

Abstract
Numerous genetic studies have provided compelling evidence to establish DNA polymerase ε (Polε) as the primary DNA polymerase responsible for leading strand synthesis during eukaryotic nuclear genome replication. Polε is a heterotetramer consisting of a large catalytic subunit that contains the conserved polymerase core domain as well as a 3′ → 5′ exonuclease domain common to many replicative polymerases. In addition, Polε possesses three small subunits that lack a known catalytic activity but associate with components involved in a variety of DNA replication and maintenance processes. Previous enzymatic characterization of the Polε heterotetramer from budding yeast suggested that the small subunits slightly enhance DNA synthesis by Polε in vitro. However, similar studies of the human Polε heterote-tramer (hPolε) have been limited by the difficulty of obtaining hPolε in quantities suitable for thorough investigation of its catalytic activity. Utilization of a baculovirus expression system for overexpression and purification of hPolε from insect host cells has allowed for isolation of greater amounts of active hPolε, thus enabling a more detailed kinetic comparison between hPolε and an active N-terminal fragment of the hPolε catalytic subunit (p261N), which is readily overexpressed in Escherichia coli. Here, we report the first pre-steady-state studies of fully-assembled hPolε. We observe that the small subunits increase DNA binding by hPolε relative to p261N, but do not increase processivity during DNA synthesis on a single-stranded M13 template. Interestingly, the 3′ → 5′ exonuclease activity of hPolε is reduced relative to p261N on matched and mismatched DNA substrates, indicating that the presence of the small subunits may regulate the proofreading activity of hPolε and sway hPolε toward DNA synthesis rather than proofreading.
Keywords: DNA polymerase epsilon, Human DNA replication, Pre-steady-state kinetics, Leading strand replication, 3′ → 5′ exonuclease activity
1. Introduction
Since its identification in budding yeast, DNA polymerase (Pol) ε has been of major interest on account of its role in a wide variety of biological processes in eukaryotes. Polε, along with Polα and Polδ, is a key eukaryotic DNA replication enzyme [1], and is believed to be the primary leading strand synthesis polymerase during nuclear genome replication based on findings in budding yeast [2], fission yeast [3], and humans [4]. In addition to its role in nuclear DNA replication, Polε has been implicated in cell cycle regulation [5–8], gene silencing [9,10], sister chromatid cohesion [11,12], and base excision repair [13].
Polε is a heterotetramer with an overall architecture that was shown to be conserved in humans [14,15], budding yeast [16], and African clawed frog [17]. The p261 catalytic subunit (Pol2 in yeast) consists of an N-terminal domain containing the conserved polymerase and 3′ → 5′ exonuclease subdomains [18,19], as well as a C-terminal domain that is required for interaction with the three small subunits, p59, p12, and p17 (Dpb2, Dpb3, and Dpb4 in yeast) [14,15,20,21]. A low-resolution (20 A) ° structure of the yeast Polε heterotetramer obtained by cryo-electron microscopy has shown that Pol2 forms a globular head-like structure, while the three small subunits associate with Pol2 to form an extended tail-like structure that is suggested to interact with newly-synthesized double-stranded DNA (dsDNA) [22]. Recently, two ternary crystal structures of the N-terminal domain of Pol2 were solved and show that Pol2 possesses a novel P domain that makes additional contacts with the double-stranded region of the DNA substrate and contributes to processive DNA synthesis [23,24].
Notably, only Pol2 and Dpb2 are essential in yeast, while deletions of Dpb3 and Dpb4 are non-lethal [25,26]. Interestingly, only the C-terminal domain of Pol2 is essential as deletions of the entire N-terminal domain are viable, albeit with a prolonged S phase [20,21]. Bioinformatics tools have revealed that the C-terminal domain contains a distantly related copy of the exonuclease-polymerase module in which both enzymatic activities are nonfunctional [27]. Such inactive polymerase domains are likely to play a key structural role in assembly of replication complexes [28,29]. Taken together, these observations suggest that a critical role of Polε in yeast DNA replication involves protein-DNA and protein-protein interactions at the replication fork mediated by the C-terminal domain of Pol2 and the Dpb2 subunit, and that the polymerase activity of Polε is important for timely replication fork progression.
Expression systems in yeast [30,31] and insect cells [32] have allowed for purification of the yeast Polε heterotetramer in sufficient quantities for studies of its catalytic properties. Furthermore, proteolysis of Polε in yeast generates a highly conserved [33] and active N-terminal fragment of Pol2 that is readily isolated from full-length Polε [31,34]. Purification of both forms has enabled investigation of the effects of the small subunits on the catalytic properties of yeast Polε. Biochemical assays have demonstrated that the Dpb3-Dpb4 dimer is able to bind dsDNA and that the yeast Polε heterotetramer contains an additional DNA binding site that has similar affinity for single-stranded DNA (ssDNA) and dsDNA [35]. Moreover, longer regions of dsDNA were previously shown to slightly increase the processivity of the yeast Polε heterotetramer, but not the Pol2 subunit alone [22,36]. These results suggest that the small subunits may contribute to the catalytic activity of yeast Polε in vivo.
