

Abstract
Aim
Aberrant methylation of the promoter, P2, and the first exon, E1, regions of the tumor suppressor gene RASSF1A, have been associated with hepatocellular carcinoma (HCC), albeit with poor specificity. This study analyzed the methylation profiles of P1, P2 and E1 regions of the gene to identify the region of which methylation most specifically corresponds to HCC and to evaluate the potential of this methylated region as a biomarker in urine for HCC screening.
Methods
Bisulfite DNA sequencing and quantitative methylation-specific polymerase chain reaction assays were performed to compare methylation of the 56 CpG sites in regions P1, P2 and E1 in DNA isolated from normal, hepatitic, cirrhotic, adjacent non-HCC, and HCC liver tissue and urine samples for the characterization of hypermethylation of the RASSF1A gene as a biomarker for HCC screening.
Results
In tissue, comparing HCC (n = 120) with cirrhosis and hepatitis together (n = 70), methylation of P1 had an area under the receiver operating characteristics curve (AUROC) of 0.90, whereas methylation of E1 and P2 had AUROC of 0.84 and 0.72, respectively. At 90% sensitivity, specificity for P1 methylation was 72.9% versus 38.6% for E1 and 27.1% for P2. Methylated P1 DNA was detected in urine in association with cirrhosis and HCC. It had a sensitivity of 81.8% for α-fetoprotein negative HCC.
Conclusion
Among the three regions analyzed, methylation of P1 is the most specific for HCC and holds great promise as a DNA marker in urine for screening of cirrhosis and HCC.
Keywords: biomarkers, cirrhosis, hepatocellular carcinoma, methylation, RASSF1A, urine
INTRODUCTION
Methylation of multiple tumor suppressor genes is implicated in hepatocarcinogenesis.1–4 These hypermethylation events also offer promise as tools to detect cancer in body fluids.5–9 Among these, aberrant hypermethylation of the RASSF1A gene (mRASSF1A) is found in 90% of liver cancer tissues.10–16 The RASSF1A gene is a member of the Ras association domain family that can regulate the cell cycle and trigger apoptosis.17–20 Many studies have demonstrated that the mRASSF1A has resulted in downregulation of gene expression. The inverse association between mRASSF1A and its RNA expression has been shown both in vitro in hepatocellular carcinoma (HCC) cell lines (HepG2 and Hep3B) and in vivo in patient liver samples from hepatitis, cirrhosis and HCC.21–24 Moreover, the increase of RASSF1A expression resulted in suppressed cancer properties such as proliferation, colony formation and apoptosis resistance in many cancerous cell lines, including HCC cell lines.25–30 Thus, the mRASSF1A has been suggested for its important role in hepatocarcinogenesis and its potential as a biomarker for HCC, but often with poor measures of specificity.10,11,31,32
Our previous studies suggested that the locations of the CpG sites analyzed affect the sensitivity and specificity of biomarkers for HCC.33,34 Yan et al.35 demonstrated by examining regions P1, P2 and E1 of the promoter and first exon of RASSF1A, that methylation of the P1 region was most specific for breast cancer detection. Although there are over 30 publications studying the association between mRASSF1A and HCC, to our knowledge, the methylation of the P1 region has not been investigated for HCC. This study compares methylation profiles of P1, P2 and E1 of the RASSF1A gene in HCC and non-HCC liver tissues and shows that methylation of the P1 region is most specific to liver carcinogenesis or HCC.
We and others have shown that urine contains DNA from the circulation and that this DNA is mostly derived from apoptotic cells.36–41 This circulation-derived urine DNA is filtered through the kidney barrier resulting in DNA fragmentation to sequences less than 300 base pairs (bp) (low molecular weight [LMW] DNA)38,42 which can then be used to detect cancer-derived genetic modifications.42–47 Encouragingly, we have also shown that the methylated P1 region detected in urine was associated with HCC development, thus suggesting that mRASSF1A can serve as a potential marker for hepatocarcinogenesis.
METHODS
Study subjects
Human samples were obtained with written informed consent from patients and were acquired under institutional review board approvals from the National Cheng-Kung University Medical Center, Taiwan, the Buddhist Tzu Chi Medical Center in Hualien, Taiwan, and Johns Hopkins University School of Medicine (Baltimore, MD, USA). DNA samples from normal tissues were purchased from Capital Biosciences (Rockville, MD, USA). Detailed sample information is provided in Tables 1–4.
Table 1.
Clinicopathological characterization of the liver tissues analyzed by BS–PCR DNA sequencing
| Characteristic | Normal (n = 3) | Hepatitis (n = 5) | Cirrhosis (n = 5) | HCC and adjacent non-HCC (n = 5) |
|---|---|---|---|---|
| Mean age (years) ± SD | 35.67 ± 19.14† | 60.6 ± 14.9 | 60 ± 10.4 | 63 ± 10.7 |
| Male/female (n) | 3/0 | 1/4 | 3/2 | 4/1 |
| HBV/HCV/both/non-viral or unknown (n) | – | 1/2/1/1 | 2/2/0/1 | 1/2/0/2 |
| Stage 1/2/3/4/unknown (n) | – | – | – | 3/2/0/0/0 |
| Grade 1/2/3/unknown (n) | – | – | – | 0/2/3/0 |
| Mean size of tumor (cm) ± SD | – | – | – | 7.1 ± 2.2 |
| AFP levels ≤20/>20 ng/mL/unknown (n) | – | – | – | 1/4/0 |
The individual age for the three normal liver subjects is 57, 20 and 30 years.
