

Abstract
Pulmonary arterial hypertension (PAH) may be suspected based on the clinical history, physical examination and electrocardiogram findings but imaging is usually central to confirming the diagnosis, establishing a cause and guiding therapy. The diagnostic pathway of PAH involves a variety of complimentary investigations of which computed tomography pulmonary angiography (CTPA) has established a central role both in helping identify an underlying cause for PAH and assessing resulting functional compromise. In particular CTPA is considered as the gold standard technique for the diagnosis of thromboembolic disease. This article reviews the CTPA evaluation in PAH, describing CTPA techniques, a systematic approach to interpretation and spectrum of key imaging findings.
Keywords: Pulmonary arterial hypertension (PAH), computed tomography (CT), echocardiography
Introduction
Pulmonary arterial hypertension (PAH) is defined as an increase in mean pulmonary arterial pressure greater than 25 mmHg at rest, or greater than 30 mmHg during exercise (1). In addition to elevated pulmonary artery pressures and pulmonary vascular resistance, post capillary pressure may also be elevated (>15 mmHg) in cases of left ventricular dysfunction. Causes of PAH are many and varied and it is important to emphasize that PAH encompasses many different patho-physiologies, often involving complex interactions between the pulmonary circulation and right heart. Historical classification systems have attempted to subdivide PAH into pathologies primarily involving the pulmonary arteries, termed primary PAH and those involving the lung parenchyma and heart, termed secondary PAH. Subsequent research has led to a more in depth understanding of the complex interrelating mechanisms at play in PAH and in 1998, a clinical classification of PAH was first established. This suggested categorizing PAH into groups which share similar pathological findings, similar haemodynamic characteristics and, similar management. The latest iteration of this system is the 2013 Nice classification which maintains the general scheme of previous systems with five main groups of disorders (Table 1).
Table 1. World Health Organization Pulmonary Hypertension Clinical Classification, 2013 (2).
| Group 1 Pulmonary arterial hypertension |
| Idiopathic pulmonary arterial hypertension |
| Heritable |
| Drug induced |
| Secondary to other diseases, such as: |
| Connective tissue disease |
| HIV |
| Portal hypertension |
| Congenital heart disease |
| Persistent pulmonary hypertension of the newborn |
| Group 1’ Pulmonary veno-occlusive disease or pulmonary capillary haemangiomatosis |
| Group 2 Pulmonary hypertension secondary to left heart disease |
| Group 3 Pulmonary hypertension secondary to lung diseases |
| Group 4 Pulmonary hypertension secondary to chronic thromboembolic disease |
| Group 5 Pulmonary hypertension with unclear multifactorial mechanisms |
PAH may initially be suggested by the clinical history, physical examination and electrocardiogram findings but imaging is usually central to suggesting or confirming the diagnosis, establishing a cause and guiding therapy. The diagnostic pathway of PAH involves a variety of complimentary investigations with the overall aim of establishing the diagnosis, identifying an underlying cause and assessing resulting functional compromise. Transthoracic echocardiography (TTE) provides a useful initial screening tool, in which the pulmonary arterial pressure is indirectly assessed via calculation of tricuspid regurgitation jet velocity (1). TTE also provides valuable information regarding cardiac morphology and function and in some cases may elicit the cause, e.g., atrial septal defect (ASD). Right heart catheterisation (RHC) is the gold standard method of quantifying the pulmonary artery pressures and can also be used to measure the pulmonary capillary wedge pressure in suspected left heart disease. RHC provides very limited information regarding the underlying cause of PAH (3). Cardiovascular magnetic resonance imaging (MRI) is the reference standard technique for assessment of left and right heart function and for detection of intracardiac shunts but currently has a limited role for assessment of the peripheral pulmonary arteries. MRI may not be suitable in some patients such as those with an implanted ferromagnetic device or claustrophobia.
Historically, the role of contrast enhanced computed tomography pulmonary angiography (CTPA) has been to exclude structural lung and proximal thromboembolic diseases as underlying causes of PAH (4-6). Recent technological advances in CT spatial and temporal resolution and developments such as dual source technology and ECG gating, now facilitate a more detailed assessment of the pulmonary vasculature and of cardiac morphology. As a result, CTPA is now able to provide a more comprehensive evaluation in patients with PAH and in many centres has become an integral investigation in the PAH diagnostic pathway. CTPA is also now considered as the gold standard technique for diagnosis of thromboembolic disease, both acute and chronic. This article reviews the CTPA evaluation in PAH, describing CTPA techniques, a systematic approach to interpretation and spectrum of key imaging findings.