In contrast, characterization of the catalytic activities of human Polε has been more limited. While the N-terminal domain of the human Polε catalytic subunit (p261N) can be readily overexpressed and purified from Escherichia coli in quantities suitable for biochemical analysis [37], thorough kinetic studies of the human heterotetramer of Polε (hereafter referred to as hPolε) have been precluded by the difficulty of expressing comparable amounts of active protein. Recently, hPolε was obtained from a baculovirus expression system and was shown to be as active as and catalytically similar to the full-length p261 subunit and the p261N fragment under the reported conditions [14]. Using a similar approach, we have reconstituted and purified fully-assembled hPolε from a baculovirus-insect cell system. We then used pre-steady-state kinetics to measure kinetic parameters of hPolε for the first time. We find that the small subunits do not appear to affect DNA synthesis by hPolε but enhance DNA binding and decrease the 3′ → 5′ exonuclease activity, suggesting a potential role in regulating the proofreading activity of hPolε.
2. Experimental procedures
2.1. Materials
Materials used for experiments described below were purchased from the following sources: [γ-32P]ATP from Perkin-Elmer Life Sciences (Boston, MA); Optikinase from USB (Cleveland, OH); and dNTPs from Bioline (Taunton, MA).
2.2. DNA substrates
All DNA substrates listed in Table 1 were purchased from Integrated DNA Technologies, Inc. (Coralville, IA) and purified as described previously [38]. The M13mp2 ssDNA template was generously provided by Dr. Zachary Pursell from the Tulane University School of Medicine. All primers were 5′-radiolabeled by incubating with [γ-32P]ATP and Optikinase for 3 h at 37 °C. Excess [γ-32P]ATP was removed by passing the reaction mixture through a Bio-Spin 6 column (Bio-Rad). The 5′-radiolabeled primers were annealed to their respective templates (41-mer in Table 1 or M13mp2 ssDNA) by incubating the primer and template in a 1:1.15 ratio for 5 min at 95 °C and then cooling slowly to room temperature over several hours.
Table 1.
DNA substrates.
| D-1 | 5′-CGCAGCCGTCCAACCAACTCA-3′ 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ |
| M-1 | 5′-CGCAGCCGTCCAACCAACTCAC-3′ 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ |
| 21-mer M13 | 5′-ACGGCTACAGAGGCTTTGAGG-3′ |
| 45-mer M13 | 5′-GCAACGGCTACAGAGGCTTTGAGGACTAAAGACTTTTTCATGAGG-3′ |
2.3. Purification of the human DNA polymerase ε heterotetramer and the p261N fragment
The cDNAs for the human Polε subunits p12 (Clone ID 5443810), p17 (Clone ID 2822216), and p59 (Clone ID 8991936) were obtained from Open Biosystems. The p261 gene was amplified from pCR-XL carrying the Polε gene [39]. The genes encoding for all full-length human Polε subunits were cloned into a pFastBac-1 transfer vector (Life Technologies). During cloning, a His6 tag was added to the N-terminus of the p59 subunit. All cloned genes were verified by DNA sequencing. Preparation of high titer baculoviruses and protein expression in insect cells were performed using the Bac-to-Bac baculovirus expression system (Life Technologies) as described previously [40]. 1.8 × 109 Sf21 cells in 1 L of shaking culture were infected simultaneously with four recombinant baculoviruses encoding for each subunit and were cultivated at 25 °C for 65 h. The wild-type Polε heterotetramer was isolated from lysate by Ni-IDA affinity chromatography (Bio-Rad), followed by a HiTrap Heparin HP affinity column, and finally by a Superose 12 size-exclusion column (GE Healthcare). The concentration of purified hPolε was determined by UV spectrometry at 280 nm. SDS-PAGE analysis revealed that hPolε was purified to near-homogeneity (Fig. 1). The pure peak fractions were combined, aliquoted, and flash-frozen in liquid nitrogen for long-term storage at −80 °C. The wild-type p261N fragment was overexpressed and purified as described previously [41].
Fig. 1.

Analysis of hPolε purity by SDS-PAGE. To evaluate the purity of hPolε, the final protein sample was run on a 12% SDS-PAGE gel: lane 1, protein marker; lane 2, eluate from Superose 12 size-exclusion column. The p12 and p17 subunits have identical mobilities and are indistinguishable.
2.4. Reaction buffers
All assays were performed at 20 °C in reaction buffer E (50 mM Tris–OAc, pH 7.4 at 20 °C, 8 mM Mg(OAc)2, 1 mM DTT, 10% Glycerol, 0.1 mg/mL BSA, and 0.1 mM EDTA).
2.5. Pre-steady-state kinetic assays
In the burst assays, a pre-incubated solution of hPolε or p261N (10 nM) and 5′-radiolabeled D-1 DNA (40 nM) in buffer E was rapidly mixed with dTTP (5 μM) and Mg2+ (8 mM). In the exonuclease assays, a pre-incubated solution of hPolε or p261N (100 nM) and 5′-radiolabeled D-1 or M-1 DNA (20 nM) in buffer E was rapidly mixed with Mg2+ (8 mM) to initiate the excision reaction. All reactions were quenched with the addition of 0.37 M EDTA. All reactions were performed using a rapid chemical quench-flow apparatus (KinTek).
