

Abstract
Physical exercise may provide protection against the cognitive decline and neuropathology associated with Alzheimer’s disease, although the mechanisms are not clear. In the present study, APP/PSEN1 double-transgenic and wild-type mice were allowed unlimited voluntary exercise for 7 months. Consistent with previous reports, wheel-running improved cognition in the double-transgenic mice. Interestingly, the average daily distance run was strongly correlated with spatial memory in the water maze in wild-type mice (r2 = .959), but uncorrelated in transgenics (r2 = .013). Proteomics analysis showed that sedentary transgenic mice differed significantly from sedentary wild-types with respect to proteins involved in synaptic transmission, cytoskeletal regulation, and neurogenesis. When given an opportunity to exercise, the transgenics’ deficiencies in cytoskeletal regulation and neurogenesis largely normalized, but abnormal synaptic proteins did not change. In contrast, exercise enhanced proteins associated with cytoskeletal regulation, oxidative phosphorylation, and synaptic transmission in wild-type mice. Soluble and insoluble Aβ40 and Aβ42 levels were significantly decreased in both cortex and hippocampus of active transgenics, suggesting that this may have played a role in the cognitive improvement in APP/PSEN1 mice. β-secretase was significantly reduced in active APP/PSEN1 mice compared to sedentary controls, suggesting a mechanism for reduced Aβ. Taken together, these data illustrate that exercise improves memory in wild-type and APP-overexpressing mice in fundamentally different ways.
Introduction
Alzheimer’s disease is the most common form of dementia, characterized by plaques of misfolded amyloid β (Aβ), neurofibrillary tangles of hyperphosphorylated tau, widespread neurodegeneration, and loss of cognitive and other functions. Considerable evidence now supports the notion that factors affecting cardiovascular function may also affect the symptoms and severity of Alzheimer's disease. Indeed, obesity, glucose intolerance, and high cholesterol are all considered risk factors for Alzheimer’s disease. Inversely, evidence suggests that physical exercise may improve memory in Alzheimer's patients and perhaps slow cognitive decline. Taken together, these results suggest that metabolic factors may play a significant role in Alzheimer's dementia.
Like Alzheimer patients, transgenic mice that overexpress mutant human amyloid precursor protein (APP) typically show improved memory when given the opportunity to exercise. The improved cognition is usually accompanied by reductions in Aβ and amyloid plaques. However, improvements in memory and pathology are not always observed, and there is little understanding of how exercise-induced changes may come about. Adlard et al. (2005) observed a substantive reduction in plaque load and total Aβ in 6-month-old TgCRND8 mice given the opportunity for voluntary wheel-running for 5 months. However, they did not observe changes in APP, α-, β-, or γ-secretase activity, APP C-terminal fragments (CTFs), or degradative enzymes. They repeated the study in a separate group of mice given access to running wheels for only 1 month, starting at 6 weeks of age. Again, Aβ was reduced in the presence of normal levels of APP, secretase activity, insulin degrading enzyme (IDE), and neprilysin. However, this time α- and β-CTFs were significantly reduced. They concluded that transient changes in APP processing occurred early in the exercise routine, resulting in long-lasting reductions in Aβ. In contrast, Maesako et al. (2012) observed reduced CTFs after 5 months of voluntary exercise in 7.5-month old J20 mice. Although the J20 and TgCRND8 transgenic lines harbor the same Swedish (K670N/M671L) and Indiana (V717F) mutations, they are driven by different promoters and the mice are on different background strains. These and other methodological differences may produce large differences in outcomes. Indeed, voluntary running, forced running (e.g., shock-motivated), and swimming have all been used to induce activity-related changes in APP-overexpressing transgenics. Exercise can also improve memory in wild-type mice in the absence of mutant APP or Aβ, so non-amyloidogenic factors may be partly responsible for, or interact with, the reductions in Aβ to improve memory. Voluntary exercise in wild-type mice has been associated with increases in neurogenesis, neurotrophic factors, synaptogenesis, and gluconeogenesis. Thus, despite the numerous papers addressing the effects of physical activity in Alzheimer models, we do not have a clear understanding of how exercise clears Aβ pathology and improves memory. In the present study we used a novel 24-h home-cage running wheel system that eliminates non-running artifacts and avoids potential nuisance factors such as aversive control or exposure to a running wheel for a short period every day. Control mice were treated identically except that running wheels were inoperable.
Methods
Subjects
The APP/PSEN1 double-transgenic mice (#004462, Jackson Laboratories, Bar Harbor, ME) incorporate a chimeric human/murine amyloid precursor protein (APP) construct bearing the Swedish double mutation (K595N/M596L) and the exon-9-deleted human presenilin 1 (PSEN1) mutation (APPSwe+PSEN1/ΔE9), both controlled by the mouse prion protein promoter (PrP) to drive expression mainly in CNS neurons. The transgenics are maintained as double-hemizygotes by periodic backcrossing to wild-types of the B6C3F1 strain (#100010, Jackson Laboratories). These double-transgenic mice have spatial memory deficits as early as 7 months of age (Bernardo et al., 2009; Reiserer et al., 2007). Mice were group-housed in standard tub cages with 3–5 per cage, until 5 months of age, at which time they were individually housed in similar caging, each outfitted with a running wheel (9–12 per group). Hippocampal and cortical tissue from the same mice were used for proteomics (7–10 per group), ELISA (7–8 per group) and Western (2–8 per group) assays. Mice were sacrificed 24 hours following water-maze probe trial.
Exercise
Our computer interfaced, automated exercise monitoring system collected data continually for 7 months, except for ~1 min. each week needed to replace the dirty cage. This system incorporates a commercially available running wheel (Fast-Trac #K3251, Bio-Serv, Frenchtown, NJ) and uses standard mouse housing cages. Half of the mice of each genotype had free access to functional running wheels (active groups), while the other half had access to otherwise identical non-functional running wheels (inactive groups) to control for potential environmental enrichment from presence of the running wheel. The running-wheel system uses infrared emitters and detectors to sense wheel motion (Fig. 1), and provides high-resolution data that can be used to study not only speed and duration of running, but multiple levels of structure and rhythms, and running direction preferences (clockwise vs. counter-clockwise). Data were collected through tracking of nine equally-spaced reflective strips attached to the outside of the running wheels, each of which provided an infra-red “pulse” input. Unlike other running wheel systems, our design eliminated false signals generated by wheel rocking and continuous sensor-detector proximity through fail-safes programmed into the software. Each running-wheel input generated a separate indexed data file which was imported into a database or spreadsheet program for analysis. The time elapsed between pulses for a single sensor provided speed data, while the order of leading edges between sensors provided direction data. The signals were processed by a LabView (National Instruments, Austin, TX) program that counted one revolution for every nine pulses in the same direction.
Figure 1. Novel running-wheel system.
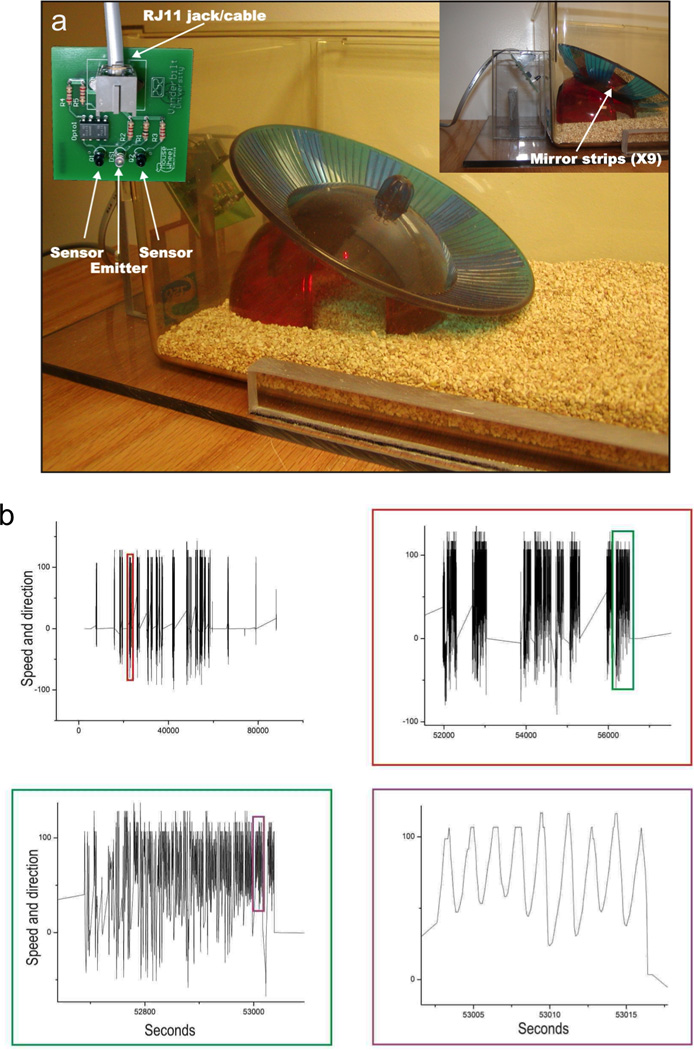
(a) Commercially-available running wheels were adapted for novel home-made electronics that allow quick cage change, and software that eliminates movement due to rocking and adventitious sensor proximity. (b) Four panels document with increasing precision the readout from the running-wheel software.
