

Abstract
This work describes several synthetic approaches to append organic functional groups to gold and silver N-heterocyclic carbene (NHC) complexes suitable for applications in biomolecule conjugation. Carboxylate appended NHC ligands (3) lead to unstable AuI complexes that convert into bis-NHC species (4). A benzyl protected carboxylate NHC-AuI complex 2 was synthesized but deprotection to produce the carboxylic acid functionality could not be achieved. A small library of new alkyne functionalized NHC proligands were synthesized and used for subsequent silver and gold metalation reactions. The alkyne appended NHC gold complex 13 readily react with benzyl azide in a copper catalyzed azide-alkyne cycloaddition reaction to form the triazole appended NHC gold complex 14. Cell cytotoxicity studies were performed on DLD-1 (colorectal adenocarcinoma), Hep-G2 (hepatocellular carcinoma), MCF-7 (breast adenocarcinoma), CCRF-CEM (human T-Cell leukemia), and HEK (human embryonic kidney). Complete spectroscopic characterization of the ligands and complexes was achieved using 1H and 13C NMR, gHMBC, ESI-MS, and combustion analysis.
Introduction
Through the substitution of ancillary ligands with varying electronic, steric, and redox-active groups, metal complexes provide a highly versatile platform for drug design that can yield a range of biological activities.1 Metallo-drugs have received considerable attention as potential anti-cancer agents. For instance, some metal complexes exhibit cytotoxicities fifteen times higher than that of cisplatin,2 and others display IC50 values (a measure of anti-proliferative efficacy) as low as 2 nM.3 Despite these benefits, metal-based anti-cancer agents still fall short in two ways: (1) complex instability in vivo and (2) lack of cell selectivity. As a result, the chemotherapeutic benefit of potent metallo-drugs is often overshadowed detrimental toxicities due to heavy metal accumulation in the body. Metal-based compounds are clearly very promising drug candidates if their stability and delivery are precisely controlled.4 This assertion has spurred a resurgence in the field of metallo-therapeutics, most noteworthy in this venture are metal N-heterocyclic carbene (NHC) complexes.2,5
Many contemporary metallo-drugs suffer from instability in vivo; demetalation and degradation result in the formation of metal aqua complexes that are difficult to excrete, causing an array of patient toxicity issues.6 However, metal-NHC complexes can provide a potential solution to this problem. Metal-NHCs form strong bonds between transition metals and the NHC carbene carbon. The robust two electron σ-donation of NHC ligands renders their metal complexes stable to air, water, acid, and heat.7,8,9 Recent reports indicate metal-NHC complexes incorporating Ag, Au, Pt, Pd, Ru, Ni, and Cu ions exhibit impressive anti-proliferative activity.5 Among the most successful of these complexes are silver, copper, and gold derivatives (Figure 1).2,10-11-13
Moreover, gold-NHC complexes have received particular attention due the gold ion's utility in anti-rheumatic and anti-viral therapies. 1,14,15,16,17 The impressive stability combined with remarkably high cytotoxicity of gold-NHC complexes warrant further investigation and development. However, like other metal-based anti-cancer agents, gold-NHC complexes still lack selectivity; they show high toxicity towards both off-target healthy cells and cancerous cells.18 While it is known that tuning ligand steric and electronic properties of metal-NHC complexes can modulate reactivity and selectivity,2,5,19 individual responses to specific drugs vary widely from one patient to the next. Therefore, one approach to reducing off-target activity is to conjugate metal-based drugs to a biomolecule that can act as a carrier.
Broadly defined, bio-conjugation is the covalent linkage of a natural or synthetic biomolecule (e.g., peptide, nucleic acid, metabolite), with another moiety, such as a small molecule, immobilized material or surface, or molecular complex. Modifying the biomolecule and the molecule/material of interest with complementary functional groups that react exclusively (bio-orthogonally) with each other is the fundamental technique employed in bio-conjugation.20 Presented in this work is the synthesis and characterization of NHC proligands bearing conjugation-ready functional groups, as well as their corresponding AgI and AuI complexes.
Results and Discussion
Ubiquitous in the field of bio-conjugation chemistry are amide bond linkages. A half-life of nearly 600 years in neutral solution at 25 °C21 coupled with relatively inert reactivity within the biochemical environment of the body, renders the amide bond the gold standard for bio-conjugation.20 Motivated by this fact, the initial target was a NHC featuring a pendant carboxylic acid group that could be condensed with an amine to form an amide bond in a subsequent bio-conjugation step. One approach involves protecting the carboxylic acid group as an ester during the NHC synthesis and metalation steps. For example, treating 1-mesityl-1H-imidazole with benzyl 2-chloroacetate provides 1-mesityl-3-(2-benzylacetyl) imidazolium chloride (1) in 80% yield as a colorless solid (Eq. 1).
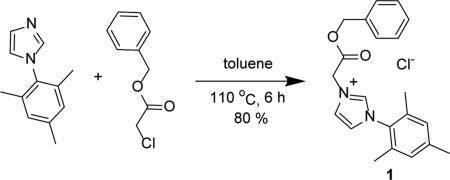 |
(1) |
Confirming its identity, the 1H NMR spectrum (CDCl3) of 1 exhibits a prototypical downfield resonance for the proton in the C2 (NCN) position of the imidazole at 10.41 ppm (see experimental section for atom labeling). Other diagnostic signals include singlets at 2.01 and 2.32 ppm attributable to the mesityl methyl groups. Two more singlets appear at 5.18 and 5.98 ppm for the benzyl and acetyl CH2 groups, respectively. A notable resonance in the 13C NMR spectrum is the carbonyl group that resonates downfield at 166.4 ppm.
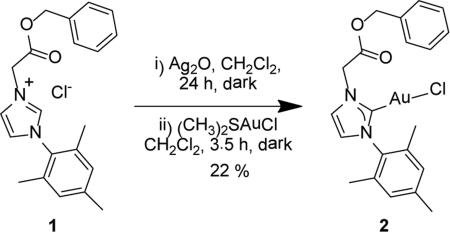 |
(2) |
Transmetalation of the ligand from silver(I) in situ provides the benzyl protected NHC-AuI complex 2 in 22.2 % yield (Eq. 2). Evidence for the identity of 2 comes from positive mode ESI-MS .analysis (calculated for [M-Cl]+ 531.1341; Found [M-Cl]+ 531.1339 m/z) and NMR spectroscopy. As in the proligand 1, the 1H NMR spectrum of 2 also exhibits four singlets for the mesityl methyls and CH2 protons at 1.99 (o-CH3), 2.32 (p-CH3), 5.14 (N-CH2), and 5.24 ppm (O-CH2). Importantly, the downfield resonance at 10.41 ppm for 1 is absent in the spectrum of 2. In the 13C{1H} NMR spectrum of 2, the carbene carbon dramatically shifts downfield from 141.3 ppm of 1 to 174.1 ppm. The carbonyl carbon remains in a similar position at 166.7 ppm suggesting the oxygen atoms of the ester group are not interacting with the AuI metal ion.
Although many NHC-AuI species are stable under aqueous, acidic, and elevated temperatures,7, 22-24 a successful ester deprotection from a NHC-AuI complex is unprecedented. Treating complex 2 for 24 h at room temperature under 1 atm of H2 over 10% Pd/C, followed by an aqueous work-up under aerobic conditions, failed to remove the benzyl group. Instead, a direct metalation with a carboxylic acid functionalized NHC was sought.