2.6. Active site titration assay
A pre-incubated solution of hPolε (50 nM) and increasing concentrations of 5′-radiolabeled D-1 DNA (10–80 nM) was rapidly mixed with dTTP (5 μM) and Mg2+ (8 mM). Each time point was quenched at 100 ms to ensure maximum product formation from completion of the burst phase. All reactions were performed on a rapid chemical quench-flow and repeated in triplicate.
2.7. Processivity assays
A pre-incubated solution of hPolε or p261N (250 nM) and 5′-radiolabeled 21- or 45-mer primer annealed to M13mp2 ssDNA (25 nM) in buffer E was mixed with all four dNTPs (100 μM each) and Mg2+ (8 mM). The reaction was quenched at various time points with the addition of 0.37 M EDTA.
2.8. Product analysis
Reaction products from the pre-steady-state kinetic assays and the active site titration assay were separated by denaturing PAGE (17% polyacrylamide, 8 M urea, and 1× TBE running buffer) and quantified using a Typhoon TRIO (GE Healthcare) and ImageQuant (Molecular Dynamics). Reaction products from the processivity assays were separated by denaturing PAGE using a 17% or an 8% gel for the 21-mer M13 and 45-mer M13 substrates, respectively.
2.9. Data analysis
All kinetic data were fit by nonlinear regression using Kaleida-Graph (Synergy Software). Data for the burst assay were fit to Eq. (1)
| (1) |
where A is the amplitude of active enzyme, k1 is the observed burst rate constant, and k2 is the observed steady-state rate constant.
Data from the active site titration assay were fit to Eq. (2)
| (2) |
where represents the equilibrium dissociation constant for the binary complex (E·DNA), E0 is the active enzyme concentration, and D0 is the DNA concentration.
Data from exonuclease assays under single-turnover conditions were fit to Eq. (3)
| (3) |
where A is the reaction amplitude and kexo is the observed DNA excision rate constant.
3. Results and discussion
3.1. Burst assays
Previously, we expressed and purified an exonuclease-deficient form of p261N from E. coli for pre-steady-state kinetic analysis [41]. We found that a single-nucleotide incorporation by p261N, like most kinetically-characterized DNA polymerases, is limited by a conformational change following nucleotide binding, while additional nucleotide incorporations are limited by dissociation of the enzyme from the E·DNA binary complex [42–49]. Both phases can be observed at once by performing a burst assay in which the enzyme is pre-incubated with an excess of the DNA substrate to allow formation of a stable E·DNA complex. To see if hPolε behaves similarly to p261N, we performed burst assays with both hPolε and p261N under identical conditions. Briefly, a pre-incubated solution of hPolε or p261N (10 nM) and 5′-radiolabeled D-1 DNA (40 nM, Table 1) was rapidly mixed with dTTP (5 μM) and Mg2+ (8 mM) at 20 °C for various durations of time. Notably, the D-1 DNA substrate used in this study is identical to the D-1 DNA substrate previously used to kinetically characterize p261N [41,50]. Both hPolε and p261N exhibited a fast burst of product formation with rate constants of 90 ± 28 s−1 and 101 ± 14 s−1, respectively (Fig. 2). Similarly, the rate constants for nucleotide incorporation by the yeast Polε catalytic subunit and holoenzyme were found to be nearly identical to each other [51]. Following the burst phase, hPolε and p261N catalyzed additional product formation characterized by slower linear phases with rate constants of 0.047 ± 0.006 s−1 and 0.018 ± 0.004 s−1, respectively. Thus, hPolε follows a similar kinetic pattern to p261N. The rate constant of the linear phase for p261N is similar to what was previously measured for the exonuclease-deficient mutant, and this rate constant was shown to be equivalent to the steady-state rate of product formation at 20 °C as well as the E·DNA dissociation rate constant (koff) [41]. Therefore, it is likely that the rate constant of the linear phase (0.047 s−1) for hPolε is identical to koff. Unexpectedly, the koff measured for hPolε is 2.6-fold higher than that of p261N. A possible source of this difference may be due in part to the high abundance of acidic residues in the C-terminal portion of the p17 subunit. Notably, the p17 subunit is also a component of the human CHRAC-15/17 histone fold complex and its negatively charged C-terminal region was previously shown to be necessary for full enhancement of nucleosome sliding by the CHRAC-15/17 complex [52]. Similarly, this C-terminal region may be essential for facilitating sliding by hPolε along the DNA substrate during highly processive DNA synthesis.
Fig. 2.

Biphasic kinetics of correct dTTP incorporation by hPolε and p261N at 20 °C. A pre-incubated solution of 10 nM hPolε (●) or p261N (■) and 40 nM 5′-radiolabeled D-1 DNA was rapidly mixed with 5 μM dTTP and 8 mM Mg2+ and quenched after various times with 0.37 M EDTA. The data were fit to Eq. (1) to yield burst phase rate constants of 90 ± 28 s−1 and 101 ± 14 s−1 and linear phase rate constants of 0.047 ± 0.006 s−1 and 0.018 ± 0.004 s−1 for hPolε and p261N, respectively.