Techniques
CTPA protocols vary between institutions depending upon the type of scanner, the ability to perform ECG-gating and type of injector pump used amongst other factors. Regardless of technique the aim of any CTPA study is to optimally opacify the right heart chambers and pulmonary arterial tree and limit cardiac and diaphragmatic motion artefact. Correct timing of contrast medium administration is critical and is usually achieved via a bolus tracking or a test bolus technique. Bolus tracking involves placing a cursor in the main pulmonary artery (MPA) and triggering of scan acquisition at a pre-defined threshold (typically 150 HU). This method is generally reliable but in some patients with severe cardiac impairment or congenital heart disease the delay time can be very long and a test bolus injection may be preferable. Test bolus involves a 20-25 mL contrast medium injection and continuous low dose scanning through the MPA to create a time-density curve from which the subsequent main scan is planned.
Non-ECG gated CTPA is best acquired in a helical fashion in a caudo-cranial direction from diaphragm to apices in held shallow inspiration to minimise diaphragm related artefact. Approximately 80 mL of contrast medium is injected at 4-5 mL/s with scan triggering 5 s after peak MPA enhancement. Image reconstructions with 1-2 mm axial sections and 1 mm overlap is suggested.
ECG-gated CTPA is usually performed using retrospective gating (continuous data acquisition through the cardiac cycle) with aggressive dose modulation to help minimise radiation exposure. Maximal exposure is typically restricted to the diastolic period of the cardiac cycle (60-70% of the R-R interval for heart rates under 60 bpm and 50-80% above 60 bpm). A cranio-caudal scan direction with triggering 2 s after MPA enhancement is used with a pitch of 0.4-0.5 depending on heart rate. Image reconstructions with 1 mm axial sections and 1 mm overlap is suggested.
Dual energy CTPA is a technique which exploits the photoelectric absorption properties of iodinated contrast media at different kilovoltage (kV) levels. Scan technique is very similar to a standard ungated CTPA but with the addition of a saline flush to minimise superior vena cava (SVA) related artefact. Simultaneous data acquisitions at both 140 and 80 kV (with dual source technology or via rapid kV switching with single source scanners) is used to generate an iodine perfusion map of the lung parenchyma via a subtraction technique. The datasets can also be reconstructed to provide standard “anatomical” grey scale images. Dual energy CTPA provides both anatomical and functional information and is providing new insights into a range of pulmonary vascular pathology including conditions associated with PAH such as chronic thromboembolic disease (CTED) (7-9).
Systematic approach to interpretation
Thoracic abnormalities associated with PAH can be broadly classified by anatomical site, namely the pulmonary vasculature, heart and lung parenchyma. Due to the many underlying causes of PAH, a systematic approach to CTPA interpretation is essential to avoid many subtle but potentially significant findings.
Pulmonary arteries
The MPA is conventionally measured at its bifurcation, perpendicular to its long axis, on an axial slice (Figure 1). Multiple CTPA findings have been described which suggest PAH regardless of cause. A MPA diameter >29 mm has traditionally been used as a threshold measurement above which PAH is suggested (10,11), stated as having 97% positive predictive value, 89% specificity and 87% sensitivity (12). Subsequent studies have called into question the specificity of this finding, especially in patients with advanced interstitial lung disease and have also shown that dilatation of the MPA above 32 mm may be a more specific cut-off value (13). It must be emphasised that normal MPA dimensions are frequently seen in cases of mild PAH and therefore a normal MPA diameter cannot exclude the diagnosis (13).
Figure 1.
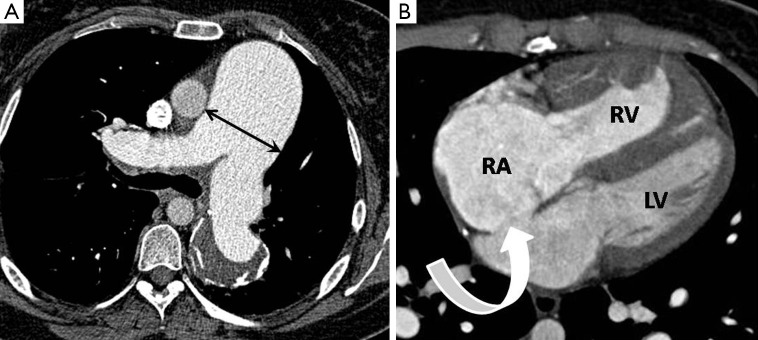
Generic CTPA signs suggesting the presence of PAH. (A) Axial image showing substantial enlargement of the MPA measured at its bifurcation (arrows); (B) axial image showing enlargement of the right heart chambers (RA >3.5 cm) with right ventricular hypertrophy (wall thickness >4 mm) which in this case is secondary to a large ostium secundum atrial septal defect (curved arrow). RA, right atrium; RV, right ventricle; LV, left ventricle; CTPA, computed tomography pulmonary angiography; PAH, pulmonary arterial hypertension.