Treating zwitterion 3 with 1.1 eq of silver(I) oxide in deoxygenated water and allowing the reaction to stir for 24 h at 50 °C in the dark, followed by addition of 1.1 eqof NaCl generates the proposed bis-NHC-silver(I) complex Na[(NHC)2Ag] in situ.25 After filtering the reaction mixture through Celite®, and adding 1.2 eq of solid (CH3)2SAuICl, the solution turns black. After 24 h, the reaction mixture was passed through a pad of Celite® and fine fritted funnel. The filtrate was reduced to 5 mL whereupon the dark purple solution was again filtered to remove gold particles that form during the reaction. The remaining solvent was removed in vacuo to give complex 4 as an off-white residue in 30% yield (Eq. 3).
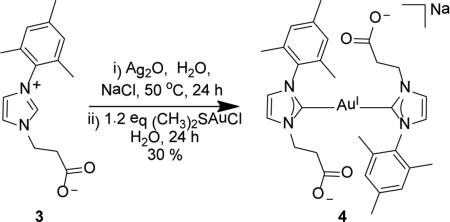 |
(3) |
A 1H NMR spectrum of complex 4 in DMSO-d6 exhibits resonances consistent with the structure depicted in equation 3. The 1H NMR resonances for the two NHC ligands are equivalent by symmetry. The ethyl fragment of the carboxylic acid arms appears as two triplets at 4.38 (t, J = 7.6 Hz, 4H) and 2.59 (t, J = 7.7, 4H). The mesityl methyls resonate at 2.41 and 1.67 ppm and the NHC ring protons are distinct and appear at 7.79 and 7.38 ppm. Most important to the characterization of 4 as a bis-NHC complex, the carbene carbon resonates at 182.8 ppm in the 13C{1H} NMR spectrum, which is diagnostic for trans-disposed NHCs bound to AuI. This contrasts with more upfield signals at approximately 165-175 ppm observed for mono-NHC AuI chloride complexes.26 Positive mode ESI-MS data also confirm the identity of 4 as the bis-NHCAuI complex. The reaction progress was monitored via 1H NMR spectroscopy. Spectra obtained at early reaction times (7 h) suggest the initial formation of a mono-NHC-AuI species that then binds a second NHC to produce a bis-NHC-AuI. Preliminary cell viability studies using CEM (T cell leukemia) and Ramos (Burkitt's lymphoma) as the trial cell lines to assess the pre-conjugation cytotoxicity of complex 4 and ligand 3 indicated negligible cell death in both cell lines, as determined by MTS assay (see supporting information). Thus no attempts were made to pursue conjugation strategies with 4.
Another bio-orthogonal conjugating method is the Huisgen cycloaddition of organo-azides and alkynes,27-29 or the versatile copper catalyzed version CuAAC.30-31 Azide-alkyne cycloaddition at the periphery of ligands bound to metal ions is known,32 as well a unique example where the azide and alkyne are directly attached to a metal ion.33-34 Also, NHC ligands featuring a pendant alkyne group can be functionalized via CuAAC prior to metalation.35-37 Considered one of the most useful reactions in bio-conjugation, the azide-alkyne click38 reaction finds use in a wide range of bioconjugation applications including labeling proteins, immobilizing peptides on a surface, and functionalizing DNA.20,39 Considering the ample precedent, an alternative NHC-metal conjugation strategy was sought that involves appending an alkyne group to an NHC metal complex.
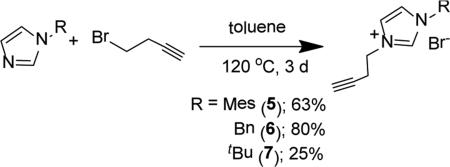 |
(4) |
Owing to the abundance of imidazolium salt derivatives in the literature, finding a synthetic strategy to achieve an alkyne appended NHC ligand proved facile. However, unlike previously reported ligands of this type,40 the target alkyne functionalized heterocycles 5-7 demanded more aggressive conditions to complete the reaction. Refluxing an imidazole with 4-bromobut-1-yne in toluene for 3 d provides the corresponding imidazolium salt (Eq. 4). Despite the high temperature and extended reaction time, the methodology permitted the assembly of a small library of new proligands 5-7. In fact, in the case of the mesityl and benzyl substituted heterocylces 5 and 6, this procedure led to moderate to high yields, 63 and 80%, respectively. The extremely hygroscopic character of the t-butyl substituted N-heterocycle 7 hampered product isolation, leading to a poor yield (<25%).
Spectroscopic characterization of compounds 5-7 by 1H and 13C{1H} NMR spectroscopy agrees with reported literature data of similar NHC proligands.41,42,43,44 The 1H NMR spectrum of each compound reveals a diagnostic downfield chemical shift (10.0-10.5 ppm), corresponding to the deshielded C2 proton flanked by two nitrogen atoms of the imidazolium. The terminal alkynyl proton of ligands 6 and 7 appear as triplets (JHH = 2.7-2.5 Hz) due to long range coupling with the methylene group distal to the heterocyclic ring and adjacent to the triple bond. This long range coupling is still present for compound 5 but is obscured in the 1H NMR spectrum due to signal overlap with a large singlet at 2.05 ppm attributable to a mesityl methyl group. 13C{1H} NMR spectra of ligands 5-7 exhibit C1 carbon resonances between 135.7 ppm and 137.9 ppm.
 |
(5) |
To demonstrate the viability of these ligands in stabilizing metal ions, the AgI complex of the mesityl derivative 5 was pursued. Treating 5 with 0.7 equiv of Ag2O in chloroform for 3 d provides the Ag1 complex 8-Mes in 65% yield as an off-white powder (Eq 5). Aliquots of the reaction mixture removed periodically throughout the duration of the reaction and subjected to 1H NMR spectroscopic analysis provides a method of monitoring the reaction progress. The proligand's C2 imidazolium proton, resonating at 10.15 ppm, decreases over time concomitant with the growth of a set of resonances in the aromatic and aliphatic regions corresponding to complex 8. Total conversion of 5 to the AgI species 8 requires stirring at room temperature for three days in the absence of light. Passing the reaction mixture through Celite®, concentrating the filtrate, and adding an excess of ether precipitates complex 8. De Fremont and co-workers43 report the preparation of a bulky dodecyl NHC-AgI species that required similar forcing conditions to achieve complete conversion. The 1H NMR data are consistent with the structure of 8. One interesting feature is the 13C{1H} NMR data suggests the AgI ion is labile in solution; the carbene carbon is not detectable in the range typical for Ag1 complexes (170 ppm to 190 ppm).43
The lability of the silver-carbene bond in solution is the fundamental feature that makes NHC-AgI complexes excellent transmetalating agents for carbene transfer reactions. 41, 45,46,47 Bond lability studies by several groups43,45,47 and correlation with efficacy in transmetalation reactions with NHC-AgI indicate that the more labile a silver-carbene bond is, the broader the 13C NMR signal of the carbene carbon. In the case of complex 8, the carbene carbon signal broadens significantly and is indistinguishable from the baseline. Gradient heteronuclear multiple bond coherence (gHMBC) experiments elucidate C-H connectivities within 8 and confirm the assignments. These data show clear long range coupling between the carbene carbon (182.5 ppm) of the imidazol-2-ylidene ring with the proximal methylene protons of the alkyne (4.38 ppm) and the C4 and C5 (C=C) of the imidazolium ring (7.36 and 6.92 ppm). Indirect detection of the carbene carbon at 182.5 ppm rather than >200 ppm precludes the possibility of a free carbene and supports the assignment of 8 as the NHC-Ag1 complex.41,43,45-46,47
Metalation of ligand 6 (R = benzyl) by the same procedure used for 5, leads to the new benzyl substituted Ag1 complex 9. Using proligand 7 led to incomplete product conversion (<40%), as determined by 1H NMR spectroscopy, even upon extended reaction times (>5 d). Complex 9 is cytotoxic to several cancer cell lines and only minimally selective compared to healthy kidney cells (see ESI): DLD-1 (colorectal adenocarcinoma; IC50 = 6.8 ± 0.8), Hep-G2 (hepatocellular carcinoma; IC50 = 6.9 ± 0.7), MCF-7 (breast adenocarcinoma; 17.1 ± 0.4), and HEK (human embryonic kidney; IC50 = 22.6 ± 0.9).
