

Introduction
Tufting Enteropathy (TE) is a rare congenital abnormality of the intestinal mucosa, characterized clinically by intestinal failure and intractable diarrhea [1], and histologically by persistent villous atrophy, with disorganization and dedifferentiation of enterocytes, leading to the “tufting” appearance of the surface epithelium [2]. Recently, the molecular pathophysiology of TE has been established as a defect/deficiency in the Epithelial Cell Adhesion Molecule (EpCAM), inherited as an autosomal recessive trait (Mendelian Inheritance in Man #613217) [3].
We describe a Caucasian male infant, who presented with intractable diarrhea, vomiting, and failure to thrive at 2 weeks of age and subsequently was diagnosed with TE, confirmed by immunohistochemistry and genetic testing. Exome sequencing revealed compound heterozygosity for a known pathogenic G>A mutation in the donor splice site of exon 4 (c.426-1G>A) [3] and a novel nonsense mutation (c.265C>T, p.Gln89X) in EPCAM.
Initial Presentation
The patient, a 16-day-old male infant, was born to non-consanguineous Caucasian parents, of European descent, at 37 weeks gestation, via elective Caesarian section, weighing 2,900g at birth. At presentation, the infant had copious amounts of watery diarrhea (>10 bowel movements daily) with mucus, but no blood. Additionally, he had persistent, non-bilious, non-bloody emesis with feeds, and weight loss of 20% from birth.
Initial evaluation and physical examination were remarkable for dehydration, failure to thrive and lack of dysmorphic or syndromic features. Newborn screening, imaging studies, stool testing, infectious, immunologic, allergic and extensive laboratory work up did not reveal an etiology for the infant’s symptoms. As part of the diagnostic workup, an upper endoscopy and flexible sigmoidoscopy, with biopsies, were performed at 3 weeks of age. Gross findings were unremarkable, but histologic findings revealed acute esophagitis, gastritis and duodenitis, as well as inflammatory eosinophilic infiltrates in the lamina propria of the colon, suggestive of an infectious or allergic etiology (Fig. 1). As the infant’s clinical condition did not improve with time, nor with an exclusive elemental formula diet, additional biopsies were obtained at 2 months of age, revealing villous atrophy in the duodenum, with coalescence of the surface epithelium into characteristic tufts in both the duodenum and the colon (Fig. 2).
Fig. 1.
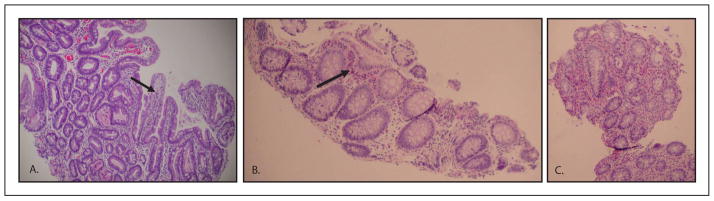
Intestinal biopsies at 3 weeks of age demonstrate mild duodenitis with slightly shortened villi, but otherwise preserved villous architecture (a); mucosal eosinophilia in the right colon, as indicated by the arrow (b); marked mucosal eosinophilia in the recto-sigmoid colon, with >50 esosinophils per high powered field (c).
Fig. 2.
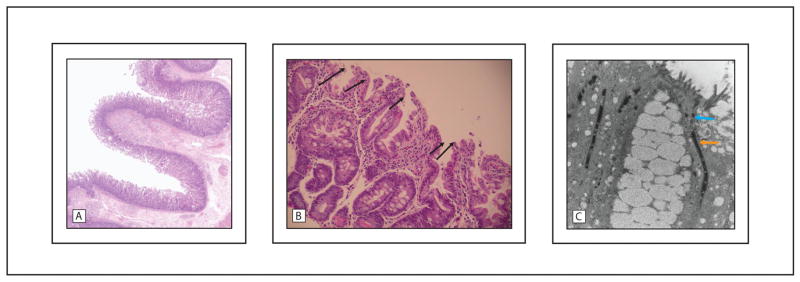
Normal architecture of healthy duodenal villi (a) compared to “tufting” of the duodenal villi (arrows) observed in follow-up biopsies at 2 months of age (b). Electron microscopy of the intestinal tissue demonstrates elongation of the desmosomes (white arrow) typical of Tufting Enteropathy, compared to normal length desmosomes (black arrow) (c).
Diagnostic Confirmation
The diagnosis of TE was established based on the infant’s clinical course and histological findings. To further support the diagnosis, immunohistochemical staining for EpCAM was performed, with absence of EpCAM confirmed in the duodenum and colon; electron microscopy demonstrated significant elongation of desmosomes in both tissues (Fig. 2c). Ultimately, genetic studies were undertaken to corroborate the diagnosis, as well as to explore a possible disease genotype-phenotype relationship in TE.