3.2. Active site titration assay
To determine if the increase in koff observed for hPolε resulted in overall weaker binding affinity for DNA, we measured the equilibrium dissociation constant of the E·DNA binary complex ( ). Observation of a burst phase is indicative of formation of a stable E·DNA complex that is able to rapidly form product upon nucleotide binding. Therefore, the amplitude of product formation during the burst phase is directly related to the concentration of the E·DNA complex. By titrating the enzyme with increasing amounts of DNA, the dependency of the burst amplitude, or rather the concentration of the E·DNA binary complex, on free DNA concentration can be determined. From this relationship, the is derived. To determine the for hPolε, a pre-incubated solution of hPolε (50 nM) and increasing concentrations of 5′-radiolabeled D-1 DNA (10–80 nM) was rapidly mixed with dTTP (5 μM) for 100 ms before quenching with 0.37 M EDTA. The reactions were performed in triplicate and the data were fit to Eq. (2) to yield a of 33 ± 5 nM and an active enzyme concentration (E0) of 9.0 ± 0.7 nM, corresponding to 18% enzyme activity (Fig. 3). A similarly low activity was observed for the p261N exonuclease-deficient mutant [41]. Interestingly, the measured for hPolε (33 nM) is 2.4-fold lower than the of 79 nM determined previously for the p261N exonuclease-deficient mutant [41]. This difference indicates that the extended structure of hPolε relative to p261N increases the DNA binding affinity of hPolε, most likely by increasing the number of contacts that hPolε is able to make with the DNA substrate. Using the koff of 0.047 s−1 estimated from the burst assay, the second-order rate constant of DNA binding ( ) is calculated to be 1.4 × 106 M−1 s−1, which is over 5-fold higher than the kon calculated for p261N [41]. Thus, the 2.6-fold increase in koff of hPolε relative to p261N is compensated by an even larger increase in kon, suggesting that the small subunits assist hPolε in associating with the primer-template more efficiently.
Fig. 3.

Active site titration of hPolε at 20 °C. A pre-incubated solution of hPolε (50 nM) and increasing concentrations of 5′-radiolabeled D-1 DNA (10–80 nM) was rapidly mixed with dTTP (5 μM). All reactions were quenched after 100 ms with the addition of 0.37 M EDTA. The data were fit to Eq. (2) to yield a of 33 ± 5 nM and d an enzyme active concentration of 9.0 ± 0.7 nM.
3.3. Processivity assays
Previously, it was shown that the small subunits of yeast Polε slightly enhanced the processivity of DNA synthesis relative to the Pol2 catalytic subunit alone, but only when the length of the dsDNA region of the singly-primed DNA substrate was 40 nucleotides or longer [22]. This observation correlated well with the extra length of yeast Polε afforded by interaction between Pol2 and the small subunits. To see if the processivity of hPolε demonstrated a similar dependence on both the presence of its small subunits and the length of the primer, we compared the processivities of hPolε and p261N during DNA synthesis on an M13mp2 ssDNA template containing a 21- or 45-nucleotide primer. A pre-incubated solution of hPolε or p261N (250 nM) and 5′-radiolabeled 21- or 45-mer M13 primer (Table 1) annealed to M13 ssDNA (25 nM) was mixed with all four nucleotides (100 μM) for various times and the products were separated by denaturing PAGE. During extension from the 21-mer M13 primer (Fig. 4A), both hPolε and p261N showed strong pauses after the addition of 22 or 23 nucleotides, indicating that the presence of the small subunits does not affect the processivity of hPolε relative to p261N on the 21-mer M13 DNA substrate. Surprisingly, hPolε and p261N show similar pausing patterns during extension from the 45-mer M13 primer as well (Fig. 4B). Thus, under these conditions, the small subunits do not appear to enhance the processivity of DNA synthesis by hPolε. However, it should be noted that the nucleotide sequence of the 21-mer M13 primer is offset by 3 nucleotides at the 5′ end relative to the 45-mer M13 primer. When the samples from both extension reactions are separated on a 12% denaturing PAGE gel (Fig. S1), the resulting pausing patterns show a slight offset that is accounted for by the aforementioned sequence offset in the two primers. Thus, the observed pausing pattern is likely a consequence of secondary structure forming at various positions in the M13mp2 ssDNA that is blocking continued DNA synthesis by both hPolε and p261N. It is evident that highly processive DNA synthesis by hPolε in vivo requires additional processivity factors, including PCNA, RFC, and RPA. Consistently, synthesis of large products on an M13 ssDNA substrate by hPolε was previously shown to be dependent on PCNA and RFC in vitro [14].
Fig. 4.

Processive DNA synthesis by hPolε and p261N on singly-primed M13mp2 ssDNA templates at 20 °C. A pre-incubated solution of hPolε or p261N (250 nM) and 5′-radiolabeled (A) 21- or (B) 45-mer primer annealed to M13 ssDNA (25 nM) was mixed with all four dNTPs (100 μM) for various times before quenching with the addition of 0.37 M EDTA. Products extended from the 21-mer primer were separated by 17% denaturing PAGE, while products extended from the 45-mer primer were separated by 8% denaturing PAGE.