The lability of 8 and 9 raises concerns for future use in biological applications, thus the NHC-AuI derivative was sought. In a sealable NMR tube, combining complex 8 with 1.2 equiv of (CH3)2SAuICl in CDCl3 yields the NHC-AuI complex 10, quantitatively (Eq. 6).
 |
(6) |
1H and 13C{1H} NMR spectra obtained at variable time intervals indicate the initial transmetalation from silver(I) to gold(I) occurs with a distinct 13C NMR signal shift from ~182.5 to 172.0 ppm. These data support the immediate formation of a single species that does not undergo subsequent mono- to bis-NHC conversion. Several reports43-44 and a study by Huynh and co-workers48 addressing the spectroscopic trends of mono- versus bis-NHC complexes of gold(I) and gold(III), support the assignment of 10 as a mono-NHC-AuI species. Attempts to isolate and purify 10 result in the formation of very fine inseparable colloidal gold. However, it is reasonable to expect that complex 10 could be employed in subsequent conjugation reactions without the need for prior isolation.
Purification problems and apparent stability issues employing the imidazole based N-heterocycle prompted an attempt to stabilize the AuI species with a benzimidazole N-heterocycle. Using the identical procedure to eq 4, the 1-methyl-3-(prop-2-yn-1-yl)-1H-benzo[d]imidazol-3-ium bromide (11) was synthesized in 78% yield. Subsequent treatment of 11 with Ag2O in chloroform for 3 d according to Scheme 1 provides the NHC-AgI complex 12 in 42% yield. A 1H NMR spectrum of complex 12 reveals a triplet resonance at 2.11 ppm for the alkynyl proton, a singlet at 4.10 ppm for the methyl protons, and triplets at 4.62 and 2.85 ppm for the CH2 protons of the V-substituted alkyne. Importantly, a resonance at 190.3 ppm in the 13C{1H} NMR spectrum of 12 is attributable to the carbene carbon bound to AgI. Complex 12 was isolable but not as a pure species; instead, 12 can be generated in situ. Treating in situ prepared 12 with one equiv of (CH3)2SAuCl in chloroform for 1 h provides the NHC-AuI complex 13 in 45% yield (Eq. 8). Unlike 12, complex 13 is isolable, stable to disproportionation, and is amenable to solution phase characterization by 1H and 13{1H} NMR spectroscopy. More importantly, single crystals deposit from a concentrated dichloromethane:hexanes mixture (1:3) of 13. Figure 2 depicts the solid state structure of 13 and all metric parameters can be found in the electronic supporting information. Complex 13 crystallizes in the orthorhombic Pbca space group with one molecule as the asymmetric unit. Considering the fact that bromide ions are present in solution during the preparation of 13, it was important to learn that the solid state structure contains a Au-Cl rather than a Au-Br bond, which is consistent with combustion analysis results. Typical for two-coordinate Au(I) complexes the C1-Au1-Cl(1) bond angle is nearly 180° (176.44(16)°). The alkyne moiety in the solid state points above the plane of the rings, but in solution the bond freely rotates to give Cs symmetry, and therefore equivalent methylene protons on C8 {(4.69 ppm, t, 2H, 3J = 7.0 Hz)} and C9 {2.85 (dt, 2H, 3J = 7.0 Hz, 4J = 2.0 Hz)}.
Scheme 1.

In situ preparation of [NHC]AgBr (12) followed by transmetalation to prepare [NCN]AuBr (13).
Complex 13 (IC50 = 16.19) is cytotoxic to the human leukemia cell line CCRF-CEM, but the ligand 11 alone (IC50 = 981.65) is not. The low toxicity of the ligand compared to the complex reconfirms that the Au ion is the active agent.
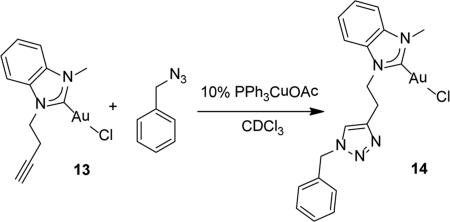 |
(7) |
Conjugation to the alkyne group on 13 is possible via copper catalyzed azide-alkyne cycloaddition. A test reaction with benzyl azide was conducted in a NMR tube and monitored via 1H NMR and IR spectroscopy. Complex 13, benzyl azide, 10% PPh3CuOAc, and CDCl3 were added to an NMR tube. After intervals of 10 min, 24 h, and 48 h, IR and 1H NMR spectra were obtained. The IR spectra indicate the absorption at 2100 cm−1 attributable to the azide stretching mode diminishes overtime and disappears after 48 h. In addition, new resonances attributable to the triazole-NHC-Au complex 14 appear in the 1H NMR spectra; most notable are the methylene resonances at 5.49 ppm, 4.77 ppm, and 3.62 ppm.
Conclusions
This report recounts efforts in developing carboxylic acid and alkyne functionalized NHC-metal complexes for biomolecule conjugation. The carboxylic acid functionalized mono NHC-AuI complex was not isolable but led to the reliable isolation of the water stable and water soluble Na[(NHC)2AuICl] complex 4. Although separation from gold particles generated in the reaction is a challenge, the bis-NHC-gold(I) complex 4, once isolated, remains stable in the solid state for months.
The pendant carboxylate group on the NHC proligand 3 imparts remarkable water solubility to complex 4, allowing the isolated bis-NHC-gold(I) product to fulfill two criteria of a viable biological agent: water stability and water solubility. Unfortunately, preliminary biological studies by MTS assay revealed no pronounced cytotoxicity of 4 towards the assayed CEM (T-cell leukemia) and Ramos (Burkitt's lymphoma) cell lines. Complex 9 is cytotoxic to both healthy and cancer cells, though it is not clear if the NHC-Ag complex remains intact, considering the lability of the Ag+ ion, as determined by 13C NMR spectroscopy.
Illustrated in the synthesis of 2 is an alternative synthetic route to a carboxylic acid functionalized gold-NHC complex. Installing a benzyl ester protecting group throughout the metalation and transmetalation steps results in the robust (NHC)AuICl species 2. However, performing the hydrogenolysis reaction revealed that complex 2 not only remains intact, but can also tolerate an aqueous and aerobic work-up. Though not the desired result, this offers further testament to the strength of the NHC-gold bond, which is an important feature for biological applications.
Four new alkyne functionalized imidazolium bromide salts 5-7, and 11, and their corresponding NHC-AgI and NHC-AuI derivatives, are described. The broadened carbene carbon resonance in the 13C{1H} NMR spectrum of 8 indicates the silver-carbene bond is labile, and proved to be an excellent transmetalation agent. The identity of the NHC-AuI complex 10 was unequivocally confirmed via one and two dimensional NMR studies. The instability of 10 towards decomposition precludes its use in cell cytotoxicity studies. However, the decomposition problem was solved by the synthesis of the stable benzimidazole gold(I) derivative 13. Finally, a conjugation test reaction between 13 and benzyl azide was successful. Conjugation to other molecules of interest and using the alkyne-functionalized ligands 5-7 and 11 with other metal ions are open areas for future investigation.
Experimental
General Considerations
Compounds 1-mesityl-1H-imidazole, 1-tert-butyl-1H-imidazole, and 3 were prepared according to previously reported procedures.25,49-50 Unless stated otherwise all syntheses and manipulations were performed under aerobic conditions. (CH3)2SAuICl and Ag2O were obtained from Sigma Aldrich and used without further purification.