For genetic testing, exome sequencing was performed under a research protocol, as previously described [4]. Sequence analysis revealed a known pathogenic splicing mutation at position −1 of exon 4 in EPCAM (c.426-1G>A, Fig. 3a) [3]. The mutation was present in 48% of the 50 reads covering the nucleotide. Familial testing revealed that the mutation was inherited paternally. A second, novel nonsense mutation of maternal origin was identified, confirming compound heterozygosity in EPCAM, consistent with a molecular diagnosis of TE. The second mutation (c.265C>T) is predicted to result in an introduction of a premature stop codon at amino acid 89 (p.Gln89X) (Fig. 3b). The mutation was found in 54% of 61 sequencing reads covering the position. It is previously unreported and absent from the exome variant server (http://evs.gs.washington.edu/EVS/), and other commonly used genomic databases (i.e, 1,000 genomes, CPGM)[4]. Both mutations were confirmed clinically by capillary sequencing.
Fig. 3.

Next-generation sequencing results of the two EPCAM mutations from the proband viewed in the Integrative Genome Viewer [8]. Mutation 1, c.426-1G>A, was predicted to impact EPCAM RNA splicing (a) and mutation 2, c.265C>T, p.Gln89X (b).
Clinical Course
The patient is now 2 years old and has had clinical improvement in symptoms, growth, and development with initiation of long-term, outpatient, parenteral nutrition (PN). He currently receives 80% of his calories from PN; the remainder, from continuous gastric feeds of elemental formula. He has had stable, mild, TPN-related cholestasis and continues to require systemic budesonide therapy for persistent eosinophilic enterocolitis. Attempts to wean budesonide have resulted in histologically-documented recurrence of intestinal eosinophilic infiltrates, and predominantly upper-tract GI symptoms (i.e. vomiting/retching).
Conclusions
Certain TE phenotypes (i.e. Maltese) appear to be associated with more favorable clinical outcomes, including successful weaning of PN [5]. Recent published reports, however, have failed to establish a clear disease histology-phenotype relationship [6]. As more causative mutations in EPCAM are described, it will become possible to evaluate whether a disease genotype-phenotype relationship exists for TE.
Herein, we describe a novel nonsense mutation in EPCAM (p.Gln89X), associated with the clinical and immunohistochemical diagnosis of TE. This finding provides new insight into the disease pathogenesis of TE and raises the possibility for a disease genotype-phenotype relationship. It is plausible that these causative mutations could help explain the unusual feature of eosinophilic enterocolitis in our patient, since they were associated with absent EpCAM expression. Although, in theory, permeability in the intestinal mucosa of patients with TE could predispose to eosinophilic infiltrates, this histologic finding is not commonly reported in the TE literature [5]. Alternatively, there may be a genetic component to duodenal vs. colonic manifestations of TE, as not all patients develop tufts in both the small and the large intestine. The reason for this differential manifestation of disease expression remains unknown. Finally, as recently suggested by Sivagnanam et. al., new information regarding EPCAM mutations, particularly nonsense mutations, may have important therapeutic implications [7]. Improved understanding of the genetic mutations in EpCAM, and increased knowledge regarding the disease genotype-phenotype relationship, could lead to more accurate disease prognostics, as well as to the identification of individualized treatment adjuncts for patients with TE; particularly, as treatment options for TE are limited, and the majority of patients remain PN-dependent and/or go on to require intestinal transplantation.
Footnotes
The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosures: This work was supported, in part, by the National Center for Research Resources and the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) through Grant Number KL2 TR000089, DLD (PI); and the National Institutes of Child Health and Human Development of the NIH through a postdoctoral training grant in Pediatric/Developmental Clinical Pharmacology (1T32HD069038-01; Gregory L. Kearns, PI) of which VS is a recipient.
References
- 1.Reifen RM, Cutz E, Griffiths AM, et al. Tufting enteropathy: a newly recognized clinicopathological entity associated with refractory diarrhea in infants. J Pediatr Gastroenterol Nutr. 1994;18(3):630–8. [PubMed] [Google Scholar]
- 2.Sherman PM, Mitchell DJ, Cutz E. Neonatal enteropathies: defining the causes of protracted diarrhea of infancy. J Pediatr Gastroenterol Nutr. 2004;38(1):16–26. doi: 10.1097/00005176-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Sivagnanam M, Mueller JL, Lee H, et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135(2):16–26. doi: 10.1053/j.gastro.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinwiddie DL, Bracken J, Bass J, et al. Molecular diagnosis of infantile onset inflammatory bowel disease by exome sequencing. Genomics. 2013;102(5–6):442–447. doi: 10.1016/j.ygeno.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeGaetano J, Sebire NJ, Hill S, et al. Mucosal Inflammation as a Component of Tufting Enteropathy. Immunogastroenterology. 2013;2(1):62–67. [Google Scholar]
- 6.Lemale J, Coulomb A, Dubern B, et al. Intractable diarrhea with tufting enteropathy: a favorable outcome is possible. J Pediatr Gastroenterol Nutr. 2001;52(6):734–739. doi: 10.1097/MPG.0b013e31820731db. [DOI] [PubMed] [Google Scholar]
- 7.Sivagnanam M, Mueller JL, Szigeti R, et al. Transcripational read-through induction treatment trial in intestinal failure induced by an EpCAM nonsense mutation. Case Rep Med. 2012 doi: 10.1155/2012/173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]