A diameter of the MPA larger than that of the adjacent ascending thoracic aorta has also been suggested as a specific finding for moderate/severe PAH (12,13). In the absence of significant underlying structural lung disease, tortuous dilatation of the segmental pulmonary arteries (segmental artery: bronchus ratio >1.25) in at least three lobes has been reported in PAH and the combination of this sign and MPA dilatation has been estimated as 100% specific (12). MPA laminated in situ thrombus with foci of calcification may also be seen in cases of severe PAH regardless of aetiology (14).
Once the MPA calibre has been measured the pulmonary arterial branch vessels should be followed in a systematic manner down to the visible segmental and sub-segmental levels. The pulmonary arterial system is best scrutinised using appropriate window settings (width 700, level 100) which optimise the sensitivity for detection of filling defects.
CTED is an important underlying cause of PAH (WHO group 4), as it is potentially treatable with pulmonary thromboendarterectomy surgery. CTED is characterised by intraluminal thrombus organization with resultant partial or complete arterial obliteration. The precise mechanism by which this occurs is incompletely understood but acute pulmonary embolism, either as a single event or as recurrent episodes is thought to be the trigger and is followed by progressive pulmonary vascular remodelling with arteriolar medial hypertrophy and in situ thrombosis (15,16). The true incidence of CTED is unknown but recent studies estimate a prevalence of up to 4% in patients who survive an episode of acute pulmonary embolism (17,18).
Characteristic CTPA features of CTED are laminated arterial filling defects forming obtuse angles with the vessel wall (as opposed to acute angles seen with acute pulmonary embolism), pulmonary arterial bands and webs and post-stenotic arterial dilatation (19,20). Review of lung windows images typically show a “mosaic” pattern of attenuation with regions of low attenuation corresponding to hypoperfused segments. Such perfusion defects become even more conspicuous when imaged with an iodine perfusion map as provided by dual energy CTPA techniques (Figure 2).
Figure 2.

Chronic thromboembolic disease. (A) Axial image showing extensive laminated, partially calcified thrombus lining the pulmonary artery bifurcation (arrows); (B) coronal image showing hypertrophied bronchial collateral vessels (arrow); (C) axial lung windows image showing a mosaic pattern of lung attenuation with multiple regions of hypoperfusion (stars); (D) coronal iodine perfusion map overlay image showing multiple perfusion defects (stars).
Thoracic aorta
The thoracic aorta should be inspected for the presence of a patent ductus arteriosus (PDA) which manifests as a communication between the MPA and aorta, just distal to the origin of the left subclavian artery and causing left to right shunting. A PDA is suspected on CTPA in the presence of focal unopacified blood or turbulent contrast mixing within the MPA (Figure 3).
Figure 3.

Patent ductus arteriosus. (A) Coronal CT image showing a communication between the undersurface of the distal aortic arch and the left MPA branch (arrow); (B) sagittal CT image again showing the communication in the typical location for a patent ductus. CT, computed tomography; MPA, main pulmonary artery.
The thoracic aorta usually gives rise to several bronchial arteries which often originate from the undersurface of the arch. In normal individuals the bronchial arteries are small (<2 mm) and can be difficult to visualise. Certain disease states including CTED can cause bronchial artery hypervascularisation (tortuous vessels with diameters >3 mm) as a means of systemic arterial supply to bypass a high pulmonary vascular resistance or diseased pulmonary arterial circulation. Other causes of bronchial hypervascularisation include bronchiectasis and any cause of chronic hypoxaemia (21).
Pulmonary veins
Analysis of the pulmonary veins can yield important insights into the underlying cause of PAH. Normal calibre, smooth and straight pulmonary veins, in the setting of PAH suggest an arteriolar or microvascular aetiology whereas dilated and tortuous veins imply a post-capillary or left heart cause (22). The anatomy and drainage pathway of the pulmonary veins should routinely be assessed. Normally, four veins drain into the left atrium (LA); namely a set of right and left superior and inferior pulmonary veins. Variations in this arrangement are common, for example, a right middle accessory vein which separately drains the right middle lobe or a common isthmus of the left sided pulmonary veins.