3.4. Excision of matched and mismatched DNA substrates by hPolε
Like many replicative DNA polymerases [53–57], hPolε possesses a 3′ → 5′ exonuclease domain which catalyzes proofreading activity [23,24] that is responsible for excising mismatched base pairs formed during DNA synthesis. To determine whether the small subunits have any effect on the proofreading activity of hPolε, we compared the 3′ → 5′ exonuclease activities of hPolε and p261N on a matched DNA substrate (D-1) and a DNA substrate containing a single mismatched base pair (M-1, Table 1). As with the D-1 DNA substrate, the M-1 DNA substrate was previously used to evaluate the contribution of proofreading to the overall fidelity of p261N [50]. Briefly, a pre-incubated solution of hPolε or p261N (100 nM) and 5′-radiolabeled D-1 or M-1 DNA (20 nM) was rapidly mixed with Mg2+ (8 mM) for various durations of time. Each plot of remaining substrate versus reaction time was fit to Eq. (3) to yield the overall excision rate constant (kexo) as a function of the rate constant of DNA transfer from the polymerase site to the exonuclease site, the rate constant of DNA dissociation and rebinding to the exonuclease site, and the true excision rate constant. For the matched D-1 substrate, hPolε and p261N catalyzed excision with measured kexo values of 0.018 ± 0.002 s−1 and 0.041 ± 0.004 s−1, respectively (Fig. 5A). This 2.3-fold decrease in kexo for hPolε relative to p261N suggests that the small subunits may somehow regulate the 3′ → 5′ exonuclease activity of hPolε against excision of correctly matched DNA, either by limiting transfer of the matched DNA substrate from the polymerase active site to the exonuclease active site or by decreasing the true rate constant of ssDNA excision. The kexo values of both enzymes were then measured in the presence of a single mismatched base pair using the M-1 DNA substrate and were determined to be 0.19 ± 0.03 s−1 and 1.4 ± 0.2 s−1 for hPolε and p261N, respectively (Fig. 5B). Interestingly, excision by p261N was enhanced by over 30-fold in the presence of a single mismatch, while hPolε only experienced a 10-fold stimulation. Notably, a similar pattern was observed for enhancement of the excision rate constant of the yeast Polε catalytic subunit and holoenzyme [51]. This result provides evidence that the excision rate constant is mostly limited by the transfer of the DNA substrate from the polymerase active site to the exonuclease active site, as the excision rate constant of ssDNA at the exonuclease site should be independent of DNA duplex stability. It is possible that the extended structure of hPolε is limiting DNA substrate transfer to the exonuclease site relative to p261N. However, investigation of DNA substrate transfer between the exonuclease and polymerase active sites of both the yeast Polε catalytic subunit and holoenzyme suggests that the additional subunits have no effect on this transfer [58]. Thus, further studies are required to completely characterize the mechanism of 3′ → 5′ exonuclease activity catalyzed by both hPolε and p261N and determine the cause of this difference.
Fig. 5.
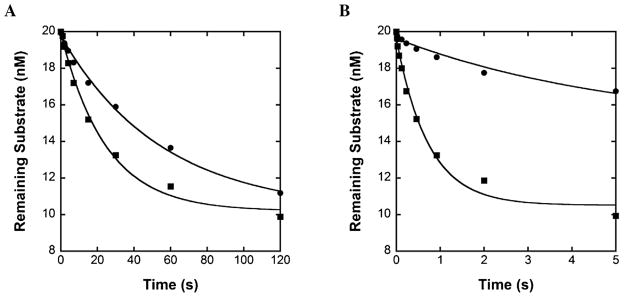
Excision of matched and mismatched DNA by hPolε at 20 °C. A pre-incubated solution of 100 nM hPolε (●) or p261N (■) and 20 nM 5′-radiolabeled (A) D-1 or (B) M-1 DNA was rapidly mixed with Mg2+ and quenched after various times with 0.37 M EDTA. The data were fit to Eq. (3) to yield kexo. For the matched D-1 DNA (A), the measured kexo values were 0.018 ± 0.002 s−1 and 0.041 ± 0.004 s−1 for hPolε and p261N, respectively. For the mismatched M-1 DNA (B), the measured kexo values were 0.19 ± 0.03 s−1 and 1.4 ± 0.2 s−1 for hPolε and p261N, respectively.
3.5. Concluding remarks
We have performed the first kinetic analysis of the four-subunit hPolε holoenzyme and compared its activity to that of the p261N catalytic fragment. We found that the small subunits increase DNA binding affinity to hPolε, but do not appear to affect the processive polymerization activity of hPolε. In contrast, the reduction of the overall excision rate constant of hPolε relative to p261N indicates that the small subunits may sway the enzyme activity toward the DNA synthesis direction. To further explore this hypothesis, we are currently performing a thorough kinetic study of the mechanisms of the 3′ → 5′ exonuclease activities of both hPolε and p261N. Finally, it is worth noting that p261N was overexpressed and purified from E. coli, while hPolε was prepared from insect cells. Therefore, it is possible that post-translational modification of the catalytic subunit during overexpression in insect cells may account for some of the differences determined in this study, and this hypothesis cannot be ruled out without performing a kinetic analysis of p261N isolated from insect cells.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Institutes of Health grants [ES024585 and ES009127to Z.S., GM101167 to T.H.T., and T32 GM008512 to W.J.Z.].