Memory
Spatial cognition was assessed using a standard reference-memory task the water maze as previously described (Bernardo et al., 2009; Dhanushkodi and McDonald, 2011). Each mouse received four trials per day in a 92-cm diameter pool with a clear acrylic platform (10-cm diam.) submerged 0.5 cm below the surface of the water. Digital images were captured and analyzed using macros especially written for the water maze, run in the public-domain software NIH Image (Bazalakova et al., 2007; Miyakawa et al., 2001). Trials were conducted in a spaced fashion, i.e., each subject received its first trial before the first subject received its second trial. A 60-s probe trial was conducted without the platform to assess memory for the former platform location, 24 hours following the final acquisition session. The amount of time spent in a 30-cm-diam. zone surrounding the former platform location was used as a measure of memory during the probe trial. This zone score was used as a more accurate measure of memory, compared to quadrant time (Bernardo et al., 2007; Dhanushkodi and McDonald, 2011; Flanigan et al., 2014; Harrison et al., 2009a). Because of the shape of the pool quadrants, some locations within the target quadrant are farther away from the platform location than other locations in neighboring quadrants. For example, in a 92-cm-diam. pool a mouse might be as far as 32 cm from the platform but still inside the target quadrant, and as near as 17 cm from the platform but in a neighboring quadrant. Comparing time spent in the 30-cm zone to equivalent zones in the other three quadrants is a more precise measure of memory because it eliminates the problems caused by the shape of the quadrant, and excludes adventitious swimming in the 8-cm periphery (Fig. 2b). We did not conduct a cued-platform control task for several reasons. First, we and others have already shown that the APP/PSEN1 mice have robust spatial learning impairments in the water maze but are unimpaired on the non-spatial cued control task (Bernardo et al., 2007; Cao et al., 2007; Cohen et al., 2009; Harrison et al., 2009b; Harrison et al., 2010; Lewis et al., 2010). Second, the transgenics also exhibit deficits in cognitive flexibility (Arrazola et al., 2009; Reiserer et al., 2007; Toledo and Inestrosa, 2010), a putatively frontally-mediated cognitive process involved in, e.g., switching learning modalities from cued to spatial. Because our focus was primarily on hippocampal pathology and proteins, we wanted the water-maze task to reflect pure spatial learning deficits as much as possible. Although we could have conducted the cued control condition following the spatial version, we wanted the neurochemistry to reflect as much as possible the changes associated with spatial learning. To do this, we had to sacrifice the mice as soon as possible after the probe trial. For these reasons, we omitted the cued-platform phase in the present study.
Figure 2. Cognitive performance is tightly associated with running in wild-type mice but not APP/PSEN1 transgenics.
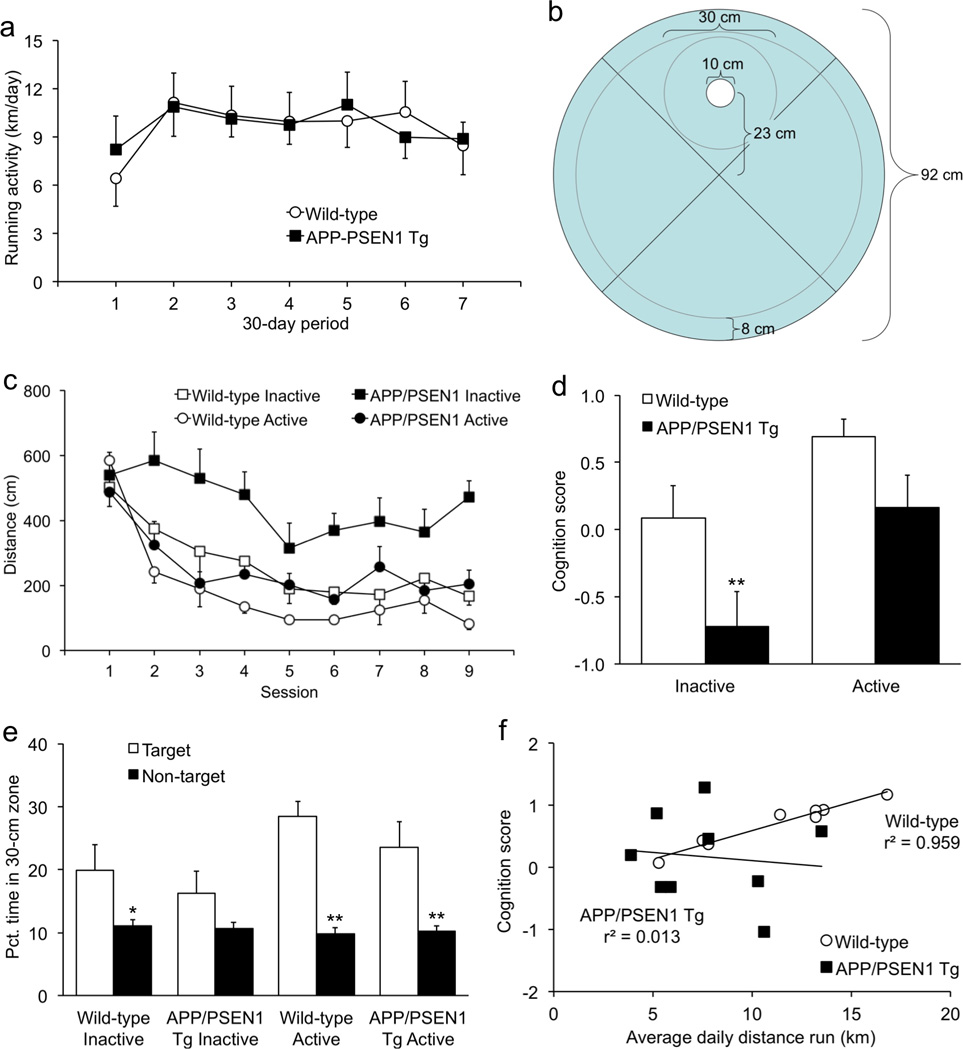
(a) During the first month of access to the running wheel, APP/PSEN1 transgenic mice ran nearly twice as much as wild-type control mice, although the difference was not statistically significant. For the rest of the study, wild-type mice increased their output to transgenic levels. (b) Diagram of water maze configured for more precise measurement of memory. A 30-cm zone is used to measure memory for the platform location. (c) Sedentary APP/PSEN1 transgenics had significantly impaired spatial learning compared with wild-type controls. Transgenics allowed to exercise freely had significantly improved performance. (d) A cognition score calculated from asymptotic performance during acquisition and precision during the probe trial was significantly lower in sedentary transgenic mice. (e) Sedentary transgenic mice did not spend more time in the target 30-cm zone than in the mean of equivalent zones in the other three quadrants. In contrast, mice in the other three groups all showed selective search for the target zone. (f) The cognition scores were significantly correlated with running in wild-type mice, but not in APP/PSEN1 transgenics. *p < .05; **p < .01.
Neurochemistry
Mice were sacrificed at 12 months of age. Brains were dissected rapidly on wet ice and bilateral hippocampi and overlying cortex were flash-frozen. Tissue was homogenized and proteins extracted from each sample. Soluble and insoluble Aβ40 and Aβ42 were measured by ELISA (#27718 & 27712, Immuno-Biological Laboratories, Minneapolis, MN) as previously described (Bernardo et al., 2009). Western blot for full-length amyloid precursor protein (APP) was conducted using 1E6 and LN27 antibodies as previously described (Bernardo et al., 2009). Westerns were also conducted for β-amyloid converting enzyme 1 (BACE1; #SAB2104731, Sigma-Aldrich, St. Louis, MO), sAPPα (gift of Dr. Fang Liao of UTHSC), protein kinase C γ (PKCγ; # SAB2104731, Sigma-Aldrich).
Proteomics sample preparation
Protein extraction was carried out by homogenizing the tissue in buffer consisting of proteinase and phosphatase buffers using Tissue Tearor (Biospec Products, Bartlesville, OK). Protein concentrations were determined with the Pierce BCA protein assay (Thermo Fisher Scientific, Waltham, MA). SDS sample buffer was added directly to the protein extracts, boiled and resolved in a short stack (5 min) by 4–12% SDS Bis-Tris PAGE (Invitrogen, Carlsbad, CA), stained overnight with colloidal blue (Invitrogen), then destained with ddH2O for 4 h. The entire stained region was excised, equilibrated in 100 mM ammonium bicarbonate, reduced, carbidomethylated, further destained, dehydrated, and finally digested with Trypsin Gold according to the manufacturer’s protocol (Promega, Madison, WI). Following digestion, peptides were concentrated under vacuum and resolubilized in 0.1% formic acid prior to 1D reverse phase nLC-ESI-MS analysis.
Peptide analysis by liquid chromatography-electrospray ionization-tandem/mass spectrometry (LC-ESI-MS/MS)
For each sample, 1 µg of peptide digest was analyzed using an LTQ XL ion-trap mass spectrometer equipped with a nano-electrospray source and a Surveyor plus binary high-pressure liquid chromatography (HPLC) pump (Thermo Scientific, San Jose, CA) using a split-flow configuration. Separations were carried out using a 150 µm × 13 cm pulled-tip Jupiter 5-µm 300-Å C18 column (Phenomenex, Torrance, CA). The HPLC was set up with two mobile phases that included solvent A (0.1% formic acid in ddH2O) and solvent B (0.1% formic acid in 85% ddH2O/15% acetonitrile), and was programmed as follows: 0% of solvent B for 15 min (2 µL/min, load), 0%–50% of solvent B for 65 min (~0.5 nL/min, analyze), and 0% of solvent B for 20 min (2 µL/min, equilibrate). The LTQ XL was operated in data-dependent triple-play mode, with a survey-scan range of 300–1200 m/z, followed by a zoom scan for charge-state determination for each MS2, which were carried out with 2.0 dalton isolation widths on the three most intense ions. MS data were collected in profile mode for all scan types. Charge-state screening and dynamic exclusion were enabled with a minimal signal intensity of 2000, a repeat count of 2, and an exclusion duration of 90 s for ions +/− 1.5 m/z of the parent ion. The automatic gain control settings were 3.0 × 104 and 5.0 × 103 ions for survey and zoom modes, respectively. Scan times were set at 25 msec and 50 msec for survey and zoom modes, respectively. For collision-induced dissociation, the activation time, activation Q, and normalized collision energy were set at 10 msec, 0.25, and 35%, respectively. The spray voltage was set at 1.9 kV with a capillary temperature of 170° C. All runs were carried out in duplicate from the same samples.