Partial anomalous pulmonary venous drainage (PAPVD) is characterised by abnormal drainage of one or more veins into the right sided circulation and has an incidence of approximately 0.05% (23). Common drainage sites include the SVC, right atrium (RA) and coronary sinus (24). PAPVD of a single vessel is normally asymptomatic and of no clinical significance as the volume of left to right shunt is low (less than 1.5:1.0), however, multiple abnormally draining veins or associated ASD increases the shunt fraction and may result in PAH. Of particular note PAPVD of the right superior pulmonary vein to the SVC is associated with sinus venosus ASD in up to 50% of cases (Figure 4) (23,24).
Figure 4.

Sinus venosus atrial septal defect with anomalous pulmonary venous drainage. (A) Axial CT image showing an interatrial communication within the superior aspect of the septum (arrow); (B) coronal CT image showing anomalous drainage of the right superior pulmonary vein into the distal superior vena cava in keeping with a left to right shunt (arrow). LA, left atrium; RA, right atrium; RV, right ventricle; CT, computed tomography.
Right heart
As a response to prolonged PAH, the right ventricle (RV) hypertrophies and dilates, eventually leading to a decline in RV systolic function and decomposition (11,25). Cardiac MRI is the gold standard in the assessment of RV volumes and function but CTPA can often identify adaptive changes secondary to PAH by observation of key morphological features. These include right atrial and ventricular enlargement (RV:LV diameter >1 at the level of the mid ventricle), RV hypertrophy (RV: free wall >4 mm) and reflux of intravenous contrast from the RA into the IVC and hepatic veins (26,27). Right atrial enlargement is also a frequent but non-specific finding (anterior-posterior diameter >3.5 cm) in patients with PAH.
Cardiac septum
In normal individuals, the interventricular septum should demonstrate a mild convex bowing into the RV cavity (28). Flattening or reverse bowing of the septum towards the left side may be seen in PAH of any cause (although this may be present in any cause of RV volume or pressure overload). ECG gated CTPA allows dynamic assessment of ventricular contraction on cine review and may show paradoxical movement of the interventricular septum (“septal bounce”) towards the LV during systole (29).
The cardiac septum should be inspected for septal defects, with resulting left to right shunting which may cause PAH if left untreated (WHO group 1). Ventricular septal defects (VSD) may be directly visualised on ECG gated CTPA studies and should be described by their location and size; muscular, perimembranous and supracristal in location (Figure 5). Signs of VSD may include a focal contrast jet passing into, or an abnormally high concentration of contrast within, the RV.
Figure 5.
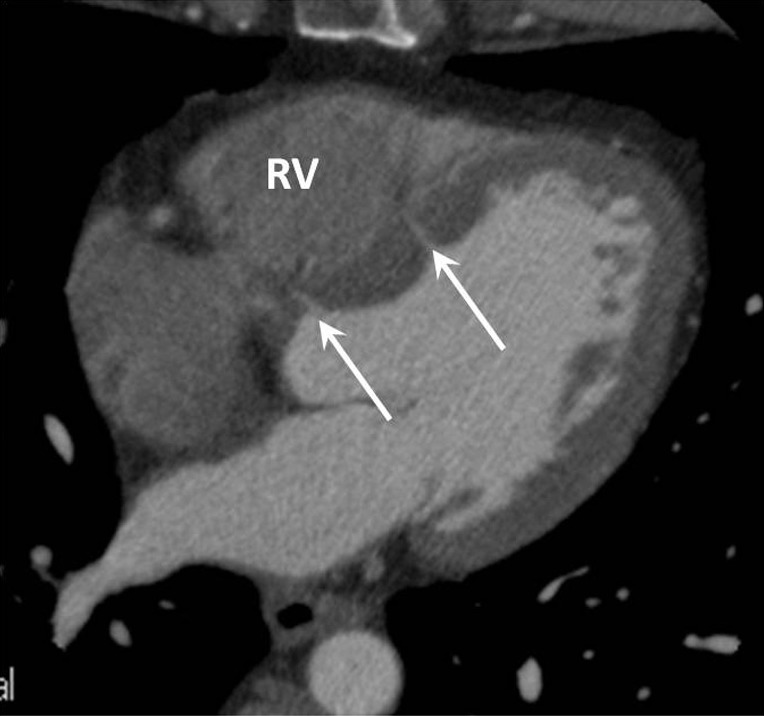
Axial CT image in a patient with mild pulmonary hypertension who had incidentally detected small membranous (adjacent to the aortic root) and muscular (adjacent to the body of the left ventricle) ventricular septal defects (arrows). RV, right ventricle; CT, computed tomography.