We are grateful to Dr. Zachary Pursell for providing us with the M13mp2 ssDNA template used for the processivity assays. We thank Jianyou Gu for the cloning of hPolε encoding sequences into the pFastBac-1 transfer vector.
Abbreviations
- Pol
polymerase
- Polε
DNA polymerase ε
- Polα
DNA polymerase α
- Polδ
DNA polymerase δ
- dsDNA
double-stranded DNA
- ssDNA
single-stranded DNA
- dNTP
3′-deoxyribonucleotide 5′-triphosphate
- DTT
dithiothreitol
- BSA
bovine serum albumin
- EDTA
ethylenediaminetetraacetic acid
- PAGE
polyacrylamide gel electrophoresis
- CHRAC-15/17
chromatin accessibility complex 15 and 17 kDa proteins
- PCNA
proliferating cell nuclear antigen
- RFC
replication factor C
- RPA
eukaryotic single-strand DNA binding protein
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.dnarep.2015.01.008.
Footnotes
Conflict of interest statement
The authors declare that there are no conflict of interest.
A figure showing the products of extension from the 21- and 45-mer M13 primers on a single 12% denaturing PAGE gel (Fig. S1) is available in the supporting information.
References
- 1.Brieba LG. Template dependent human DNA polymerases. Curr Top Med Chem. 2008;8:1312–1326. doi: 10.2174/156802608786141098. [DOI] [PubMed] [Google Scholar]
- 2.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyabe I, Kunkel TA, Carr AM. The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS Genet. 2011;7:e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shinbrot E, Henninger EE, Weinhold N, Covington KR, Goksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny DM, Gibbs RA, et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014;24 doi: 10.1101/gr.174789.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Navas TA, Zhou Z, Elledge SJ. DNA polymerase epsilon links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- 6.Muramatsu S, Hirai K, Tak YS, Kamimura Y, Araki H. CDK-dependent complex formation between replication proteins Dpb11, Sld2 Pol (epsilon}, and GINS in budding yeast. Genes Dev. 2010;24:602–612. doi: 10.1101/gad.1883410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL. Mrc1 and DNA polymerase epsilon function together in linking DNA replication and the S phase checkpoint. Mol Cell. 2008;32:106–117. doi: 10.1016/j.molcel.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kesti T, McDonald WH, Yates JR, 3rd, Wittenberg C. Cell cycle-dependent phosphorylation of the DNA polymerase epsilon subunit Dpb2, by the Cdc28 cyclin-dependent protein kinase. J Biol Chem. 2004;279:14245–14255. doi: 10.1074/jbc.M313289200. [DOI] [PubMed] [Google Scholar]
- 9.Iida T, Araki H. Noncompetitive counteractions of DNA polymerase epsilon and ISW2/yCHRAC for epigenetic inheritance of telomere position effect in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:217–227. doi: 10.1128/MCB.24.1.217-227.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith JS, Caputo E, Boeke JD. A genetic screen for ribosomal DNA silencing defects identifies multiple DNA replication and chromatin-modulating factors. Mol Cell Biol. 1999;19:3184–3197. doi: 10.1128/mcb.19.4.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carson DR, Christman MF. Evidence that replication fork components catalyze establishment of cohesion between sister chromatids. Proc Natl Acad Sci U S A. 2001;98:8270–8275. doi: 10.1073/pnas.131022798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards S, Li CM, Levy DL, Brown J, Snow PM, Campbell JL. Saccharomyces cerevisiae DNA polymerase epsilon and polymerase sigma interact physically and functionally, suggesting a role for polymerase epsilon in sister chromatid cohesion. Mol Cell Biol. 2003;23:2733–2748. doi: 10.1128/MCB.23.8.2733-2748.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parlanti E, Locatelli G, Maga G, Dogliotti E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007;35:1569–1577. doi: 10.1093/nar/gkl1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bermudez VP, Farina A, Raghavan V, Tappin I, Hurwitz J. Studies on human DNA polymerase epsilon and GINS complex and their role in DNA replication. J Biol Chem. 2011;286:28963–28977. doi: 10.1074/jbc.M111.256289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Pursell ZF, Linn S. Identification and cloning of two histone fold motif-containing subunits of HeLa DNA polymerase epsilon. J Biol Chem. 2000;275:23247–23252. doi: 10.1074/jbc.M002548200. [DOI] [PubMed] [Google Scholar]