All NMR spectra were collected on either a Varian Mercury Broad Band 300 MHz or Varian Inova 500 MHz spectrometer. 1H and 13C chemical shifts are reported in 5 (ppm) with the solvent peak referenced as an internal reference (CDCl3 δ = 7.26 ppm for 1H and 77.00 ppm for 13C, DMSO-d6 δ = 2.54 ppm for 1H and 39.54 ppm for 13C).
Electrospray Ionization Mass Spectrometry (ESI-MS) data for positive mode were obtained according to the following procedure. Depending on solubility, the sample was dissolved in methylene chloride, chloroform, or water and then directly injected into an auto-sampler. After injection, it was subjected to ESI with methanol as the mobile phase. The ions were detected with an Agilent 6210 TOF-MS instrument and the data were processed using MassHunterTMsoftware.
Synthesis of 1-mesityl-3-(2-benzylacetyl)imidazolium chloride (1)
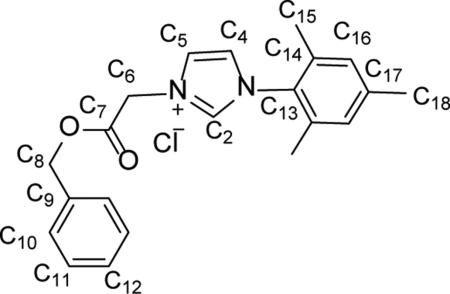
1-mesityl-3-(2-benzylacetyl) imidazolium chloride (1) was prepared by adding 1-mesityl-1H-imidazole (0.640 g, 3.44 mmol), a magnetic stir bar, and 4 mL of dry toluene to a 250 mL round bottom flask. The mixture was allowed to stir for 2 min followed by the dropwise addition of benzyl 2-chloroacetate (0.53 mL, 3.44 mmol). The system was placed under Ar and heated to 110 °C for 6 h while stirring and then stirred for an additional 12 h at room temperature. The solvent was removed in vacuo and the resulting colorless solid was triturated with ether (1.02 g, 79.9%). 1H NMR(500 MHz, CDCl3) δ = 10.41 (s, 1H, HC2), 7.94 (s, 1H, HC5), 7.34 (m, 5H, HC10, HC11, HC12), 7.08 (s, 1H, HC4), 6.95 (s, 2H, HC16), 5.98 (s, 2H, H2C8), 5.18 (s, 2H, H2C6), 2.32 (s, 3H, H3C18), 2.01 (s, 6H, H3C15). 13C NMR (126 MHz, CDCl3) δ = 166.4 (C7), 141.3 (C2), 139.7 (C13), 139.7 (C17), 134.3 (C14), 134.3 (C9), 130.6 (C16), 129.7 (C10), 128.7 (C12), 128.6 (C11), 124.4 (C4), 122.3 (C5), 68.4 (C8), 50.6 (C6), 21.1 (C18), 17.4 (C15). Anal. Calcd. for C21H23ClN2O2 (370.88 g/mol): C: 68.01%; H: 6.25%; N: 7.55%, Found; C: 67.99%; H: 6.43 N: 7.61%.
Synthesis of 1-mesityl-3-(2-benzylacetyl)imidazole-2-ylidene gold(I) chloride (2)
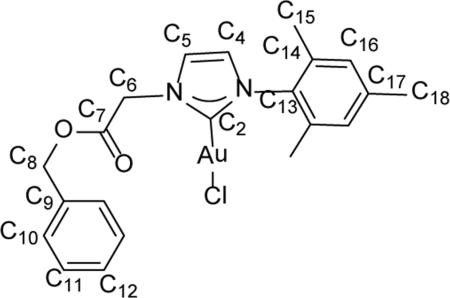
1-mesityl-3-(2-benzyleacetyl)imidazolium gold(I) chloride 2 was prepared via an in situ transmetalation from silver(I). Proligand 1 (0.201 g, 0.543 mmol) and Ag2O (0.150 g, 0.647 mmol) were suspended in 5 mL of dichloromethane and allowed to stir for 24 h in the absence of light. The resulting suspension was passed through a Celite® pad and fine frit filter directly into a stirring suspension of (CH3)2SAuICl (0.075 g, 0.255 mmol) in 5 mL of dicholoromethane. The reaction mixture was allowed to stir at room temperature for 3.5 h in darkness. The mixture was then passed through a Celite® pad and medium fritted funnel to yield an amber colored filtrate. The solvent was concentrated and then treated with an excess of pentanes to precipitate 2 as a fine off-white powder (0.032 g, 22.2% ). 1H NMR (500 MHz, CDCl3) δ = 7.38 (m, 5H, HC10, HC11, HC12), 7.23 (s, 1H, HC5), 6.95 (s, 2H, HC16), 6.92 (s, 1H, HC4), 5.24 (s, 2H, H2C8), 5.14 (s, 2H, H2C6), 2.33 (s, 3H, H3C18), 2.00 (s, 6H, H3C15). 13C NMR (126 MHz CDCl3) δ = 174.1 (C2), 166.7 (C7), 139.8 (C13), 139.8 (C17), 134.8 (C14), 134.4 (C9), 129.4 (C16), 128.8 (C10), 128.8 (C12), 128.7 (C11), 122.5 (C4), 121.7 (C5), 68.2 (C8), 51.9 (C6), 21.1 (C18), 17.7 (C15). ESI-MS (positive ion, calculated for M = C21H22AuClN2O2) Theor: 1155.1963 m/z [2M+Na]+, 1097.2377 m/z [2M-Cl]+, 589.0928 m/z [M+Na]+, 531.1341 m/z [M-Cl]+. Found: 1155.1945 m/z [2M+Na]+, 1097.2374 m/z [2M-Cl]+, 589.0926 m/z [M+Na]+, 531.1339 m/z [M-Cl]+.
Synthesis of sodium bis-(1-mesityl-3-(2-carboxylatoethyl)imidazol-2-ylidene)- gold(I) (4)
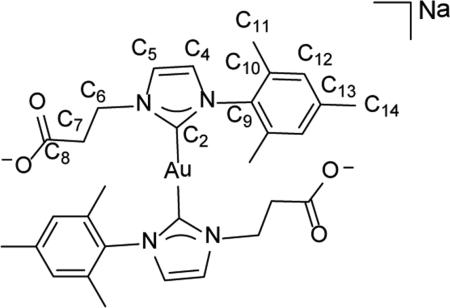
Bis(1-mesityl-3-(2-carboxylatoethyl)imidazol-2-ylidene) gold(I) sodium salt 4 was prepared via an in situ transmetalation reaction. To a 100 mL Schlenk flask under an atmosphere of Ar, 138.0 mg (0.534 mmol) of 3, 130.0 mg (0.561 mmol) of Ag2O, a magnetic stir bar, and 20 mL of deoxygenated water were added and left to stir for 24 h at 50 °C in darkness. After 24 h, the reaction mixture was allowed to cool to ambient temperature whereupon 32.0 mg (0.548 mmol) of NaCl was added and allowed to stir for an additional 30 min at room temperature. The solution was passed through a Celite® pad and fine fritted funnel. To the filtrate, 160.0 mg (0.543 mmol) of (CH3)2SAuICl was added and allowed to stir for 24 h at room temperature in the absence of light, under an atmosphere of Ar. After 24 h the reaction mixture was passed through a pad of Celite® and fine fritted funnel. The filtrate volume was reduced to 5 mL whereupon the dark purple solution was filtered to remove any gold particles. The remaining solvent was removed from the filtrate in vacuo to give 4 as an off-white residue (62.8 mg, 30.1%). 1H NMR (500 MHz, DMSO-d6) δ = 7.79 (d, 2H, J = 1.9 Hz, HC5), 7.38 (d, 2H, J = 1.9 Hz, HC4), 6.95 (s, 4H, HC12), 4.38 (t, J = 7.6 Hz, 4H, H2C6), 2.59 (t, J = 7.7, 4H, H2C7), 2.41 (s, 6H, HC14), 167 (s, 12H, H3C11). 13C NMR (126 MHz, DMSO-d6) δ = 182.8 (C2), 174.1 (C8), 138.2 (C13), 134.4 (C9), 134.1 (C10), 128.4 (C12), 122.5 (C5), 122.5 (C4), 48.2 (C6), 40.2 (C7), 20.6 (C14), 16.8 (C11). ESI-MS (positive ion, calculated for M = C30H34N4O4Au). Theor: 757.2036 m/z [M+2Na]+, 735.2216 m/z [M-H+Na]+, 713.2397 m/z [M+2H]+. Found: 757.2023 m/z [M+2Na]+, 735.2215 m/z [M-H+Na]+, 713. 2386 m/z [M+2H]+.