ASDs are classified according to their location and size; ostium secundum (most common, occurring in the region of the fossa ovalis), sinus venosus type (communication between the SVC or IVC and LA), ostium primum and coronary sinus type ASD. Sinus venous ASD warrants particular mention as it has a strong association with PAH and is a particularly challenging diagnosis to make with TTE as the defect lies outside conventional scan planes (30). As mentioned previously there is a frequent association with PAPVR of the right upper lobe into the SVC (80-90% of cases) which further enhances the volume of left to right shunting. Recently MDCT has been shown as a reliable technique for establishing the diagnosis and defining pulmonary venous anatomy which helps with planning of surgical correction (Figure 4).
Left heart
TTE remains the diagnostic standard in the assessment of cardiac function and morphology, however, inspection of the left ventricle on CTPA may provide a clue to an underlying cause of left ventricular dysfunction, which is important cause of PAH (WHO group 2). Left ventricular dysfunction may be caused by a wide range of pathologies. Aortic or mitral valvular thickening and calcification can indicate valvular stenosis; studies have shown that the degree of calcification of both the aortic and mitral valves correlates with the degree of valvular stenosis identified at TTE (31,32). Left ventricular dilatation can occur secondary to coronary artery ischemia, mitral regurgitation and dilated cardiomyopathy. Left ventricular hypertrophy may be seen secondary to hypertension, aortic stenosis and cardiomyopathic infiltration.
Left atrial enlargement is a common finding in patients with mitral stenosis and may also be seen in PAH secondary to any cause of chronically elevated left ventricular filling pressures (Figure 6). An antero-posterior left atrial diameter >4.5 cm is a useful guide when assessing for enlargement on axial CT images.
Figure 6.
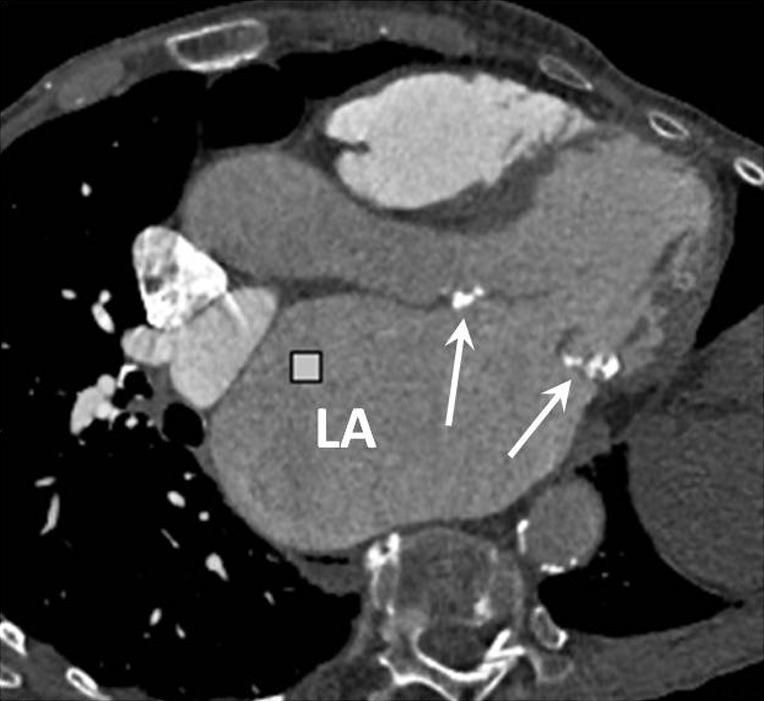
Axial oblique CT image in an elderly patient with mitral stenosis secondary to rheumatic fever. The anterior and posterior mitral valve leaflets are thickened and calcified (arrows) and there is massive enlargement of the left atrium. LA, left atrium; CT, computed tomography.
Pericardium
Pericardial effusions may be seen in PAH of any underlying aetiology. Thickening of the pericardium (>4 mm) or pericardial calcification, in the presence of left ventricular diastolic dysfunction, may indicate constrictive pericarditis. Pericardial calcification in isolation is however non-specific and is most commonly the result of previous pericarditis.
Lung parenchyma
Analysis of the lung parenchyma on lung window settings (width 1,500, level -500) will demonstrate the presence of any underlying parenchymal lung disease (WHO group 3), such as emphysema, interstitial lung disease, sarcoidosis or Langerhans cell histiocytosis (33).