Protein identification and quality control
The XCalibur RAW output files were centroided, converted to MzXML format, and the resulting mgf files were then created using both ReAdW and MzXML2Search respectively (http://sourceforge.net/projects/sashimi/). The data were searched using SEQUEST (v.27 rev12, .dta files), set for two missed cleavages, a precursor mass window of 0.45 Da, tryptic enzyme, variable modification M @ 15.9949, and static modifications C @ 57.0293. Searches were performed with a mouse subset of the UniRef100 database, which included common contaminants such as digestion enzymes and human keratins. Identified peptides were filtered, grouped, and quantified using ProteoIQ (Premierbiosoft, Palo Alto, CA). Only peptides with charge state of ≥ 2+, a minimum peptide length of 6 amino acids were accepted for analysis. ProteoIQ incorporates the two most common methods for statistical validation of large proteome datasets, false discovery rate (FDR), and protein probability (Keller et al., 2002; Nesvizhskii et al., 2003; Weatherly et al., 2005). The false discovery rate (FDR) was set at < 1% cut-off, with a peptide probability of ≥ 0.5, a protein group probability of ≥ 0.7, and number of unique peptides per protein of ≥ 2. Relative quantification was performed via spectral counting, and spectral count abundances were normalized between samples.
Statistical analysis
Most behavioral, histological, and neurochemical data were analyzed using one-and two-way factorial analyses of variance (ANOVAs), followed when appropriate with orthogonal paired comparisons. Tukey's HSD was used for non-orthogonal post-hoc comparisons to restrict familywise error. To correlate neurochemical with behavioral data, a composite cognition score was computed from the mean of the last four days of water-maze acquisition, when mice were performing asymptotically, and the time spent in the 30-cm target zone during the probe trial. To calculate the composite measure, the inverse of the acquisition score was taken so that "good" cognition for each measure was a higher number. Each mouse's score was then converted to a z score based on the mean and standard deviation of all mice for the particular measure. Finally, the mean of the two z scores was taken as the composite cognition score, with a population mean of 0.0 and a standard deviation (SD) of 1.0. The mean distance (km) run per day for the last 30 days was used as the activity score for each mouse. Time-series data were analyzed using hierarchical linear modeling, with time as a continuous repeated measure and subject as a nominal random factor nested within exercise group and genotype.
Filtered data for 1460 proteins were first analyzed using 2 × 2 factorial ANOVAs, with genotype and running-wheel status as between-subjects factors. Paired follow-up comparisons were conducted using Fisher’s protected LSD test on those proteins having significant main or interaction effects on the omnibus ANOVA. To avoid spurious Type I errors, follow-up comparisons were restricted to a priori hypotheses of interest, i.e., inactive wild-type vs. inactive APP/PSEN1, active wild-type vs. inactive wild-type, and active APP/PSEN1 vs. inactive APP/PSEN1. Initial ELISA data were analyzed using multivariate ANOVAs (MANOVAs) fitted to identity matrices. Pearson product-moment correlations were conducted among running, cognition, and Aβ measures. All statistical tests were two-tailed with α set at .05.
Results
Mice ran for 7 months on the running wheels, and data were collected every day. There were no significant differences in the distance run per month [Fig. 2a; genotype F(17) < 0.1, p = .934; Genotype × Month F(1,107) < 0.1, p = .875]. The time run per day and speed of running also did not differ by genotype [F’s < 1.6, p’s > .227], and were both significantly correlated with distance [r’s > .595, p’s < .012]. Consistent with our previous studies showing spatial learning deficits in these transgenics, the inactive APP/PSEN1 mice performed significantly worse than their inactive wild-type counterparts during water-maze acquisition [Fig. 2c, F(1,36) = 10.8, p = .0022; Genotype × Session F(1,316) = 2.3, p = .0301]. There was also a significant main effect for running-wheel status, demonstrating that mice with active running wheels performed better in the water maze than the sedentary groups [F(1,36) = 13.3, p = .0008]. Genotype × Activity interactions were not significant (F's < 2.7; p's > .112). Follow-up analyses showed that exercise significantly improved learning only in the transgenic mice [F(1,20) = 11.0, p = .0035]. Although enhancement of learning in wild-type mice was substantive, inactive wild-type controls were already performing proficiently and thus exercise-induced improvement was not statistically significant [F(1,16) = 3.9, p = .067]. Swim speed did not differ significantly by genotype [F's < 0.5; p's > .51] or running-wheel status [F's < 4.1; p's > .052].
On the probe trial, the main effects for genotype and running-wheel status were not statistically significant with respect to time spent in the 30-cm target zone compared to the mean time in equivalent zones in the other three quadrants [Fig. 2e; F’s < 4.1; p’s >.052]. However, there was a significant Zone × Activity interaction [F(1,34) = 4.7, p = .0364]. Follow-up tests by group showed that the inactive wild-type mice spent significantly more time in the target zone than the mean of the other three zones (p = .0323). This was not the case for the inactive APP/PSEN1 mice (p = .144), consistent with the memory deficits previously demonstrated in these mice (Bernardo et al., 2009; Reiserer et al., 2007). When given the opportunity for exercise, both wild-type and APP/PSEN1 groups showed selective search for the target zone (p's < .003). On the composite cognition score, there were significant main effects for genotype [F(1,34) = 8.1, p = .0075] and running-wheel status [Fig. 2d; F(1,34) = 10.1, p = .0031]; the interaction effect was not significant [F(1,34) = 0.4, p = .554]. To determine whether the improvement in memory was related to the extent that mice ran, we conducted correlations between the composite cognition score and the average distance per day during the last month on the running wheels. Figure 2f shows that cognition correlated tightly with the amount of exercise in wild-type mice [r(7) = .9791, p < .0001] but not in transgenics [r(6) = −.114, p = .770].
Factorial analyses of filtered protein data revealed that 194 proteins differed significantly with respect to genotype, running-wheel status, and/or the interaction between the two. Follow-up analyses on these protein showed that 42 proteins differed between the inactive wild-type and APP/PSEN1 groups; 30 proteins differed significantly between the wild-type active and inactive groups; and 27 proteins differed between the APP/PSEN1 active and inactive groups. Notably, only two of the 30 proteins that changed in response to running-wheel activity in the wild-type mice also changed in the active transgenics. Initial follow-up analyses were conducted to determine baseline differences between wild-type and transgenic animals with non-functional running wheels, i.e., to determine which changes are specifically relevant to APP overexpression and Aβ-associated neuropathology in the absence of wheel running. Of the 42 proteins that differed between the two inactive groups, 30 were overexpressed and 12 were reduced (Table 1). Most of the proteins expressed differentially between wild-type and APP/PSEN1 inactive groups clustered into pathways involved in synaptic transmission, cytoskeletal regulation, neurogenesis, oxidative phosphorylation, protein processing, and apoptosis. Three proteins differed with respect to glycolysis/ gluconeogenesis, all involved in pyruvate metabolism. Glial fibrillary acidic protein (GFAP) and heat shock 90-α (HSP90-α) were also significantly changed in APP/PSEN1 mice, indicative of the elevated neuroinflammation previously reported in these transgenics (Bernardo et al., 2009). Most prominent among the significantly-different proteins were a 20-fold increase in voltage-dependent anion channel 1 (VDAC1), an 18.6-fold increase in protein kinase A regulatory subunit 2β (PKA2β), and a 10-fold increase in protein kinase C γ (PKCγ). These proteins are involved in synaptic transmission, mitochondrial dysfunction, and apoptosis, and together suggest dysregulation of calcium signaling in the APP/PSEN1 mice. We validated these proteins using Western blot to confirm the proteomics results. Figure 3a shows that among sedentary mice, all three of the proteins were more highly expressed in APP/PSEN1 transgenics. The Genotype × Exercise interaction was significant for PKCγ [F(1,14) = 4.9, p = .0437], as was the main effect for genotype [F(1,14) = 5.8, p = .0310]. The main effect for running-wheel status was not significant [F(1,14) = 3.9, p = .069]. Tukey follow-up test showed that the inactive wild-type mice had significantly lower PKCγ values than the other three groups [Fig. 3b; critical t = 2.1, p's < .05]. Although VDAC1 was several-fold overexpressed in sedentary transgenics compared to their wild-type counterparts, none of the main or interaction effects was statistically significant [Fig. 3c; F’s < 2.3, p’s > .178]. Transgenics of both exercise groups had significantly elevated PKA2β compared to wild-type controls [Fig. 3d; F(1,7) = 9.9, p = .0163]. The effect of running-wheel status and its interaction with genotype were not significant [F’s < 0.3; p’s > .616].
Table 1.