- 16.Ohya T, Maki S, Kawasaki Y, Sugino A. Structure and function of the fourth subunit (Dpb4p) of DNA polymerase epsilon in Saccharomyces cerevisiae. Nucleic Acids Res. 2000;28:3846–3852. doi: 10.1093/nar/28.20.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shikata K, Sasa-Masuda T, Okuno Y, Waga S, Sugino A. The DNA polymerase activity of Pol epsilon holoenzyme is required for rapid and efficient chromosomal DNA replication in Xenopus egg extracts. BMC Biochem. 2006;7:21. doi: 10.1186/1471-2091-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kesti T, Frantti H, Syvaoja JE. Molecular cloning of the cDNA for the catalytic subunit of human DNA polymerase epsilon. J Biol Chem. 1993;268:10238–10245. [PubMed] [Google Scholar]
- 19.Morrison A, Araki H, Clark AB, Hamatake RK, Sugino A. A third essential DNA polymerase in S. cerevisiae. Cell. 1990;62:1143–1151. doi: 10.1016/0092-8674(90)90391-q. [DOI] [PubMed] [Google Scholar]
- 20.Dua R, Levy DL, Campbell JL. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol epsilon and its unexpected ability to support growth in the absence of the DNA polymerase domain. J Biol Chem. 1999;274:22283–22288. doi: 10.1074/jbc.274.32.22283. [DOI] [PubMed] [Google Scholar]
- 21.Kesti T, Flick K, Keranen S, Syvaoja JE, Wittenberg C. DNA polymerase epsilon catalytic domains are dispensable for DNA replication DNA repair, and cell viability. Mol Cell. 1999;3:679–685. doi: 10.1016/s1097-2765(00)80361-5. [DOI] [PubMed] [Google Scholar]
- 22.Asturias FJ, Cheung IK, Sabouri N, Chilkova O, Wepplo D, Johansson E. Structure of Saccharomyces cerevisiae DNA polymerase epsilon by cryo-electron microscopy. Nat Struct Mol Biol. 2006;13:35–43. doi: 10.1038/nsmb1040. [DOI] [PubMed] [Google Scholar]
- 23.Hogg M, Osterman P, Bylund GO, Ganai RA, Lundstrom EB, Sauer-Eriksson AE, Johansson E. Structural basis for processive DNA synthesis by yeast DNA polymerase epsilon. Nat Struct Mol Biol. 2014;21:49–55. doi: 10.1038/nsmb.2712. [DOI] [PubMed] [Google Scholar]
- 24.Jain R, Rajashankar KR, Buku A, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Crystal structure of yeast DNA polymerase epsilon catalytic domain. PLOS ONE. 2014;9:e94835. doi: 10.1371/journal.pone.0094835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawasaki Y, Sugino A. Yeast replicative DNA polymerases and their role at the replication fork. Mol Cells. 2001;12:277–285. [PubMed] [Google Scholar]
- 26.Hogg M, Johansson E. DNA polymerase epsilon. Subcell Biochem. 2012;62:237–257. doi: 10.1007/978-94-007-4572-8_13. [DOI] [PubMed] [Google Scholar]
- 27.Tahirov TH, Makarova KS, Rogozin IB, Pavlov YI, Koonin EV. Evolution of DNA polymerases: an inactivated polymerase-exonuclease module in Pol epsilon and a chimeric origin of eukaryotic polymerases from two classes of archaeal ancestors. Biol Direct. 2009;4:11. doi: 10.1186/1745-6150-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rogozin IB, Makarova KS, Pavlov YI, Koonin EV. A highly conserved family of inactivated archaeal B family DNA polymerases. Biol Direct. 2008;3:32. doi: 10.1186/1745-6150-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baranovskiy AG, Babayeva ND, Liston VG, Rogozin IB, Koonin EV, Pavlov YI, Vassylyev DG, Tahirov TH. X-ray structure of the complex of regulatory subunits of human DNA polymerase delta. Cell Cycle. 2008;7:3026–3036. doi: 10.4161/cc.7.19.6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chilkova O, Jonsson BH, Johansson E. The quaternary structure of DNA polymerase epsilon from Saccharomyces cerevisiae. J Biol Chem. 2003;278:14082–14086. doi: 10.1074/jbc.M211818200. [DOI] [PubMed] [Google Scholar]
- 31.Maki S, Hashimoto K, Ohara T, Sugino A. DNA polymerase II (epsilon) of Saccharomyces cerevisiae dissociates from the DNA template by sensing single-stranded DNA. J Biol Chem. 1998;273:21332–21341. doi: 10.1074/jbc.273.33.21332. [DOI] [PubMed] [Google Scholar]
- 32.Dua R, Edwards S, Levy DL, Campbell JL. Subunit interactions within the Saccharomyces cerevisiae DNA polymerase epsilon (pol epsilon) complex. Demonstration of a dimeric pol epsilon. J Biol Chem. 2000;275:28816–28825. doi: 10.1074/jbc.M002376200. [DOI] [PubMed] [Google Scholar]
- 33.Uitto L, Halleen J, Hentunen T, Hoyhtya M, Syvaoja JE. Structural relationship between DNA polymerases epsilon and epsilon* and their occurrence in eukaryotic cells. Nucleic Acids Res. 1995;23:244–247. doi: 10.1093/nar/23.2.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamatake RK, Hasegawa H, Clark AB, Bebenek K, Kunkel TA, Sugino A. Purification and characterization of DNA polymerase II from the yeast Saccharomyces cerevisiae. Identification of the catalytic core and a possible holoenzyme form of the enzyme. J Biol Chem. 1990;265:4072–4083. [PubMed] [Google Scholar]
- 35.Tsubota T, Maki S, Kubota H, Sugino A, Maki H. Double-stranded DNA binding properties of Saccharomyces cerevisiae DNA polymerase epsilon and of the Dpb3p-Dpb4p subassembly. Genes Cells. 2003;8:873–888. doi: 10.1046/j.1365-2443.2003.00683.x. [DOI] [PubMed] [Google Scholar]