Synthesis of 3-(but-3-yn-1-yl)-1-mesityl-1H-imidazol-3-ium bromide (5)
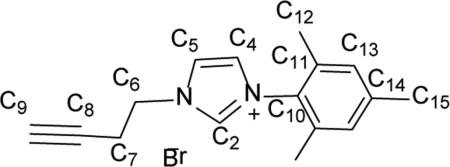
Compound 3-(but-3-yn-1-yl)-1-mesityl-1H-imidazol-3-ium bromide (5) was prepared by dissolving 1.11 g (5.95 mmol) of 1-mesityl-1H-imidazole in 10 mL of toluene in a 100 mL round bottom flask. To this stirring solution, 0.84 mL (8.92 mmol) of cold 4-bromobut-1-yne was added dropwise. The reaction mixture was allowed to reflux at 120 °C for 3 d, during which time an off-white film formed on the inside of the reaction vessel. Approximately 20 mL of diethyl ether was added directly to the reaction mixture and the contents were stirred vigorously for 3 h at room temperature. The suspension was filtered and the solid was washed with 3 × 10 mL of diethyl ether and dried under vacuum for 24 h to provide 5 as an off-white solid (0.70 g, 63.4%). gHMBC was used to determine C-H connectivities and confirm peak assignments. 1H NMR (500 MHz, CDCl3) δ = 10.15 (s, 1H, HC2), 8.23 (s, 1H, HC5), 7.17 (s, 1H, HC4), 6.98 (s, 2H, HC13), 4.89 (t, J = 6.0 Hz, 2H, HC6), 2.99 (td, J = 6.3, 2.5 Hz, 2H, HC7), 2.31 (s, 3H, HC15), 2.06 (t, J = 3.4, HC9), 2.05 (s, 6H, HC12).13C NMR (126 MHz, CDCl3) 5 141.3 (C14), 137.9 (C2), 134.2 (C9), 130.6 (C10), 129.8 (C13), 123.9 (C5), 122.7 (C4), 79.1 (C8), 72.5 (C9), 48.6 (C6), 21.1 (C7), 21.1 (C15), 17.6 (C12). Anal. Calcd. for C16H19BrN2 (319.25 g/mol): C: 60.20%; H: 6.00%; N: 8.78%, Found; C: 60.03%; H: 6.01 N: 8.86%.
Synthesis of 1-benzyl-3-(but-3-yn-1-yl)-1H-imidazol-3-ium bromide (6)
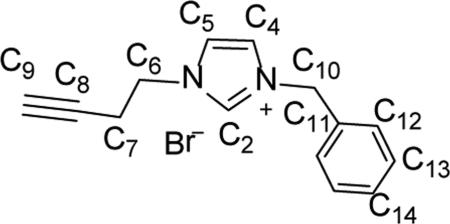
Compound 1-benzyl-3-(but-3-yn-1-yl)-1H-irmdazol-3-ium bromide (6) was prepared by a similar synthetic procedure as 5 using 1.00 g (6.32 mmol) of 1-benzyl-1H-imidazole, 0.71 mL (7.59 mmol) of 4-bromobut-1-yne, and 15 mL of toluene. Compound 6 was isolated as an off-white powder (1.48 g, 80.2%). gHMBC was used to determine C-H connectivities and confirm peak assignments. 1H NMR (500 MHz, CDCl3) δ = 10.45 (t, J = 1.5 Hz, 1H, HC2), 7.75 (t, J = 1.7 Hz, 1H, HC5), 7.45 (m, 2H, HC12), 7.42 (t, J = 1.5 Hz, 1H, HC4), 7.34 (m, 3H, HC13, HC14), 5.56 (s, 2H, H2C10) 4.51 (t, J = 6.4 Hz, 2H, H2C6), 2.84 (td, J = 6.1 Hz, 2.5 Hz, 2H, H2C7), 2.08 (t, J = 2.6 Hz, 1H, HC9). 13C NMR (126 MHz, CDCl3) δ = 137.1 (C2), 132.8 (C11), 129.5 (C14), 129.4 (C13), 128.9 (C12), 122.9 (C5), 121.6 (C4), 78.7 (C8), 72.9 (C9), 53.4 (C10), 48.4 (C6), 20.8 (C7). ESI-MS (positive ion, calculated for M = C14H15BrN2). 327.0070 m/z [M+Cl]+, 291.0484 m/z [M+H]+, 211.1238 m/z [M+Br]+. Anal. Calcd. for C14H15BrN2 (291.19 g/mol): C: 57.75%; H: 5.19%; N: 9.62%, Found; C: 57.67%; H: 5.13 N: 9.57%.
Synthesis of 3-(but-3-yn-1-yl)-1-(tert-butyl)-1.H-iinidazol-3-ium bromide (7)
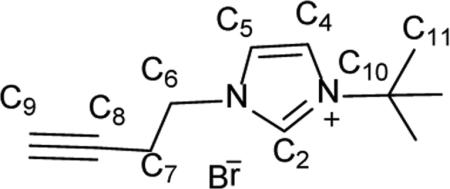
Compound 3-(but-3-yn-1 -yl)-1 -(tert-butyl)-1H-imidazol-3-ium bromide (7) was prepared by a similar synthetic procedure as 5 but with a slightly modified work-up. The synthesis was performed using 1.11 mL (8.05 mmol) of 1-(tert-butyl)-1H-imidazole, 1.13 mL (12.1 mmol) of 4-bromobut-1-yne, and 10 mL of toluene. After the reaction was complete, the mixture was allowed to cool to room temperature and 20 mL of diethyl ether was added directly to the reaction vessel. The mixture was stirred for 10 min and the supernatant was decanted. This process was repeated three more times using 10 mL of fresh diethyl ether each time. After the final decanting, the solid was suspended in 2 mL of diethyl ether and transferred directly to a vial. The remaining solvent was removed in vacuo and the resulting off-white, very hygroscopic solid 7 was dried under vacuum for 24 h before transferring to an Ar filled glovebox for storage (0.453 g, 21.9%). 1H NMR (500 MHz, CDCl3) δ = 10.47 (s, 1H, HC2), 7.75 (s, 1H, HC5), 7.55 (s, 1H, HC4), 4.62 (t, J = 6.2 Hz, 2H, H2C6), 2.88 (td, J = 6.4, 1.7 Hz, 2H, H2C7), 2.07 (t, J = 1.9 Hz,1H, HC9), 1.68 (s, 9H, H3C11). 13C NMR (126 MHz, CDCl3) δ = 135.7 (C2), 122.9 (C5), 119.2 (C4), 79.1 (C8), 72.4 (C9), 60.4 (C10), 48.0 (C6), 30.0 (C11), 20.8 (C7). Anal. Calcd. for C11H17BrN2 (257.17 g/mol): C: 51.37%; H: 6.66%; N: 10.89%, Found; C: 51.03%; H: 6.88 N: 11.11%.