Proteins differentially expressed between Inactive APP/PSEN1 and Inactive Wild-type (control) mice.
| Accession number |
Protein | Gene | Mol. wt. (kDa) |
Peptides | Fold change |
p | Pathway |
|---|---|---|---|---|---|---|---|
| Q2NKI4 | Protein kinase C γ (PKCγ) | Prkcg | 72.9 | 8 | 10.0 | <.05 | Synaptic transmission |
| Q9CZV8 | F-box/LRR-repeat 20 | Fbxl20 | 48.3 | 2 | 3.6 | <.05 | |
| Q3TYH2 | Ras-related protein Rab-15 | Rab15 | 24.3 | 3 | 2.9 | <.05 | |
| A2ALV3 | Endophilin-A1 | Sh3gl2 | 48.2 | 10 | 1.7 | <.01 | |
| Q9JIS5 | Synaptic vesicle glycoprotein 2A | Sv2a | 82.6 | 8 | 1.7 | <.05 | |
| B0QZN5 | Vesicle-associated membrane 2 | Vamp2 | 17.8 | 3 | 1.6 | <.05 | |
| P80316 | T-complex protein 1 subunit ε | Cct5 | 59.6 | 3 | 2.9 | <.05 | Cytoskeletal regulation |
| A2ASS6 | Titin | Ttn | 3904.0 | 34 | 2.1 | <.01 | |
| Q3TGW0 | Actin-related protein 3 | Actr3 | 47.3 | 8 | 1.6 | <.05 | |
| Q3TIZ0 | Tubulin α1C | Tuba1c | 49.9 | 22 | 1.2 | <.05 | |
| Q6ZWR6 | Nesprin-1 | Syne1 | 1009.3 | 7 | - | <.01 | |
| E9Q7P1 | Collagen, type XXII, α1 | Col22a1 | 159.8 | 2 | - | <.01 | |
| B3KXD8 | Formin-like 3 | Fmnl3 | 66.7 | 2 | 0.8 | <.05 | |
| Q9JKC6 | Cell cycle exit and neuronal differentiation 1 | Cend1 | 15.0 | 2 | 8.6 | <.01 | Neurogenesis/neuroproliferation |
| Q61488 | Desert hedgehog | Dhh | 43.5 | 2 | 5.7 | <.05 | |
| P70388 | DNA repair RAD50 | Rad50 | 153.4 | 3 | - | <.05 | |
| E9QKH0 | CLIP-associating protein 1 isoform 1 | Clasp1 | 169.2 | 4 | 0.5 | <.05 | |
| P04259 | Keratin, type II cytoskeletal 6B (cytokeratin 6B) | Krt6b | 60.0 | 11 | 0.3 | <.05 | |
| B4E1T1 | Keratin, type II cytoskeletal 5 (cytokeratin 5) | Krt5 | 58.8 | 10 | 0.3 | <.05 | |
| Q3TL54 | Tripartite motif-containing 43A | Trim43a | 52.1 | 3 | 5.7 | <.05 | Protein processing |
| Q9ER38 | Torsin-3A | Tor3a | 43.8 | 2 | - | <.01 | |
| Q3TIF8 | 40S ribosomal S24 | Rps24 | 15.4 | 2 | - | <.05 | |
| Q9D2H5 | Tripartite motif-containing 42 | Trim42 | 82.9 | 2 | 0.3 | <.05 | |
| Q6ZPR1 | Cilia and flagella associated protein 97 | Kiaa1430 | 60.8 | 2 | 0.0 | <.05 | |
| P31324 | cAMP-dependent protein kinase II-β regulatory subunit (PKA2β) | Prkar2b | 46.1 | 4 | 18.6 | <.001 | Apoptosis |
| P97350 | Plakophilin-1 | Pkp1 | 80.8 | 5 | 5.2 | <.01 | |
| P68254 | 14-3-3 θ | Ywhaq | 27.7 | 12 | 0.6 | <.01 | |
| Q3THL7 | Voltage-dependent anion channel 1 | VDAC1 | 30.7 | 6 | 20.0 | <.001 | Oxidative phosphorylation |
| Q9D6R2 | Isocitrate dehydrogenase [NAD] subunit α, mitochondrial | Idh3a | 39.6 | 9 | 2.8 | <.01 | |
| P14094 | Sodium/potassium-transporting ATPase subunit β-1 | Atp1b1 | 35.2 | 10 | 1.3 | <.05 | |
| Q3THG0 | ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit | Atp5o | 23.4 | 4 | - | <.01 | |
| Q68FL4 | Adenosylhomocysteinase 3 | Ahcyl2 | 66.8 | 4 | 0.2 | <.05 | |
| O70250 | Phosphoglycerate mutase 2 | Pgam2 | 28.8 | 2 | 3.4 | <.01 | Glycolysis/gluconeogenesis |
| P40142 | Transketolase | Tkt | 67.6 | 14 | 0.7 | <.05 | |
| P45376 | Aldose reductase | Akr1b1 | 35.7 | 3 | 0.2 | <.01 | |
| P03995 | Glial fibrillary acidic protein | Gfap | 49.9 | 20 | 12.1 | <.01 | Neuroinflammation |
| P07901 | Heat shock 90-α | Hsp90aa1 | 84.7 | 35 | 0.7 | <.01 | |
| D3Z4R5 | Uncharacterized | 42.4 | 4 | 5.7 | <.05 | Unknown | |
| F6WSS0 | GTP-binding protein 10 | Gtpbp10 | 8.3 | 2 | 3.6 | <.05 | |
| D3YXI3 | uncharacterized 3 | 50.0 | 16 | 1.2 | <.05 | ||
| F6VUT6 | Protein Gm8251 | Gm8251 | 225.8 | 3 | - | <.05 | |
| D3Z4Y4 | Keratin 5 | Krt5 | 60.3 | 9 | 0.4 | <.01 |
All proteins are listed that had significantly different main or interaction effects using 2 × 2 ANOVAs followed by Fisher's protected LSD; df = 15 for each comparison. A 0-fold change indicates that the protein was not expressed at detectable levels in the inactive APP/PSEN1 mice; a dash indicates that the protein was not expressed at detectable levels in the inactive wild-type mice.
Figure 3. Validation of highly-overexpressed proteins in sedentary APP/PSEN1 transgenic mice.
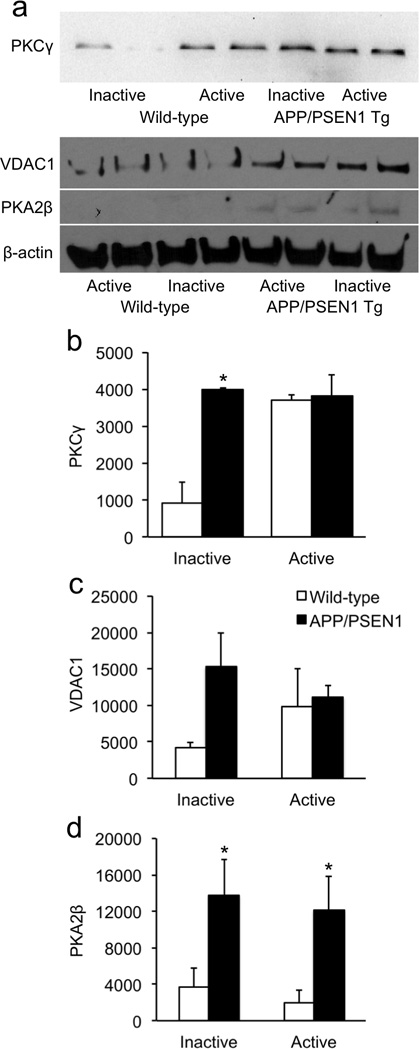
(a) Western blots of three of the most over-expressed proteins from the proteomics data in Table 1. Protein levels were elevated in sedentary transgenics compared to sedentary wild-type controls. (b) PKCγ was significantly elevated in sedentary transgenic mice. In response to exercise, PKCγ levels increased in wild-type mice but were unchanged in transgenics. (c) VDAC1 was elevated several fold in sedentary APP/PSEN1 mice compared to sedentary wild-types, although the difference was not statistically significant. (d) PKA2β was significantly elevated in both transgenic groups, compared to their wild-type counterparts. *p < .05.
Table 2 shows the proteomics changes induced by long-term running activity in wild-type mice. Chronic exercise significantly altered proteins involved in oxidative phosphorylation, cytoskeletal regulation, and protein processing. Additional changes were observed in pathways known to be affected by exercise, such as glucose metabolism and synaptic transmission. Indeed, some of the same proteins altered in sedentary APP/PSEN1 transgenics in these pathways (Table 1) were affected by exercise in wild-type mice, suggesting potential avenues for improving cognition and/or attenuation of neuropathology. Interestingly, of the 30 proteins significantly overexpressed in sedentary APP/PSEN1 transgenics (Table 1), nine were reduced back to wild-type levels by voluntary exercise (Table 3). These include two uncharacterized proteins (Accession #D3Z4R5 & #F6VUT6), as well as clusters of proteins involved in neurogenesis and cytoskeletal regulation. Indeed, all four of the cytoskeletal proteins reduced in active APP/PSEN1 mice (Table 3) were significantly overexpressed in sedentary transgenics compared to sedentary wild-type mice (Table 1). None of the proteins expressed at lower levels in sedentary transgenics was significantly altered by running-wheel activity. Notably, the running-induced changes in transgenic mice were largely different from those of wild-type mice. Specifically, exercise induced broad changes in proteins associated with oxidative phosphorylation in wild-type mice, but this pathway was minimally changed in active APP/PSEN1 mice. The converse was true with respect to neurogenesis and neuroproliferation; this represented the largest cluster of proteins that were changed by exercise in transgenic mice, but there were few running-induced changes in this pathway in wild-type mice. Although proteins involved in cytoskeletal regulation were altered by running in mice of both genotypes, the effects were in the opposite direction—largely increased for wild-type mice and uniformly decreased for transgenics. Remarkably, the disruptions in synaptic proteins observed in sedentary APP/PSEN1 transgenics were unaltered by 7 months of physical exercise. Only two proteins were affected by exercise in mice of both genotypes, glutathione s-transferase µ1 (Gstm1) and α1C tubulin (Tuba1c). Gstm1 is involved in glutathione metabolism and was downregulated 20% in active mice of both genotypes. Tuba1c is a cytoskeletal protein that was significantly over-expressed in sedentary transgenics (Table 1). In active mice, hippocampal Tuba1c levels were increased in wild-types but reduced in APP/PSEN1 transgenics. This pattern of genotypic changes was observed in a number of proteins in the cytoskeletal regulatory pathway.