As mentioned previously a mosaic pattern of lung attenuation is an important observation in the setting of PAH. It is defined as a patchwork of regions of differing attenuation within the lungs and raises the possibility of CTED but can also be seen in a variety of other conditions including hypersensitivity pneumonitis and air trapping due to asthma or other forms of small airways disease (34). These entities can be differentiated on CT by correlating inspiratory with expiratory images and by evaluating the appearance of the pulmonary vasculature.
Patchy ill-defined centrilobular ground glass nodularity is a feature associated with several causes of PAH including idiopathic PAH (formerly known as primary pulmonary hypertension) in which histological studies have shown the nodules to reflect cholesterol granulomas at the site of obliterative small vessel changes (Figure 7). Such nodularity can also been seen in cases of long standing left to right heart shunts as well as a variety of interstitial lung diseases including subacute hypersensitivity pneumonitis and smoking related respiratory bronchiolitis. Centrilobular ground glass nodularity in association with PAH also raises the possibility of pulmonary capillary haemangiomatosis (PCH), a rare vascular proliferative condition characterised by multiple angiomatous capillary lesions (Figure 8). PCH carries a very poor prognosis without curative lung or heart-lung transplantation and conventional vasodilator therapy (which is widely used to treat other forms of PAH) should be avoided due to a risk of inducing fatal pulmonary oedema. Pulmonary venocclusive disease (PVOD), which is characterised by extensive fibrotic small venous occlusions (WHO group 1 diseases) can also manifest as diffuse ill-defined centrilobular ground glass nodularity. Key CT discrimators favouring PVOD over PCH and idiopathic PAH are the presence of widespread smoothly thickened interlobular septa and pleural effusions (35).
Figure 7.
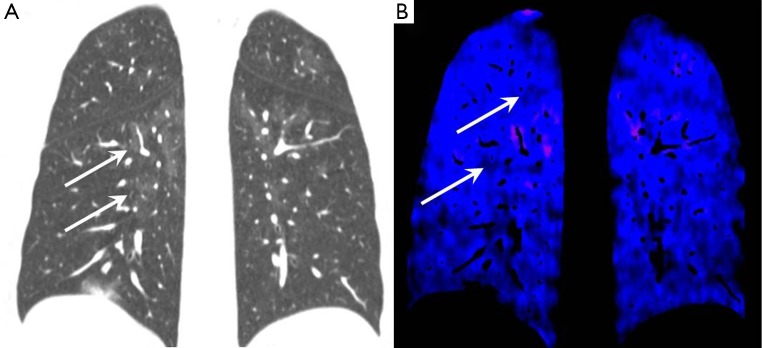
Idiopathic pulmonary arterial hypertension. (A) Coronal lung windows CT image showing multiple subtle ill-defined ground glass opacities throughout both lungs (arrows); (B) coronal dual energy iodine perfusion map showing regions of hypoperfusion corresponding to the nodular areas (arrows). CT, computed tomography.
Figure 8.

Range of parenchymal lung findings on CTPA associated with pulmonary hypertension. (A) Mosaic attenuation pattern may be associated with chronic thromboembolic disease; (B) diffuse fine nodularity with right upper lobe consolidation in sarcoidosis; (C) nodularity and septal thickening in a case of pulmonary capillary haemangiomatosis; (D) basal subpleural honeycomb change in a case of usual interstitial pneumonia interstitial lung disease. CTPA, computed tomography pulmonary angiography.
Sarcoidosis most often appears as upper lobe predominant peri-lymphatic distribution fine nodularity, typically along the fissures, bronchi and in the subpleural regions (Figure 8). There may also be also more confluent “mass like” lesions and frequently there will be enlarged hilar and mediastinal lymphadenopathy, often containing fine “egg-shell” like calcifications. The most common forms of diffuse interstitial lung disease associated with PAH are usual interstitial pneumonia (UIP) which classically has a lower lobe predominance with subpleural honeycomb formation and non-specific interstitial pneumonia (NSIP) which manifests as diffuse regions of ground glass infiltration and scattered subpleural reticular changes (Figure 8).
Conclusions
CTPA is established as a core imaging investigation in the work-up of patients with known or suspected PAH. PAH may also initially be suspected based on specific CTPA features. CTPA enables a comprehensive assessment of both the lung parenchyma and pulmonary vasculature and the development of ECG-gating has enabled improved assessment of cardiac morphology including depiction of ASDs and PAPVR. Radiologists should be familiar with the role of CTPA in this setting and have a systematic approach to interpretation of these examinations in order to streamline the patient pathway.