Synthesis of 1-(but-3-yn-1-yl)-3-mesityl-1H-imidazol-2(3H)-ylidene)silver(I) bromide (8)
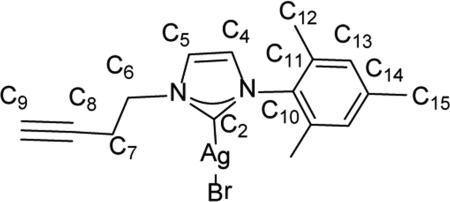
Inside an Ar filled glovebox, 303.0 mg (0.949 mmol) of compound 5 was added to a vial and dissolved in 4 mL of chloroform. Silver(I) oxide (151.1 mg, 0.652 mmol) was added directly as a solid to the stirring solution of 5. The reaction vessel was wrapped in aluminum foil and allowed to stir for 3 d at room temperature. The reaction mixture was passed through a pad of Celite® atop a filter paper fitted glass pipette to remove any insoluble gray particulates. The amber colored filtrate was reduced in vacuo to 1 mL whereupon an excess of diethyl ether (~5 mL) was added to precipitate an off-white solid. The supernatant was decanted, the solid was suspended in 2 mL of diethyl ether, and the supernatant was decanted again (this was repeated 2 more times with 2 × 2 mL of diethyl ether). The solid was dried completely under vacuum to give an 8 as an off-white solid (264.5 mg, 65.4%). Heteronuclear multiple bond coherence (gHMBC) was applied to determine C-H connectivities and confirm peak assignments. 1H NMR (500 MHz, CDCl3) δ = 7.36 (d, 1H, J = 1.6 Hz, HC5), 6.94 (s, 2H, HC13), 6.92 (d, 1H, J = 1.6 Hz, HC4), 4.38 (t, J = 6.2 Hz, 2H, HC6), 2.76 (td, J = 6.7, 2.4 Hz, 2H, HC7), 2.33 (s, 3H, HC15), 2.04 (t, J = 2.9, HC9), 1.94 (s, 6H, HC12).13C NMR (126 MHz, CDCl3) 5 182.5 (C2), 139.2 (C14), 135.1 (C10), 134.6 (C11), 129.1 (C13), 122.3 (C4), 121.2 (C5), 79.2 (C8), 71.8 (C9), 49.9 (C6), 21.6 (C7), 20.8 (C15), 17.5 (C12). Anal. Calcd. for C16H18AgBrN2 (426.11 g/mol): C: 45.10%; H: 4.26%; N: 6.57%, Found; C: 45.26%; H: 4.13 N: 6.89%.
Synthesis of 1-(but-3-yn-1-yl)-3-benzyl-1H-imidazol-2(3H)-ylidene)silver(I) bromide (9)
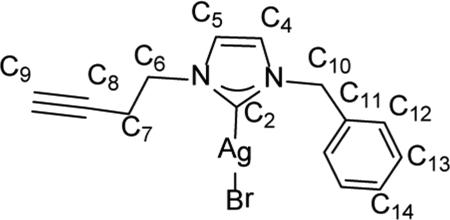
Compound 9 was prepared in a similar procedure to 8 using 300 mg (1.03 mmol) of 6, 167.1 mg (0.72 mmol) of Ag2O, and 4 mL of chloroform to yield an off-white powder (82.3 mg, 40.1%). 1H NMR (500 MHz, CDCl3) δ = 7.34 (m, 3H, HC13, HC14), 7.23 70 (m, 2H, HC12), 7.14 (d, J = 1.8 Hz, 1H, HC5), 6.90 (d, J = 1.7 Hz, 1H, HC4), 5.32 (s, 2H, H2C10), 4.32 (t, J = 6.4 Hz, 2H, H2C6), 2.71 (dt, J = 6.3, 2.4 Hz, 2H, H2C7), 2.07 (t, J = 2.6 Hz, 1H, HC9). 13C NMR (126 MHz, CDCl3) δ = 181.9 (C2), 135.6 (C11), 129.1 (C14), 128.6 (C13), 127.8 (C12), 121.9 (C5), 120.9 (C4), 80.1 (C8), 75 72.1 (C9), 55.8 (C10), 50.3 (C6), 21.8 (C7).
In situ preparation of 1-(but-3-yn-1-yl)-3-mesityl-1H-imidazol-2(3H)-ylidene)gold(I) bromide (10)
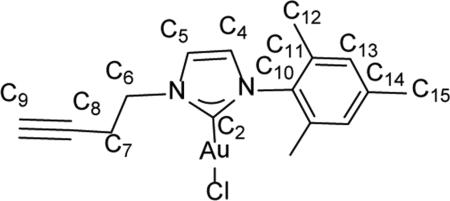
Inside an Ar filled glovebox, 1-(but-3-yn-1-yl)-3-mesityl-1H-imidazol-2(3H)-ylidene)gold(I) bromide (10) was prepared directly in a sealable NMR tube by dissolving 30 mg (0.0704 mmol) of complex 8 in 0.5 mL of CDCl3 and then adding 15.9 mg (0.054 mmol) of solid (CH3)2SAuCl directly to the solution. The tube was capped and inverted several times to thoroughly mix the reagents. The progress of the reaction was monitored by 1H and 13C NMR spectroscopy. gHMBC was used to determine C-H connectivities and confirm peak assignments. 1H NMR (500 MHz, CDCl3) δ = 7.31 (d, 1H, J = 1.9 Hz, HC5), 6.94 (d, 1H, J = 1.9 Hz, HC4), 6.87 (s, 2H, HC13), 4.41 (t, J = 6.4 Hz, 2H, HC6), 2.85 (td, J = 6.4, 2.6 Hz, 2H, HC7), 2.31 (s, 3H, HC15), 2.07 (t, J = 2.5, HC9), 2.00 (s, 6H, HC12).13C NMR (126 MHz, CDCl3) 5 172.0 (C2), 139.7 (C14), 134.7 (C10), 134.7 (11), 129.4 (C13), 121.8 (C4), 121.4 (C5), 79.5 (C8), 72.0 (C9), 49.7 (C6), 21.4 (C7), 21.1 (C15), 17.8 (C12). Isolation of complex 10 was hampered by decomposition. Solution phase NMR characterization is provided.
Synthesis of 1-methyl-3-(prop-2-yn-1-yl)-1H-benzo[d]imidazol-3-ium bromide (11)
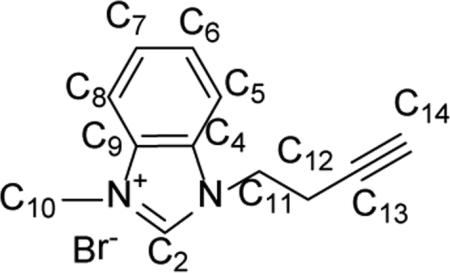
Compound 1 -methyl-3-(prop-2-yn-1-yl)-1H-benzo [d] imidazol-3-ium bromide (11) was prepared by a similar synthetic procedure as 5 using 1.06 g (8.00 mmol) of 1-methyl-1H-benzo[d]imidazole, 1.58 g (12.00 mmol) of 4-bromobut-1-yne, and 10 mL of toluene. Compound 11 was isolated as an off-white powder (1.56 g, 78%). 1H NMR (CDCl3, 500 MHz) δ = 11.11 (s, 1H, HC2), 7.66-7.90(m, 4H, HC5, HC6 HC7 HC8), 4.86 (t, J=6.5 Hz, 2H, HC11), 4.31 (s, 3H, HC10), 3.04 (dt, J = 6.5 Hz, J = 2.6 Hz, 2H, HC12), 2.08 (t, J = 2.6 Hz, 1H, HC14). 13C{1H} NMR (CDCl3, 500 MHz): δ = 143.2 (C2), 131.8 (C9), 131.3 (C4), 127.3 (C6), 127.3 (C7), 113.4 (C5), 113.0 (C8), 78.9 (C13), 73.1 (CM), 46.1 (C11), 34.1 (C10), 20.4 (C12). Anal. Calcd. for C12H13BrN2 (265.15 g/mol): C: 54.36%; H: 4.94%; N: 10.57%, Found: C: 54.25%; H: 5.18 N: 10.61%.