Table 2.
Proteins differentially expressed between Active and Inactive (control) Wild-type mice.
| Accession number |
Protein | Gene | Mol. wt. (kDa) |
Peptides | Fold change |
p | Pathway |
|---|---|---|---|---|---|---|---|
| Q3TTA7 | E3 ubiquitin ligase CBL-B | Cblb | 109.2 | 3 | 7.1 | <.001 | Oxidative phosphorylation |
| Q9D6R2 | Isocitrate dehydrogenase [NAD] subunit α, mitochondrial | Idh3a | 39.6 | 9 | 2.8 | <.01 | |
| Q60597 | 2-oxoglutarate dehydrogenase, mitochondrial | Ogdh | 116.4 | 7 | 1.9 | <.05 | |
| O08749 | Dihydrolipoamide dehydrogenase, mitochondrial | Dld | 54.2 | 12 | 1.7 | <.05 | |
| Q99KI0 | Aconitate hydratase, mitochondrial | Aco2 | 85.4 | 34 | 0.8 | <.05 | |
| P97807 | Fumarate hydratase, mitochondrial | Fh | 54.3 | 13 | 0.7 | <.01 | |
| A2AKV3 | ATP synthase, γ subunit, mitochondrial | Atp5c1 | 20.6 | 6 | 0.3 | <.05 | |
| Q8BUR4 | Dedicator of cytokinesis 1 | Dock1 | 214.9 | 3 | 5.7 | <.05 | Cytoskeletal regulation |
| Q8C928 | Erythrocyte protein band 4.1-like 2 | Epb4.1l2 | 89.8 | 4 | 5.0 | <.05 | |
| Q3U9U3 | Tubulin, β6 class V | Tubb6 | 42.1 | 13 | 1.4 | <.05 | |
| Q3TIZ0 | Tubulin, α1C | Tuba1c | 49.9 | 22 | 1.1 | <.05 | |
| Q8CEH8 | Profilin 1 | Pfn1 | 14.9 | 6 | 0.5 | <.05 | |
| Q8BWQ5 | Serine/threonine kinase DCLK3 | Dclk3 | 88.2 | 3 | 8.6 | <.01 | Protein processing |
| Q9D2F7 | Nuclear pore membrane glycoprotein 210-like | Nup210l | 208.6 | 2 | - | <.01 | |
| A2AH75 | Kinesin family member 1B | Kif1b | 130.0 | 3 | - | <.05 | |
| Q9D2H5 | Tripartite motif-containing protein 42 | Trim42 | 82.9 | 2 | 0.3 | <.05 | |
| Q2NKI4 | Protein kinase C γ (PKCγ) | Prkcg | 72.9 | 8 | 12.9 | <.05 | Synaptic transmission |
| P04627 | Serine/threonine kinase A-Raf | Araf | 67.5 | 2 | - | <.05 | |
| P01831 | Thy-1 membrane glycoprotein | Thy1 | 18.1 | 5 | 0.3 | <.01 | |
| P45376 | Aldose reductase | Akr1b1 | 35.7 | 3 | 0.5 | <.05 | Glycolysis/gluconeogenesis |
| O89116 | Vesicle transport through interaction with t-SNAREs homolog 1A | Vti1a | 25.0 | 2 | 8.6 | <.01 | Ion Transport |
| A8DUV1 | α-globin | Hbat1 | 15.1 | 6 | 1.5 | <.05 | |
| Q8VH31 | Ret finger protein-like 4A | Rfpl4a | 32.3 | 2 | - | <.05 | |
| Q9JKC6 | Cell cycle exit and neuronal differentiation 1 | Cend1 | 15.0 | 2 | 22.9 | <.001 | Neurogenesis neuroproliferation |
| E9QKH0 | CLIP-associating protein 1 | Clasp1 | 169.2 | 4 | 0.4 | <.05 | |
| P10649 | Glutathione S-transferase μ 1 | Gstm1 | 25.9 | 16 | 0.8 | <.05 | Glutathione metabolism |
| O70472 | Transmembrane protein 131 | Tmem131 | 200.3 | 2 | - | <.05 | Neuroinflammation |
| B7U582 | Heat shock protein 70-2 | Hspa2 | 69.7 | 11 | 0.8 | <.05 | MAPK/PI3K signaling |
| Q9CQQ8 | U6 snRNA-associated Sm-like LSm7 | Lsm7 | 11.6 | 2 | 9.3 | <.01 | RNA degradation |
| Q8BGH1 | Uncharacterized | 73.3 | 2 | 3.6 | <.05 | Unknown |
All proteins are listed that had significantly different main or interaction effects using 2 × 2 ANOVAs followed by Fisher's protected LSD; df = 13 for each comparison. A 0-fold change indicates that the protein was not expressed at detectable levels in the active wild-type mice; a dash indicates that the protein was not expressed at detectable levels in the inactive wild-type mice.
Table 3.
Proteins differentially expressed between Active and Inactive (control) APP/PSEN1 mice.
| Accession number |
Protein | Gene | Mol. wt. (kDa) |
Peptides | Fold change |
p | Pathway |
|---|---|---|---|---|---|---|---|
| A3KFM7 | Chromodomain helicase DNA binding 6 | Chd6 | 305.2 | 7 | - | <.01 | Neurogenesis/neuroproliferation |
| P97477 | Aurora kinase A | Aurka | 47.1 | 4 | - | <.05 | |
| Q3UJT6 | Replication factor C (activator 1) | Rfc1 | 64.6 | 3 | - | <.05 | |
| Q64522 | Histone H2A type 2-B | Hist2h2ab | 14.0 | 3 | 0.5 | <.05 | |
| Q61488 | Desert hedgehog | Dhh | 43.5 | 2 | 0.0 | <.01 | |
| P70388 | DNA repair RAD50 | Rad50 | 153.4 | 3 | 0.0 | <.05 | |
| B1AY10 | Transcriptional repressor NF-X1 | Nfx1 | 123.7 | 4 | 0.0 | <.05 | |
| Q3TIZ0 | Tubulin α1C | Tuba1c | 249.9 | 2 | 0.9 | <.05 | Cytoskeletal regulation |
| Q3TGW0 | Actin-related 3 | Actr3 | 47.3 | 8 | 0.6 | <.05 | |
| P80316 | T-complex protein 1 subunit ε | Cct5 | 59.6 | 3 | 0.2 | <.05 | |
| E9Q7P1 | Collagen, type XXII, α1 | Col22a1 | 159.8 | 2 | 0.1 | <.05 | |
| Q5SXR6 | Clathrin, heavy chain | Cltc | 9191.8 | 91 | 0.8 | <.05 | Endocytosis |
| Q80ZI6 | E3 ubiquitin ligase LRSAM1 | Lrsam1 | 83.9 | 2 | 0.0 | <.05 | |
| Q8CBD1 | Nuclear receptor-interacting protein 1 | Nrip1 | 126.2 | 2 | - | <.05 | Protein processing |
| A2RT70 | Tenascin-R | Tnr | 1149.5 | 11 | 4.2 | <.01 | Neuritogenesis |
| Q3TJ43 | Vacuolar protein sorting-associated protein 35 | Vps35 | 91.6 | 7 | 3.2 | <.05 | |
| P08249 | Malate dehydrogenase, mitochondrial | Mdh2 | 135.6 | 6 | 1.3 | <.01 | Oxidative phosphorylation |
| P14094 | Sodium/potassium-transporting ATPase subunit β-1 | Atp1b1 | 135.2 | 10 | 0.7 | <.001 | |
| P10649 | Glutathione S-transferase μ 1 | Gstm1 | 125.9 | 6 | 0.8 | <.05 | Glutathione metabolism |
| E9Q3B9 | Monoglyceride lipase | Mgll | 35.2 | 2 | 0.0 | <.05 | Glycerolipid metabolism |
| B4DZ37 | Solute carrier family 9, isoform 10 | Slc9a10 | 51.3 | 2 | 0.0 | <.05 | Ion transport |
| A2ANU2 | Polypeptide N-acetylgalactosaminyltransferase 12 | Galnt12 | 33.1 | 2 | 3.9 | <.05 | O-Glycan biosynthesis |
| D0JYX1 | Peptidase S1 and S6, chymotrypsin/Hap | 51.5 | 2 | 3.5 | <.05 | Unknown | |
| Q6PNC0 | DmX-like protein 1 | Dmxl1 | 335.8 | 4 | - | <.05 | |
| D3Z7K0 | Ubiquitin thioesterase | Outb1 | 11.2 | 5 | 0.3 | <.05 | |
| D3Z4R5 | Uncharacterized | 42.4 | 4 | 0.0 | <.01 | ||
| F6VUT6 | Protein Gm8251 | Gm8251 | 225.8 | 3 | 0.0 | <.05 |
All proteins are listed that had significantly different main or interaction effects using 2 × 2 ANOVAs followed by Fisher's protected LSD; df = 13 for each comparison. A 0-fold change indicates that the protein was not expressed at detectable levels in the active APP/PSEN1 mice; a dash indicates that the protein was not expressed at detectable levels in the inactive APP/PSEN1 mice.
