bhadra S, Ellington A, Anslyn E. ChemBioChem. 2011;12:2021–2024. doi: 10.1002/cbic.201100046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Kawasaki A, Casper M, Freier S, Lesnik E, Zounes M, Cummins L, Gonzalez C, Cook P. J Med Chem. 1993;36:831–841. doi: 10.1021/jm00059a007. [DOI] [PubMed] [Google Scholar]; b) Green S, Jellinek D, Bell C, Beebe L, Feistner B, Gill S, Jucker F, Janjíc N. Chem Biol. 1995;2:683–695. doi: 10.1016/1074-5521(95)90032-2. [DOI] [PubMed] [Google Scholar]; c) Bell C, Lynam E, Landfair D, Janjic N, Wiles M. In Vitro Cell Dev Biol Anim. 1999;35:533–542. doi: 10.1007/s11626-999-0064-y. [DOI] [PubMed] [Google Scholar]
- 15.Cerchia L, Giangrande P, McNamara J, de Franciscis V. In: Nucleic Acid and Peptide Aptamers. Mayer G, editor. Humana Press; New York: 2009. pp. 59–78. [Google Scholar]
- 16.Ullrich A, Coussens L, Hayflick J, Dull T, Gray A, Tam A, Lee J, Yarden Y, Liebermann T, Schlessinger J, et al. Nature. 1984;309:418–425. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 17.Kempiak S, Yip S, Wang Q, Zhang G, Drebin J, Murali R, Greene M. J Cell Biol. 2003;162:781–788. [Google Scholar]
- 18.Li N, Nguyen H, Byrom M, Ellington A. PLoS One. 2011;6:e20299. doi: 10.1371/journal.pone.0020299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Magalhães M, Byrom M, Yan A, Kelly L, Li N, Furtado R, Palliser D, Levy M. Mol Ther. 2012;20:616–624. doi: 10.1038/mt.2011.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madsen J, Dupont D, Andersen T, Nielsen A, Sang L, Brix D, Jensen J, Broos T, Hendrickx M, Christensen A, et al. Biochemistry. 2010;49:4103–4115. doi: 10.1021/bi100066j. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Kuan C, Mi J, Zhang X, Clary B, Bigner D, Sullenger B. Biol Chem. 2009;390:137–144. doi: 10.1515/BC.2009.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lupold S, Hicke B, Lin Y, Coffey D. Cancer Res. 2002;62:4029–4033. [PubMed] [Google Scholar]
- 23.Stewart S, Ivy M, Anslyn E. Chem Soc Rev. 2013;43:70–84. doi: 10.1039/c3cs60183h. [DOI] [PubMed] [Google Scholar]
- 24.Chu T. PhD thesis. The University of Texas; Austin (USA): 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.