- 36.Aksenova A, Volkov K, Maceluch J, Pursell ZF, Rogozin IB, Kunkel TA, Pavlov YI, Johansson E. Mismatch repair-independent increase in spontaneous mutagenesis in yeast lacking non-essential subunits of DNA polymerase epsilon. PLoS Genet. 2010;6:e1001209. doi: 10.1371/journal.pgen.1001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korona DA, Lecompte KG, Pursell ZF. The high fidelity and unique error signature of human DNA polymerase epsilon. Nucleic Acids Res. 2010;39:1763–1773. doi: 10.1093/nar/gkq1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiala KA, Suo Z. Pre-steady-state kinetic studies of the fidelity of Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2106–2115. doi: 10.1021/bi0357457. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Asahara H, Patel VS, Zhou S, Linn S. Purification, cDNA cloning, and gene mapping of the small subunit of human DNA polymerase epsilon. J Biol Chem. 1997;272:32337–32344. doi: 10.1074/jbc.272.51.32337. [DOI] [PubMed] [Google Scholar]
- 40.Gu J, Babayeva ND, Suwa Y, Baranovskiy AG, Price DH, Tahirov TH. Crystal structure of HIV-1 Tat complexed with human P-TEFb and AFF4. Cell Cycle. 2014;13:1788–1797. doi: 10.4161/cc.28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zahurancik WJ, Klein SJ, Suo Z. Kinetic mechanism of DNA polymerization catalyzed by human DNA polymerase epsilon. Biochemistry. 2013;52:7041–7049. doi: 10.1021/bi400803v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown JA, Suo Z. Elucidating the kinetic mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase B1. Biochemistry. 2009;48:7502–7511. doi: 10.1021/bi9005336. [DOI] [PubMed] [Google Scholar]
- 43.Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 44.Dahlberg ME, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow fragment): identification of a second conformational change and evaluation of the internal equilibrium constant. Biochemistry. 1991;30:4835–4843. doi: 10.1021/bi00234a002. [DOI] [PubMed] [Google Scholar]
- 45.Fiala KA, Suo Z. Mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2116–2125. doi: 10.1021/bi035746z. [DOI] [PubMed] [Google Scholar]
- 46.Hsieh JC, Zinnen S, Modrich P. Kinetic mechanism of the DNA-dependent DNA polymerase activity of human immunodeficiency virus reverse transcriptase. J Biol Chem. 1993;268:24607–24613. [PubMed] [Google Scholar]
- 47.Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow) Biochemistry. 1987;26:8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 48.Patel SS, Wong I, Johnson KA. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991;30:511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 49.Washington MT, Prakash L, Prakash S. Yeast DNA polymerase eta utilizes an induced-fit mechanism of nucleotide incorporation. Cell. 2001;107:917–927. doi: 10.1016/s0092-8674(01)00613-4. [DOI] [PubMed] [Google Scholar]
- 50.Zahurancik WJ, Klein SJ, Suo Z. Significant contribution of the 3′ → 5′ exonuclease activity to the high fidelity of nucleotide incorporation catalyzed by human DNA polymerase. Nucleic Acids Res. 2014;42:13853–13860. doi: 10.1093/nar/gku1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ganai RA, Osterman P, Johansson E. Yeast DNA polymerase epsilon catalytic core and holoenzyme have comparable catalytic rates. J Biol Chem. 2014 doi: 10.1074/jbc.M114.615278. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kukimoto I, Elderkin S, Grimaldi M, Oelgeschlager T, Varga-Weisz PD. The histone-fold protein complex CHRAC-15/17 enhances nucleosome sliding and assembly mediated by ACF. Mol Cell. 2004;13:265–277. doi: 10.1016/s1097-2765(03)00523-9. [DOI] [PubMed] [Google Scholar]
- 53.Beese LS, Steitz TA. Structural basis for the 33′ – 5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 55.Freemont PS, Friedman JM, Beese LS, Sanderson MR, Steitz TA. Cocrystal structure of an editing complex of Klenow fragment with DNA. Proc Natl Acad Sci U S A. 1988;85:8924–8928. doi: 10.1073/pnas.85.23.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savino C, Federici L, Johnson KA, Vallone B, Nastopoulos V, Rossi M, Pisani FM, Tsernoglou D. Insights into DNA replication: the crystal structure of DNA polymerase B1 from the archaeon Sulfolobus solfataricus. Structure. 2004;12:2001–2008. doi: 10.1016/j.str.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 58.Ganai RA, Bylund GO, Johansson E. Switching between polymerase and exonuclease sites in DNA polymerase epsilon. Nucleic Acids Res. 2014;43 doi: 10.1093/nar/gku1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.