Analyses of Aβ levels showed that active APP/PSEN1 transgenics had significantly lower total, soluble, and insoluble Aβ [Fig. 4a–c; λ's < .25, F's > 7.5, p's < .0045]. Follow-up analyses showed that exercise reduced all Aβ measures significantly except for cortical insoluble Aβ42 (p = .389). The average daily distance that mice ran was inversely correlated with total hippocampal Aβ40 [Fig. 4d; r(6) = −.854, p = .0070] and to a lesser extent with total hippocampal Aβ42 [r(6) = −.711, p = .0481]. Cortical Aβ levels, although significantly reduced in active transgenics, were not correlated with running (r's < .48, p's > .239). Hippocampal total Aβ42 was positively correlated with the composite cognition score [r(6) = .729, p = .0404], but total Aβ40 was unrelated [r(6) = .448, p = .265]. Neither soluble nor insoluble hippocampal Aβ42 was specifically associated with memory [r's < .631, p's > .093]. Cortical Aβ levels were also unrelated to cognition in active transgenics [r's < .501, p's > .206], and no Aβ measure was significantly associated with cognition score in sedentary APP/PSEN1 mice [|r|'s < .709, p's > .074]. Western blot analyses using two separate antibodies revealed that full-length APP levels were significantly increased in active APP/PSEN1 mice compared to their inactive counterparts [Fig. 4e–g; λ = .07, F(1,2) = 26.6, p = .0357] However, BACE1 protein levels were significantly lower in active compared to sedentary transgenics (p = .0413), suggesting that reduced activity at the β- secretase cleavage site was at least partially responsible for reduced levels of Aβ (Fig. 4h). sAPPα did not differ between running-wheel groups [Fig. 4i; F(1,4) = .0002, p = .989].
Figure 4. Exercise-induced reductions in Aβ are associated with reduced BACE1.
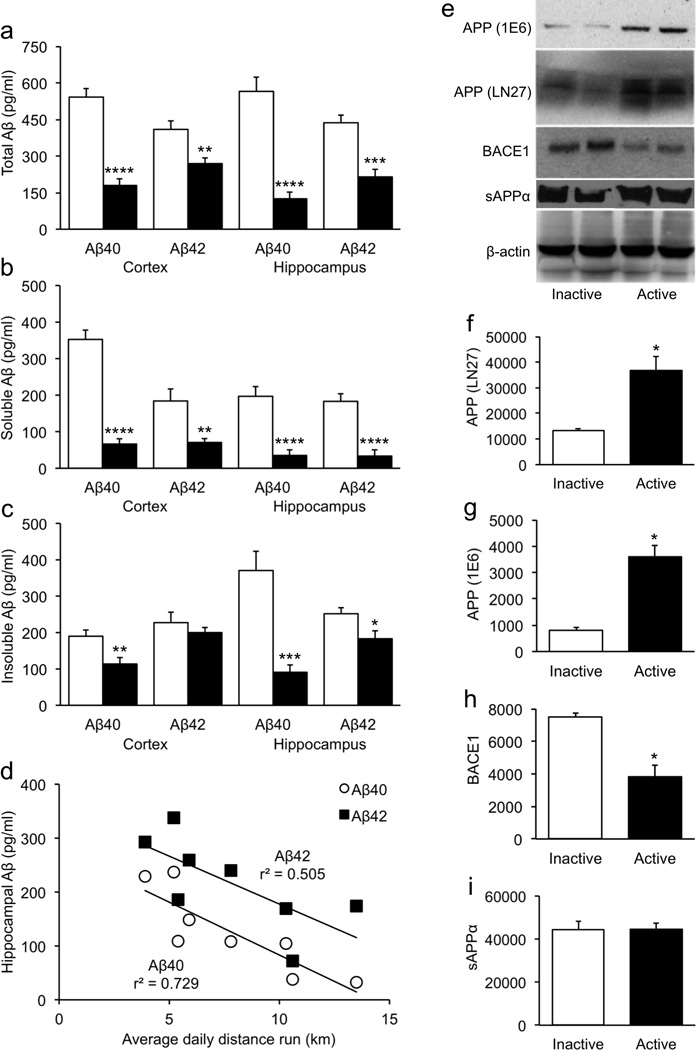
(a–c) Voluntary exercise reduced both soluble and insoluble Aβ in APP/PSEN1 transgenics, in both in hippocampus and overlying cortex. (d) The reduction in Aβ in active transgenics was significantly correlated with the amount of running. Running was not associated with cortical Aβ levels. (e–g) Full-length amyloid precursor protein (APP) was measured using two different antibodies. Both immunoblots indicated that APP was significantly elevated in active APP/PSEN1 mice. (e,h–i) In contrast to APP, BACE1 was significantly reduced in active transgenics. (i) Levels of sAPPβ were unchanged in active transgenics, compared to their sedentary counterparts. *p < .05; **p < .01; ***p < .001; ****p <.0001.
Discussion
Consistent with previous reports, we show that voluntary exercise improves memory in APP-overexpressing transgenic mice and reduces Aβ aggregation. The cognitive deficits normally observed in APP/PSEN1 transgenics were ameliorated after 7 months of voluntary exercise. Interestingly, cognition was strongly associated with the amount of running in wild-type controls but not in transgenics. Using a classical proteomics approach, we analyzed differential protein expression associated with changes in long-term physical activity. Our data suggest that running-induced changes in proteins associated with synaptic transmission, oxidative phosphorylation, and cytoskeletal regulation in wild-type mice. Exercise altered cytoskeletal proteins in APP/PSEN1 mice as well, but in the opposite direction. Synaptic proteins were largely unaffected by exercise in the double-transgenics, but neurogenesis-related proteins were significantly altered. Soluble and insoluble Aβ were broadly lowered by running, and hippocampal Aβ was negatively correlated with exercise levels. Lower levels of β-secretase in active APP/PSEN1 mice may have contributed to the reduced Aβ. Together, these results suggest that exercise may improve cognition in fundamentally different ways in wild-type and APP/PSEN1 transgenic mice.
Studies examining the effects of exercise on Aβ-associated neuropathology have produced a variety of results. This is not surprising given the different mutant lines, promoters, background strains, exercise regimens, age at initiation, diet, experimental environments, and housing conditions. Nevertheless, some exercise-induced changes occur with sufficient frequency in spite of methodological differences that they may inform us about the most robust and reliable effects. Chief among them is a reduction in Aβ and plaque formation. Consistent with most studies of voluntary exercise, we demonstrate significantly decreased Aβ in active transgenic mice. Although full-length APP protein levels were elevated in the active transgenics relative to sedentary controls, BACE1 protein was significantly reduced. This suggests that reduced amyloidogenic β-secretase cleavage in active APP/PSEN1 transgenics was at least in part responsible for the lower Aβ levels. To our knowledge this is the first report of exercise-induced changes in APP or BACE1 in APP-overexpressing mice. Three studies reported decreased α- and β-CTFs following voluntary exercise in TgCRND8 or PDGF transgenics but normal α-, β-, and γ-secretase activity (Adlard et al., 2005; Maesako et al., 2013; Maesako et al., 2012). Maesako et al. also reported increased activity of neprilysin, suggesting increased degradation of Aβ. Thus there may be multiple mechanisms by which exercise can reduce Aβ.
Many of the genotype differences noted in sedentary mice are established characteristics of APP-overexpressing mice are associated with impaired mitochondrial dysfunction and calcium dysregulation. Changes in five proteins suggest abnormal oxidative phosphorylation, which can result from mitochondrial dysfunction. This in turn alters the tricarboxylic acid cycle and directly targets Aβ (Blass, 2000; Karuppagounder et al., 2009). Synaptic proteins were also significantly altered. VDAC1 is a γ-secretase-associated protein localized to detergent-resistant membranes (DRMs), concentrated in synapses and the outer mitochondrial membrane (Hur et al., 2012; Sampson et al., 1996; Yoo et al., 2001). DRMs are rich in gangliosides and cholesterol, and the site of APP processing and Aβ aggregation (Ariga et al., 2008, 2010, 2013). Consistent with this, VDAC1 is associated with increased production of Aβ40 and Aβ42 and is elevated in brains of Alzheimer patients and APP-overexpressing transgenic mice (Cuadrado-Tejedor et al., 2011; Ramirez et al., 2009; Reddy, 2013; Yoo et al., 2001). In one study, shRNA-mediated knock-down of VDAC1 reduced Aβ levels by 70% (Hur et al., 2012). Thus it is not surprising that VDAC1 was increased in the sedentary transgenics relative to wild-type mice. The fact that physical exercise did not significantly alter VDAC1 or other synaptic proteins suggests that this did not substantively contribute to the reduced Aβ or improved cognition observed in the active APP/PSEN1 mice.