Acknowledgments
Disclosure: The authors declare no conflict of interest.
References
- 1.Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez-Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G, ESC Committee for Practice Guidelines (CPG) . Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493-537. [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62:D34-41. [DOI] [PubMed] [Google Scholar]
- 3.Hemnes AR, Forfia PR, Champion HC. Assessment of pulmonary vasculature and right heart by invasive haemodynamics and echocardiography. Int J Clin Pract Suppl 2009;(162):4-19. [DOI] [PubMed] [Google Scholar]
- 4.Chaouat A, Naeije R, Weitzenblum E.Pulmonary hypertension in COPD. Eur Respir J 2008;32:1371-85. [DOI] [PubMed] [Google Scholar]
- 5.Girgis RE, Mathai SC. Pulmonary hypertension associated with chronic respiratory disease. Clin Chest Med 2007;28:219-32, x. [DOI] [PubMed] [Google Scholar]
- 6.Polomis D, Runo JR, Meyer KC. Pulmonary hypertension in interstitial lung disease. Curr Opin Pulm Med 2008;14:462-9. [DOI] [PubMed] [Google Scholar]
- 7.Coursey CA, Nelson RC, Boll DT, Paulson EK, Ho LM, Neville AM, Marin D, Gupta RT, Schindera ST. Dual-energy multidetector CT: how does it work, what can it tell us, and when can we use it in abdominopelvic imaging? Radiographics 2010;30:1037-55. [DOI] [PubMed] [Google Scholar]
- 8.Hoey ET, Mirsadraee S, Pepke-Zaba J, Jenkins DP, Gopalan D, Screaton NJ. Dual-energy CT angiography for assessment of regional pulmonary perfusion in patients with chronic thromboembolic pulmonary hypertension: initial experience. AJR Am J Roentgenol 2011;196:524-32. [DOI] [PubMed] [Google Scholar]
- 9.Meinel FG, Graef A, Thierfelder KM, Armbruster M, Schild C, Neurohr C, Reiser MF, Johnson TR. Automated quantification of pulmonary perfused blood volume by dual-energy CTPA in chronic thromboembolic pulmonary hypertension. Rofo 2014;186:151-6. [DOI] [PubMed] [Google Scholar]
- 10.Frazier AA, Galvin JR, Franks TJ, Rosado-De-Christenson ML. From the archives of the AFIP: pulmonary vasculature: hypertension and infarction. Radiographics 2000;20:491-524; quiz 530-1, 532. [DOI] [PubMed]
- 11.Peña E, Dennie C, Veinot J, Muñiz SH. Pulmonary hypertension: how the radiologist can help. Radiographics 2012;32:9-32. [DOI] [PubMed] [Google Scholar]
- 12.Tan RT, Kuzo R, Goodman LR, Siegel R, Haasler GB, Presberg KW. Utility of CT scan evaluation for predicting pulmonary hypertension in patients with parenchymal lung disease. Medical College of Wisconsin Lung Transplant Group. Chest 1998;113:1250-6. [DOI] [PubMed] [Google Scholar]
- 13.Alhamad EH, Al-Boukai AA, Al-Kassimi FA, Alfaleh HF, Alshamiri MQ, Alzeer AH, Al-Otair HA, Ibrahim GF, Shaik SA. Prediction of pulmonary hypertension in patients with or without interstitial lung disease: reliability of CT findings. Radiology 2011;260:875-83. [DOI] [PubMed] [Google Scholar]
- 14.Frazier AA, Galvin JR, Franks TJ, Rosado-De-Christenson ML. From the archives of the AFIP: pulmonary vasculature: hypertension and infarction. Radiographics 2000;20:491-524; quiz 530-1, 532. [DOI] [PubMed]
- 15.Auger WR, Kim NH, Kerr KM, Test VJ, Fedullo PF. Chronic thromboembolic pulmonary hypertension. Clin Chest Med 2007;28:255-69. [DOI] [PubMed] [Google Scholar]
- 16.Lang IM. Chronic thromboembolic pulmonary hypertension--not so rare after all. N Engl J Med 2004;350:2236-8. [DOI] [PubMed] [Google Scholar]
- 17.Pengo V, Lensing AW, Prins MH, Marchiori A, Davidson BL, Tiozzo F, Albanese P, Biasiolo A, Pegoraro C, Iliceto S, Prandoni P, Thromboembolic Pulmonary Hypertension Study Group . Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004;350:2257-64. [DOI] [PubMed] [Google Scholar]