Synthesis of 1-methyl-3-(prop-2-yn-1-yl)-1H-benzo[d]imidazol-2(3H)-ylidene)gold(I) chloride (13)
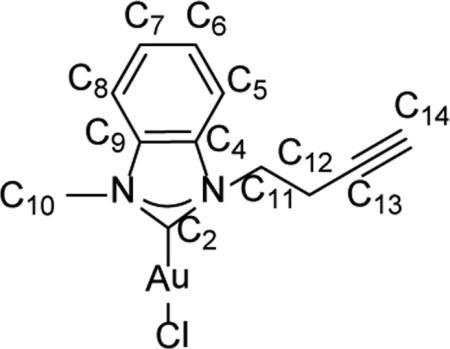
In a glovebox, solid ligand 1-methl-3-(prop-2-yn-1-yl)-1H-benzoimidazole-3-ium bromide (11) (159.09 mg, 6.00 mmol., 1 equiv.) and Ag2O (97.32 mg, 4.00 mmol., 0.7 equiv.) was dissolved in chloroform (10 mL) and stirred for 2 days to provide the NHC-AgI complex 12 in-situ. To the reaction mixture was added (CH3)2SAuCl (176.73mg, 6.00 mmol, 1 equiv.) and stirred for 1 h. Then the reaction mixture was filtrated through Celite®. The filtrate was collected and reduced under vacuum to 1 ml of solution. Diethyl ether was added to precipitate an off-white powder. The solid was collected by filtration and dried under vacuum for 2 h to provide the NHC-AuI complex 13 (112.5 mg, Yield = 45%). 1H NMR (300 MHz, CDCl3): δ = 7.44-7.57 (m, 4H, HC5 HC6 HC7 HC8), 4.69 (t, 2H, 3J = 7.0 Hz, HC11), δ4.03 (s, 3H, HC10), 2.85 (dt, 2H, 3J = 7.0 Hz, 4J = 2.0 Hz, HC12), and 1.97 (t, 1H, 4J = 2.0 Hz, HC14) ppm. 13C{1H} NMR (500 MHz, CDCl3): 5= 178.6 (C2) , 133.6 (C9), 131.1 (C4), 124.8 (C6) , 124.8 (C7), 111.6 (C5), 111.4 (C8), 76.9 (C13), 72.2 (CM), 46.9 (Cu), 35.4 (C10) and 20.5 (C9) ppm. Anal. Calcd. for C12H12AuClN2 (416.66 g/mol): C: 34.59%; H: 2.90%; N: 6.72%, Found; C: 33.98%; H: 2.92%; N: 6.49%.
NMR tube conjugation test reaction between benzyl azide and 13
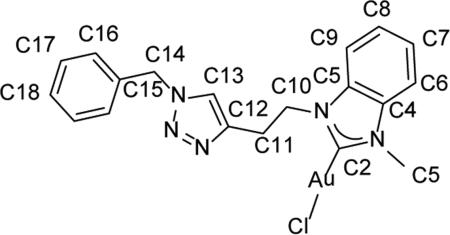
In an NMR tube, 13 (42 mg, 0.1 mmol, 1 equiv) and benzyl azide (15 mg, 0.11mmol, 1.1equiv) were dissolved in 1 ml of CDCl3. To the mixture was 10% mol catalyst PPh3Cu(OAc) was added. The NMR tube was placed in anaerobic environment for 48 h to generate 14 in situ. 1H NMR (500 MHz, CDCl3) δ = 7.72 (s, 1H, HC13), 7.33-7.42 (m, 4H, HC6, HC7, HC8, HC9), 6.85-7.16 (m, 5H, HC16, HC17, HC18), 5.49 (s, 2H, HC14), 4.77 (t, J = 7.0 Hz, 2H, HC10), 3.62 (t, J = 7.0 Hz, 2H, HC11), 3.49 (s, 3H, HC4).
Biological Studies
Cell culture
The cell lines DLD-1 (human colorectal adenocarcinoma), CEM (T cell leukemia) and Ramos (Burkitt's Lymphoma) were cultured according to ATCC specifications in RPMI-1640 medium. The cell lines MCF-7 (human breast adenocarcinoma), HEK (human embryonic kidney), and Hep-G2 (human hepatocellular carcinoma) were cultured according to ATCC specifications in DMEM medium. Both media were supplemented with 10% fetal bovine serum (Invitrogen) and the cells were incubated at 37 °C in 5% CO2.
Cytotoxicity studies using MTS assay
All cell lines studied were treated at the following concentrations of 9 (dissolved in DMSO to obtain a stock concentration of 20 mM): 0.5 μM, 1.0 μM, 2.0 μM, 4.0 μM, 8.0 μM, 16 μM, 32 μM, and 64 μM, respectively, for 48 h at 37 °C. In addition to treatments, cells were treated with two controls: (a) cells in media and (b) DMSO (0.01%). Approximately 100 uL of 2,000-5,000 (depending on the growth characteristics of the cell line) freshly collected cells were added to the inner 60 wells of its own 96-well plate. After 24 h incubation, 80 μL of old media was removed from each well and then 100 uL of each concentration of 9 was added to six wells of each of the 96-well plates. Each of the plates was subjected to this treatment and incubated for 48 h at 37 °C in 5% CO2. After 48 h, 100 μL of the drug treatment was removed and replaced with 120 uL of MTS dye (MTS = 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) diluted with PBS buffer (to 15% MTS). The assay was allotted 2 h at 37 °C for development. After incubation with the MTS dye, the 490 nm absorbance of each well was read on a Tecan plate 110 reader. Each cell measurement had the treatment background subtracted before analysis. Quantitative and statistical analyses were performed using the Origin 8.5 software. The same procedures were used to study 6 to assess whether the proligand displayed any cytotoxicity.
Supplementary Material
Fig. 1.

A few examples of the cytotoxicity of metal-NHC complexes towards breast cancer cell lines.2,10
Fig. 2.

Solid state structure of 13 with ellipsoids drawn and the 50% probability level.
Acknowledgements
The authors thank UF for financial support. The Paul Tarrant Faculty Fellowship awarded to ASV contributed financial support to the work. This work is also supported by the National Institutes of Health (GM079359 and CA133086). MEG thanks the UF Science for Life Program and Howard Hughes Medical Institute for providing funding to support this work.