When allowed to run freely, wild-type mice exhibited protein changes associated with enhanced cellular function, energy metabolism, and ATP production. Notably, all seven of the proteins appearing in both Tables 1 and 2 changed in the same direction, i.e., those elevated in sedentary transgenics were increased by running in wild-type mice, and those reduced in sedentary transgenics were decreased by running in wild-type mice. This suggests putative compensatory processes in the sedentary transgenics that were not sufficient to ameliorate the cognitive deficits. With respect to cognition, the most relevant protein changes in wild-type mice were those involved with synaptic transmission. Cognition was tightly associated with running in wild-type mice but uncorrelated in transgenics. In contrast to robust changes observed in wild-type mice, synaptic proteins were scarcely changed by exercise in transgenic mice. This is consistent with a number of papers showing an inability of APP-overexpressing mice to enhance pathways associated with synaptic plasticity in response to increased physical or cognitive demands (Caccamo et al., 2010; Dineley et al., 2010; Hu et al., 2013).
In contrast to synaptic proteins, running reversed many of the other protein abnormalities in the transgenic mice. Table 1 shows that cytoskeletal proteins may be unstable in sedentary APP/PSEN1 mice. When the transgenics were allowed to exercise, we observed a shift toward stable expression of these proteins. Cytoskeletal integrity is important for proper memory function, and is disrupted in Alzheimer's disease (Castegna et al., 2002; Castegna et al., 2003). Thus the normalization of these proteins by exercise in the present study may have contributed to the cognitive improvement in the APP/PSEN1 transgenics. Proteins associated with neuroproliferation and neurogenesis also returned to wild-type levels in APP/PSEN1 transgenics allowed to exercise. In a number of mouse lines harboring human APP and PSEN1 transgenes, neuroproliferation, neurogenesis, and survival of new neurons are diminished relative to wild-type controls (Biscaro et al., 2009; Biscaro et al., 2012; Hamilton and Holscher, 2012; Li et al., 2014). In contrast, proliferation of neural stem cells (NSCs) is reported to be either normal or elevated in the APP/PSEN1 transgenic line used in the present study (He et al., 2013; Kamphuis et al., 2012; Niidome et al., 2008; Taniuchi et al., 2007). However, the NSCs in this transgenic line are fated to become glia and the number of new neurons generated is reduced relative to wild-type controls (Lazarov and Marr, 2010; Winner et al., 2011).
With exercise, these proteins are returned to wild-type levels, suggesting that neuroproliferation has normalized. Impaired neurogenesis can also be induced by intracranial injections of Aβ42 (Zheng et al., 2013). Thus the normalization of proteins associated with neuroproliferation and neurogenesis in the active PSEN1 transgenics may be secondary to the reduction in Aβ42.
Total hippocampal Aβ42 was positively associated with cognition in active transgenics despite markedly lower levels overall in this group and a negative correlation with the amount of exercise. This is counter-intuitive given the well-established role of small Aβ42 oligomers in disrupting synaptic architecture and plasticity (Klyubin et al., 2005; Marcello et al., 2007; Selkoe, 2008). However, there is some indication that endogenous Aβ may serve a neuroprotective role, given that it is generated in response to toxic conditions such as oxidative stress or hypoxia (D'Alton and George, 2011). Indeed, a number of studies have shown that Aβ monomers are neuroprotective and improve memory (Giuffrida et al., 2009; O'Nuallain et al., 2011; Scopes et al., 2012). Aβ42 may enhance cognition through its ability to facilitate neurogenesis at low concentrations. Sotthibundhu et al. (Sotthibundhu) demonstrated concentration-dependent enhancement of neurogenesis by oligomeric Aβ42 in vitro up to a marginally-toxic 5 µM. In vivo, they showed that intraventricular injections of low concentrations of Aβ42, but not Aβ16, increased neurogenic activity in the subventricular zone of wild-type mice. Others have shown that this enhancement is specific to the 1–42 sequence and is not observed with 1–40 or 25–35 (Calafiore et al., 2006; Heo et al., 2007; Jin et al., 2004; López-Toledano and Shelanski, 2004). Our data are consistent with this. In sedentary transgenics whose Aβ42 levels were high, memory was poor. When Aβ42 concentrations were lower following exercise, proteins associated with neurogenesis were normalized and Aβ42 was positively correlated with cognition. In addition to the positive effects of Aβ42, APP may have exerted beneficial effects on the cognitive status of the active transgenics. Elevated 3–4 fold in active transgenics, APP has been shown to protect against synaptic damage, excitotoxicity, and oxidative stress in the absence of high levels of Aβ (Masliah et al., 1997; Mucke et al., 1994).
Our data illustrate several potential avenues for mechanistic investigation into the ways in which voluntary physical exercise may ameliorate Alzheimer-like neuropathology and improve memory. The enhancement of synaptic plasticity by exercise has been demonstrated previously, and may have played a role in the cognitive performance of wild-type mice in the present study. In contrast, synaptic proteins did not appreciably change in the active transgenics. Instead, the large improvement in memory in these mice was associated with reduced Aβ and normalization of proteins involved in neurogenesis and cytoskeletal regulation. Unlike previous reports, the exercise-induced Aβ reductions in the present study were accompanied by lower β- secretase levels. Taken together, these data show that multiple mechanisms may exist by which running may reduce Aβ, but that some well-established abnormalities exhibited by these double-transgenics, such as synaptic transmission and calcium dysregulation, are not substantially altered by 7 months of exercise.
Highlights.
Voluntary exercise improved cognition in APP/PSEN1 double-transgenic mice.
The distance run was correlated with memory in wild-type (r2 = .96) but not APP/PSEN1 (r2 = .01) mice.
Proteomics revealed genotype differences in sedentary mice and exercise-induced changes.
Soluble and insoluble Aβ40 and Aβ42 levels were significantly reduced in active transgenics.
Exercise increased full-length APP and decreased BACE1 in APP/PSEN1 mice.
Acknowledgements
Proteomic analysis was conducted at the UAB Comprehensive Cancer Center Mass Spectrometry/Proteomics Shared Facility. Funding was provided by the National Institutes of Health (AG031253, AG022439, & P30CA13148).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:4217–4221. doi: 10.1523/JNEUROSCI.0496-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga T, Itokazu Y, McDonald MP, Hirabayashi Y, Ando S, Yu RK. Brain gangliosides of a transgenic mouse model of Alzheimer's disease with deficiency in GD3-synthase: expression of elevated levels of a cholinergic-specific ganglioside, GT1aalpha. ASN neuro. 2013;5:141–148. doi: 10.1042/AN20130006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga T, McDonald MP, Yu RK. Role of ganglioside metabolism in the pathogenesis of Alzheimer's disease. J Lipid Res. 2008;49:1157–1175. doi: 10.1194/jlr.R800007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga T, Yanagisawa M, Wakade C, Ando S, Buccafusco JJ, McDonald MP, Yu RK. Ganglioside metabolism in a transgenic mouse model of Alzheimer's disease: expression of Chol-1alpha antigens in the brain. ASN neuro. 2010;2 doi: 10.1042/AN20100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrazola MS, Varela-Nallar L, Colombres M, Toledo EM, Cruzat F, Pavez L, Assar R, Aravena A, Gonzalez M, Montecino M, Maass A, Martinez S, Inestrosa NC. Calcium/calmodulin-dependent protein kinase type IV is a target gene of the Wnt/beta-catenin signaling pathway. J Cell Physiol. 2009;221:658–667. doi: 10.1002/jcp.21902. [DOI] [PubMed] [Google Scholar]
- Bazalakova MH, Wright J, Schneble EJ, McDonald MP, Heilman CJ, Levey AI, Blakely RD. Deficits in acetylcholine homeostasis, receptors and behaviors in choline transporter heterozygous mice. Genes Brain Behav. 2007;6:411–424. doi: 10.1111/j.1601-183X.2006.00269.x. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Harrison FE, McCord M, Zhao J, Bruchey A, Davies SS, Jackson Roberts L, 2nd, Mathews PM, Matsuoka Y, Ariga T, Yu RK, Thompson R, McDonald MP. Elimination of GD3 synthase improves memory and reduces amyloid-beta plaque load in transgenic mice. Neurobiol Aging. 2009;30:1777–1791. doi: 10.1016/j.neurobiolaging.2007.12.022. [DOI] [PubMed] [Google Scholar]
- Bernardo A, McCord M, Troen AM, Allison JD, McDonald MP. Impaired spatial memory in APP-overexpressing mice on a homocysteinemia-inducing diet. Neurobiology of Aging. 2007;28:1195–1205. doi: 10.1016/j.neurobiolaging.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Biscaro B, Lindvall O, Hock C, Ekdahl CT, Nitsch RM. Abeta immunotherapy protects morphology and survival of adult-born neurons in doubly transgenic APP/PS1 mice. J Neurosci. 2009;29:14108–14119. doi: 10.1523/JNEUROSCI.2055-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscaro B, Lindvall O, Tesco G, Ekdahl CT, Nitsch RM. Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer's disease. Neurodegener Dis. 2012;9:187–198. doi: 10.1159/000330363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass JP. The mitochondrial spiral. An adequate cause of dementia in the Alzheimer's syndrome. Annals of the New York Academy of Sciences. 2000;924:170–183. doi: 10.1111/j.1749-6632.2000.tb05576.x. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:22687–22692. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafiore M, Battaglia G, Zappala A, Trovato-Salinaro E, Caraci F, Caruso M, Vancheri C, Sortino MA, Nicoletti F, Copani A. Progenitor cells from the adult mouse brain acquire a neuronal phenotype in response to beta-amyloid. Neurobiol Aging. 2006;27:606–613. doi: 10.1016/j.neurobiolaging.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Cao D, Lu H, Lewis TL, Li L. Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2007;282:36275–36282. doi: 10.1074/jbc.M703561200. [DOI] [PubMed] [Google Scholar]
- Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M, Vilarino M, Cabodevilla F, Del Rio J, Frechilla D, Perez-Mediavilla A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer's disease transgenic mice: an insight into the pathogenic effects of amyloid-beta. Journal of Alzheimer's disease : JAD. 2011;23:195–206. doi: 10.3233/JAD-2010-100966. [DOI] [PubMed] [Google Scholar]
- D'Alton S, George DR. Changing perspectives on Alzheimer's disease: thinking outside the amyloid box. J Alzheimers Dis. 2011;25:571–581. doi: 10.3233/JAD-2011-110089. [DOI] [PubMed] [Google Scholar]
- Dhanushkodi A, McDonald MP. Intracranial V. cholerae sialidase protects against excitotoxic neurodegeneration. PloS one. 2011;6:e29285. doi: 10.1371/journal.pone.0029285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J Neurosci Res. 2010;88:2923–2932. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanigan TJ, Xue Y, Kishan Rao S, Dhanushkodi A, McDonald MP. Abnormal vibrissa-related behavior and loss of barrel field inhibitory neurons in 5xFAD transgenics. Genes Brain Behav. 2014 doi: 10.1111/gbb.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A, Holscher C. The effect of ageing on neurogenesis and oxidative stress in the APP(swe)/PS1(deltaE9) mouse model of Alzheimer's disease. Brain Res. 2012;1449:83–93. doi: 10.1016/j.brainres.2012.02.015. [DOI] [PubMed] [Google Scholar]
- Harrison FE, Allard J, Bixler R, Usoh C, Li L, May JM, McDonald MP. Antioxidants and cognitive training interact to affect oxidative stress and memory in APP/PSEN1 mice. Nutr Neurosci. 2009a;12:203–218. doi: 10.1179/147683009X423364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison FE, Hosseini AH, McDonald MP, May JM. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharmacol Biochem Behav. 2009b;93:443–450. doi: 10.1016/j.pbb.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison FE, May JM, McDonald MP. Vitamin C deficiency increases basal exploratory activity but decreases scopolamine-induced activity in APP/PSEN1 transgenic mice. Pharmacol Biochem Behav. 2010;94:543–552. doi: 10.1016/j.pbb.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Jin WL, Lok KH, Wang Y, Yin M, Wang ZJ. Amyloid-beta(1-42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 2013;4:e924. doi: 10.1038/cddis.2013.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo C, Chang KA, Choi HS, Kim HS, Kim S, Liew H, Kim JA, Yu E, Ma J, Suh YH. Effects of the monomeric, oligomeric, and fibrillar Abeta42 peptides on the proliferation and differentiation of adult neural stem cells from subventricular zone. J Neurochem. 2007;102:493–500. doi: 10.1111/j.1471-4159.2007.04499.x. [DOI] [PubMed] [Google Scholar]
- Hu YS, Long N, Pigino G, Brady ST, Lazarov O. Molecular mechanisms of environmental enrichment: impairments in Akt/GSK3beta, neurotrophin-3 and CREB signaling. PLoS One. 2013;8:e64460. doi: 10.1371/journal.pone.0064460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur JY, Teranishi Y, Kihara T, Yamamoto NG, Inoue M, Hosia W, Hashimoto M, Winblad B, Frykman S, Tjernberg LO. Identification of novel gamma-secretase-associated proteins in detergent-resistant membranes from brain. The Journal of biological chemistry. 2012;287:11991–12005. doi: 10.1074/jbc.M111.246074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A. 2004;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Orre M, Kooijman L, Dahmen M, Hol EM. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer's disease mouse model. Glia. 2012;60:615–629. doi: 10.1002/glia.22295. [DOI] [PubMed] [Google Scholar]
- Karuppagounder SS, Pinto JT, Xu H, Chen HL, Beal MF, Gibson GE. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer's disease. Neurochemistry International. 2009;54:111–118. doi: 10.1016/j.neuint.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Marr RA. Neurogenesis and Alzheimer's disease: at the crossroads. Exp Neurol. 2010;223:267–281. doi: 10.1016/j.expneurol.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL, Cao D, Lu H, Mans RA, Su YR, Jungbauer L, Linton MF, Fazio S, LaDu MJ, Li L. Overexpression of human apolipoprotein A–I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem. 2010;285:36958–36968. doi: 10.1074/jbc.M110.127829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Guo F, Zhang Q, Huo T, Liu L, Wei H, Xiong L, Wang Q. Electroacupuncture decreases cognitive impairment and promotes neurogenesis in the APP/PS1 transgenic mice. BMC Complement Altern Med. 2014;14:37. doi: 10.1186/1472-6882-14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Toledano MA, Shelanski ML. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J Neurosci. 2004;24:5439–5444. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Iwata A, Kubota M, Watanabe K, Uemura M, Noda Y, Asada-Utsugi M, Kihara T, Takahashi R, Shimohama S, Kinoshita A. Continuation of exercise is necessary to inhibit high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. PloS one. 2013;8:e72796. doi: 10.1371/journal.pone.0072796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Hayashida N, Asada-Utsugi M, Watanabe K, Uemura M, Kihara T, Takahashi R, Shimohama S, Kinoshita A. Exercise is more effective than diet control in preventing high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. The Journal of biological chemistry. 2012;287:23024–23033. doi: 10.1074/jbc.M112.367011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A, Epis R, Borroni B, Cattabeni F, Sala C, Padovani A, Di Luca M. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J Neurosci. 2007;27:1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L. Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience. 1997;78:135–146. doi: 10.1016/s0306-4522(96)00553-2. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci. 2001;21:5239–5250. doi: 10.1523/JNEUROSCI.21-14-05239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Niidome T, Taniuchi N, Akaike A, Kihara T, Sugimoto H. Differential regulation of neurogenesis in two neurogenic regions of APPswe/PS1dE9 transgenic mice. Neuroreport. 2008;19:1361–1364. doi: 10.1097/WNR.0b013e32830e6dd6. [DOI] [PubMed] [Google Scholar]
- O'Nuallain B, Klyubin I, Mc Donald JM, Foster JS, Welzel A, Barry A, Dykoski RK, Cleary JP, Gebbink MF, Rowan MJ, Walsh DM. A monoclonal antibody against synthetic Abeta dimer assemblies neutralizes brain-derived synaptic plasticity-disrupting Abeta. J Neurochem. 2011;119:189–201. doi: 10.1111/j.1471-4159.2011.07389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez CM, Gonzalez M, Diaz M, Alonso R, Ferrer I, Santpere G, Puig B, Meyer G, Marin R. VDAC and ERalpha interaction in caveolae from human cortex is altered in Alzheimer's disease. Molecular and Cellular Neurosciences. 2009;42:172–183. doi: 10.1016/j.mcn.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Reddy PH. Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer's disease? Biochimica et Biophysica Acta. 2013;1832:67–75. doi: 10.1016/j.bbadis.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impaired spatial learning in the APPSwe+PSEN1ΔE9 bigenic mouse model of Alzheimer's disease. Genes Brain Behav. 2007;6:54–65. doi: 10.1111/j.1601-183X.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- Sampson MJ, Lovell RS, Davison DB, Craigen WJ. A novel mouse mitochondrial voltage-dependent anion channel gene localizes to chromosome 8. Genomics. 1996;36:192–196. doi: 10.1006/geno.1996.0445. [DOI] [PubMed] [Google Scholar]
- Scopes DI, O'Hare E, Jeggo R, Whyment AD, Spanswick D, Kim EM, Gannon J, Amijee H, Treherne JM. Abeta oligomer toxicity inhibitor protects memory in models of synaptic toxicity. Br J Pharmacol. 2012;167:383–392. doi: 10.1111/j.1476-5381.2012.01973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotthibundhu A, Li QX, Thangnipon W, Coulson EJ. Abeta(1-42) stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol Aging. 2009;30:1975–1985. doi: 10.1016/j.neurobiolaging.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Taniuchi N, Niidome T, Goto Y, Akaike A, Kihara T, Sugimoto H. Decreased proliferation of hippocampal progenitor cells in APPswe/PS1dE9 transgenic mice. Neuroreport. 2007;18:1801–1805. doi: 10.1097/WNR.0b013e3282f1c9e9. [DOI] [PubMed] [Google Scholar]
- Toledo EM, Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer's disease. Mol Psychiatry. 2010;15:272–285. 228. doi: 10.1038/mp.2009.72. [DOI] [PubMed] [Google Scholar]
- Weatherly DB, Atwood JA, 3rd, Minning TA, Cavola C, Tarleton RL, Orlando R. A Heuristic method for assigning a false-discovery rate for protein identifications from Mascot database search results. Mol Cell Proteomics. 2005;4:762–772. doi: 10.1074/mcp.M400215-MCP200. [DOI] [PubMed] [Google Scholar]
- Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur J Neurosci. 2011;33:1139–1151. doi: 10.1111/j.1460-9568.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- Yoo BC, Fountoulakis M, Cairns N, Lubec G. Changes of voltage-dependent anion-selective channel proteins VDAC1 and VDAC2 brain levels in patients with Alzheimer's disease and Down syndrome. Electrophoresis. 2001;22:172–179. doi: 10.1002/1522-2683(200101)22:1<172::AID-ELPS172>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Zheng M, Liu J, Ruan Z, Tian S, Ma Y, Zhu J, Li G. Intrahippocampal injection of Abeta1-42 inhibits neurogenesis and down-regulates IFN-gamma and NF-kappaB expression in hippocampus of adult mouse brain. Amyloid. 2013;20:13–20. doi: 10.3109/13506129.2012.755122. [DOI] [PubMed] [Google Scholar]