- 18.Tapson VF, Humbert M. Incidence and prevalence of chronic thromboembolic pulmonary hypertension: from acute to chronic pulmonary embolism. Proc Am Thorac Soc 2006;3:564-7. [DOI] [PubMed] [Google Scholar]
- 19.Auger WR, Fedullo PF, Moser KM, Buchbinder M, Peterson KL. Chronic major-vessel thromboembolic pulmonary artery obstruction: appearance at angiography. Radiology 1992;182:393-8. [DOI] [PubMed] [Google Scholar]
- 20.Bergin CJ, Sirlin CB, Hauschildt JP, Huynh TV, Auger WR, Fedullo PF, Kapelanski DP. Chronic thromboembolism: diagnosis with helical CT and MR imaging with angiographic and surgical correlation. Radiology 1997;204:695-702. [DOI] [PubMed] [Google Scholar]
- 21.Castañer E, Gallardo X, Ballesteros E, Andreu M, Pallardó Y, Mata JM, Riera L. CT diagnosis of chronic pulmonary thromboembolism. Radiographics 2009;29:31-50; discussion 50-3. [DOI] [PubMed] [Google Scholar]
- 22.Tsai IC, Tsai WL, Wang KY, Chen MC, Liang KW, Tsai HY, Liao WC. Comprehensive MDCT evaluation of patients with pulmonary hypertension: diagnosing underlying causes with the updated Dana Point 2008 classification. AJR Am J Roentgenol 2011;197:W471-81. [DOI] [PubMed] [Google Scholar]
- 23.Gustafson RA, Warden HE, Murray GF, Hill RC, Rozar GE. Partial anomalous pulmonary venous connection to the right side of the heart. J Thorac Cardiovasc Surg 1989;98:861-8. [PubMed] [Google Scholar]
- 24.Demos TC, Posniak HV, Pierce KL, Olson MC, Muscato M. Venous anomalies of the thorax. AJR Am J Roentgenol 2004;182:1139-50. [DOI] [PubMed] [Google Scholar]
- 25.D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991;115:343-9. [DOI] [PubMed] [Google Scholar]
- 26.Barbosa EJ, Jr, Gupta NK, Torigian DA, Gefter WB. Current role of imaging in the diagnosis and management of pulmonary hypertension. AJR Am J Roentgenol 2012;198:1320-31. [DOI] [PubMed] [Google Scholar]
- 27.Resten A, Maître S, Humbert M, Sitbon O, Capron F, Simoneau G, Musset D.Pulmonary arterial hypertension: thin-section CT predictors of epoprostenol therapy failure. Radiology 2002;222:782-8. [DOI] [PubMed] [Google Scholar]
- 28.Bruzzi JF, Rémy-Jardin M, Delhaye D, Teisseire A, Khalil C, Rémy J. When, why, and how to examine the heart during thoracic CT: Part 1, basic principles. AJR Am J Roentgenol 2006;186:324-32. [DOI] [PubMed] [Google Scholar]
- 29.Hoey ET, Gopalan D, Agrawal SK, Screaton NJ. Cardiac causes of pulmonary arterial hypertension: assessment with multidetector CT. Eur Radiol 2009;19:2557-68. [DOI] [PubMed] [Google Scholar]
- 30.Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation 2006;114:1645-53. [DOI] [PubMed] [Google Scholar]
- 31.Koos R, Mahnken AH, Sinha AM, Wildberger JE, Hoffmann R, Kühl HP. Aortic valve calcification as a marker for aortic stenosis severity: assessment on 16-MDCT. AJR Am J Roentgenol 2004;183:1813-8. [DOI] [PubMed] [Google Scholar]
- 32.Mahnken AH, Mühlenbruch G, Das M, Wildberger JE, Kühl HP, Günther RW, Kelm M, Koos R. MDCT detection of mitral valve calcification: prevalence and clinical relevance compared with echocardiography. AJR Am J Roentgenol 2007;188:1264-9. [DOI] [PubMed] [Google Scholar]
- 33.Ryu JH, Krowka MJ, Pellikka PA, Swanson KL, McGoon MD. Pulmonary hypertension in patients with interstitial lung diseases. Mayo Clin Proc 2007;82:342-50. [DOI] [PubMed] [Google Scholar]
- 34.Ridge CA, Bankier AA, Eisenberg RL. Mosaic attenuation. AJR Am J Roentgenol 2011;197:W970-7. [DOI] [PubMed] [Google Scholar]
- 35.Grosse C, Grosse A.CT findings in diseases associated with pulmonary hypertension: a current review. Radiographics 2010;30:1753-77. [DOI] [PubMed] [Google Scholar]