Footnotes
† Electronic Supplementary Information (ESI) available: NMR spectra and experimental procedures. CCDC reference numbers 1033701 (13). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b000000x/
Notes and references
- 1.Berners-Price SJ, Filipovska A. Metallomics. 2011;3:863–873. doi: 10.1039/c1mt00062d. [DOI] [PubMed] [Google Scholar]
- 2.Teyssot M-L, Jarrousse A-S, Manin M, Chevry A, Roche S, Norre F, Beaudoin C, Morel L, Boyer D, Mahiou R, Gautier A. Dalton Trans. 2009:6894–6902. doi: 10.1039/b906308k. [DOI] [PubMed] [Google Scholar]
- 3.Sun RW-Y, Lok C-N, Fong TT-H, Li CK-L, Yang ZF, Zou T, Siu AF-M, Che C-M. Chem. Sci. 2013;4:1979–1988. [Google Scholar]
- 4.Che C-M, Sun RW-Y. Chem. Commun. 2011;47:9554–9560. doi: 10.1039/c1cc10860c. [DOI] [PubMed] [Google Scholar]
- 5.Liu W, Gust R. Chem. Soc. Rev. 2013;42:755–773. doi: 10.1039/c2cs35314h. [DOI] [PubMed] [Google Scholar]
- 6.Yao X, Panichpisal K, Kurtzman N, Nugent K. Am. J. Med. Sci. 2007;334:115–124. doi: 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- 7.Diez-Gonzalez S, Marion N, Nolan SP. Chem. Rev. 2009;109:3612–3676. doi: 10.1021/cr900074m. [DOI] [PubMed] [Google Scholar]
- 8.Droege T, Glorius F. Angew. Chem. Int. Ed. 2010;49:6940–6952. doi: 10.1002/anie.201001865. [DOI] [PubMed] [Google Scholar]
- 9.Jacobsen H, Correa A, Poater A, Costabile C, Cavallo L. Coord. Chem. Rev. 2009;253:2784–2784. [Google Scholar]
- 10.Weaver J, Gaillard S, Toye C, Macpherson S, Nolan SP, Riches A. Chem. Eur. J. 2011;17:6620–6624. doi: 10.1002/chem.201100321. [DOI] [PubMed] [Google Scholar]
- 11.Nobili S, Mini E, Landini I, Gabbiani C, Casini A, Messori L. Med. Res. Rev. 2010;30:550–580. doi: 10.1002/med.20168. [DOI] [PubMed] [Google Scholar]
- 12.Mullick AB, Chang YM, Ghiviriga I, Abboud KA, Tan WH, Veige AS. Dalton Trans. 2013;42:7440–7446. doi: 10.1039/c3dt32844a. [DOI] [PubMed] [Google Scholar]
- 13.Streciwilk W, Cassidy J, Hackenberg F, Muller-Bunz H, Paradisi F, Tacke M. J. Organomet. Chem. 2014;749:88–99. [Google Scholar]
- 14.Alessio E, editor. Bioinorganic Medicinal Chemistry. Wiley-VCH Verlag GmbH & Co.; Weinheim, Germany: 2011. [Google Scholar]
- 15.Higby GJ. Gold Bull. 1982;15:130–140. doi: 10.1007/BF03214618. [DOI] [PubMed] [Google Scholar]
- 16.Zhao HZ, Ning YT. Gold Bull. 2001;34:24–29. [Google Scholar]
- 17.Benedek TG. J. Hist. Med. Allied. Sci. 2004;59:50–89. doi: 10.1093/jhmas/jrg042. [DOI] [PubMed] [Google Scholar]
- 18.Chen D, Milacic V, Frezza M, Dou QP. Curr. Pharm. Des. 2009;15:777–791. doi: 10.2174/138161209787582183. [DOI] [PubMed] [Google Scholar]
- 19.Cazin CSJ, editor. N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis. Springer; United Kingdom: 2011. [Google Scholar]
- 20.Kalia J, Raines RT. Curr. Org. Chem. 2010;14:138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radzicka A, Wolfenden R. J. Am. Chem. Soc. 1996;118:6105–6109. [Google Scholar]
- 22.Hashmi ASK, Riedel D, Rudolph M, Rominger F, Oeser T. Chemistry – A European Journal. 2012;18:3827–3830. doi: 10.1002/chem.201200217. [DOI] [PubMed] [Google Scholar]
- 23.Hashmi ASK, Lothschütz C, Graf K, Häffner T, Schuster A, Rominger F. Adv. Synth. Catal. 2011;353:1407–1412. [Google Scholar]
- 24.Hashmi ASK, Lothschütz C, Böhling C, Hengst T, Hubbert C, Rominger F. Adv. Synth. Catal. 2010;352:3001–3012. [Google Scholar]
- 25.Moore LR, Cooks SM, Anderson MS, Schanz H-J, Griffin ST, Rogers RD, Kirk MC, Shaughnessy KH. Organometallics. 2006;25:5151–5158. [Google Scholar]
- 26.de Frémont P, Scott NM, Stevens ED, Nolan SP. Organometallics. 2005;24:2411–2418. [Google Scholar]
- 27.Huisgen R. In: 1,3-Dipolar Cycloaddition Chemistry. Padwa A, editor. Vol. 1. Wiley; New York: 1984. pp. 1–176. [Google Scholar]
- 28.Huisgen R. P. Chem. Soc. London. 1961:357–395. [Google Scholar]
- 29.Huisgen R. Angew. Chem. Int. Ed. 1963;2:565–598. [Google Scholar]
- 30.Tornoe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 31.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 32.Clough MC, Zeits PD, Bhuvanesh N, Gladysz JA. Organometallics. 2012;31:5231–5234. [Google Scholar]
- 33.Del Castillo TJ, Sarkar S, Abboud KA, Veige AS. Dalton Trans. 2011;40:8140–8144. doi: 10.1039/c1dt10787a. [DOI] [PubMed] [Google Scholar]
- 34.Powers AR, Yang X, Del Castillo TJ, Ghiviriga I, Abboud KA, Veige AS. Dalton Trans. 2013;42:14963–14966. doi: 10.1039/c3dt52105b. [DOI] [PubMed] [Google Scholar]
- 35.Gogolieva G, Bonin H, Durand J, Dechy-Cabaret O, Gras E. Eur. J. Inorg. Chem. 2014;2014:2088–2094. [Google Scholar]
- 36.Vuong KQ, Timerbulatova MG, Peterson MB, Bhadbhade M, Messerle BA. Dalton Trans. 2013;42:14298–14308. doi: 10.1039/c3dt51440d. [DOI] [PubMed] [Google Scholar]
- 37.Li LY, Wang JY, Wu T, Wang RH. Chem. Eur. J. 2012;18:7842–7851. doi: 10.1002/chem.201103631. [DOI] [PubMed] [Google Scholar]
- 38.Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 39.Lahann J, editor. Click Chemistry for Biotechnology and Materials Science. Wiley & Sons; Ann Arbor, USA: 2009. [Google Scholar]
- 40.He Y, Lv M.-f., Cai C. Dalton Trans. 2012;41:12428–12433. doi: 10.1039/c2dt31609a. [DOI] [PubMed] [Google Scholar]
- 41.Wang HMJ, Lin IJB. Organometallics. 1998;17:972–975. [Google Scholar]
- 42.Lin IJB, Vasam CS. Can. J. Chem. 2005;83:812–825. [Google Scholar]
- 43.de Fremont P, Scott NM, Stevens ED, Ramnial T, Lightbody OC, Macdonald CLB, Clyburne JAC, Abernethy CD, Nolan SP. Organometallics. 2005;24:6301–6309. [Google Scholar]
- 44.de Fremont P, Scott NM, Stevens ED, Nolan SP. Organometallics. 2005;24:2411–2418. [Google Scholar]
- 45.Lee KM, Wang HMJ, Lin IJB. J. Chem. Soc., Dalton Trans. 2002:2852–2856. [Google Scholar]
- 46.Herrmann WA, Schneider SK, Ofele K, Sakamoto M, Herdtweck E. J. Organomet. Chem. 2004;689:2441–2449. [Google Scholar]
- 47.Kascatan-Nebioglu A, Panzner MJ, Garrison JC, Tessier CA, Youngs WJ. Organometallics. 2004;23:1928–1931. [Google Scholar]
- 48.Han Vinh H, Guo S, Wu W. Organometallics. 2013;32:4591–4600. [Google Scholar]
- 49.Kumar N, Jain R. J. Heterocycl. Chem. 2012;49:370–374. [Google Scholar]
- 50.Occhipinti G, Jensen VR, Tornroos KW, Froystein NA, Bjorsvik H-R. Tetrahedron. 2009;65:7186–7194. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.