AFP, α-fetoprotein; BS–PCR, bisulfite polymerase chain reaction; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; SD, standard deviation.
Table 4.
Clinicopathological characteristics of the urine analyzed in this study
| Characteristic | Hepatitis (n = 44) | Cirrhosis (n = 50) | HCC (n = 78) |
|---|---|---|---|
| Mean age ± SD, years | 51.4 ± 9.8 | 58.7 ± 10.7 | 58.8 ± 12.2 |
| Male/female | 25/18/1 | 33/17/0 | 58/20 |
| HBV/HCV/both/none/unknown | 2/1/21/20/0/0 | 11/24/4/10/0 | 39/19/2/9/9 |
| Stage 1/2/3/4/unknown | – | – | 21/32/18/2/5 |
| Grade 1/2/3/unknown | – | – | 9/47/18/4 |
| Mean size of tumor ± SD, cm | – | – | 5.2 ± 3.2 |
| AFP levels, ng/mL, ≤20/>20/unknown | – | – | 44/30/4 |
AFP, α-fetoprotein; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; SD, standard deviation.
DNA isolation, urine collection, LMW urine DNA fractionation and bisulfite (BS) treatment
Tissue DNA was isolated using the Qiagen (Valencia, CA, USA) DNeasy Tissue kit according to the manufacturer's instructions. Freshly collected urine was immediately mixed with 0.5 mol/L ethylenediaminetetraacetic acid (EDTA), pH 8.0, to a final concentration of 10 mmol/L EDTA, and stored at −70°C. Total urine DNA was isolated by adding an equal volume of 6 mol/L guanidine thiocyanate (Sigma, St Louis, MO, USA) to thawed urine as described previously.38 The LMW urine DNA, DNA less than 1 kb, was obtained from total urine DNA using carboxylated magnetic beads (Agentcourt Bioscience, Beverly, MA, USA), as previously developed by us.42 BS treatment was performed using Qiagen EpiTect Bisulfite conversion kits following the manufacturer's guidelines.
Preparation of reconstituted standards of methylated and unmethylated DNA for BS polymerase chain reaction (PCR) sequencing
A reconstituted standard consisted of a known amount of methylated DNA (M), Bisulfite-converted Universal Methylated Human DNA Standard (Zymo Research, Irvine, CA, USA), in a background of unmethylated DNA (UM), HeLa DNA, as shown by Yeo et al.12 On the basis of quantification by BS-actin PCR,33 reconstituted sample sets were prepared in the following ratios: (i) 0% M, 100% UM; (ii) 10% M, 90% UM; (iii) 25% M, 75% UM; (iv) 50% M, 50% UM; and (v) 100% M, 0% UM.
BS–PCR and sequencing
Bisulfite-treated DNA was amplified by PCR for P1, P2 and E1 regions. The primer sequences and annealing temperatures for each PCR reaction are listed in Table 5 and locations are indicated in Figure 1(a). Sequencing was performed at the NAPCore Facility at the Children's Hospital of Philadelphia (Philadelphia, PA, USA). Sequencing results were analyzed using ClustalW software (available at http://www.ch.embnet.org/) and Finch TV version 1.4.0 (Geospiza, Seattle, WA, USA).
Table 5.
Primer and probe sequences used for bisulfite DNA sequencing and MSP for detecting the P1, P2 and E1 regions of the RASSF1A gene (GenBank accession no. DQ444319.1)
| Assay | Sequence | Annealing (°C) | CpG (#)† | Ref. |
|---|---|---|---|---|
| P1 BSP | F: 5′-gtaggttaagtgtgttgtttt-3′ | 54 | 1–12 | 26 |
| R: 5′-ttacccttccttccctcctt-3′ | ||||
| P2 BSP | F: 5′-aggagggaaggaagggtaag-3′ | 53 | 13–30 | 26 |
| R: 5′-taactttaaacgctaacaaa-3′ | ||||
| E1 BSP | F: 5′-aagtcggggttcgttttgtggttt-3′ | 53 | 24–59 | 26 |
| R: 5′-ccccaaataaaatcgccacaaaaa-3′ | ||||
| P1 MSP | F: 5′-agaaatacgggtattttcgc-3′ | 56 | 3–11 | – |
| R: 5′-caccccgaacgaccacaa-3′ | ||||
| Probe: 6FAM-accacaacgacgacgaccgc-BHQ1 | ||||
| P2 MSP | F: 5′-gggttttgcgagagcgcg-3′ | 56 | 14–20 | – |
| R: 5′-aaaccgcgcaataaaaacc-3′ | ||||
| Probe: 6FAM-cgcgaaccgaacgaa-BHQ1 | ||||
| E1 MSP | F: 5′-gtgttaacgcgttgcgtatc-3′ | 60 | 42–54 | 17 |
| R: 5′-aaccccgcgaactaaaaacga-3′ | ||||
| P1 short-amplicon MSP | Step 1 | 57 | 3–9 | – |
| F: 5′-aaatacgggtattttcgc-3′ | ||||
| R: 5′-gctcttcgtggtgtggtggaccacaacgacgacgac-3′ | ||||
| Step 2 | 58 | |||
| F: 5′-acgggtattttcgcgtg-3′ | ||||
| R: 5′-ttcgtggtgtggtggac-3′ |
CpG site numbers refer to the numbering in Fig. 1(a). The artificial sequences for the first step PCR of the P1 short-amplicon MSP are underlined.
BSP, bisulfite-specific polymerase chain reaction; F, forward; MSP, methylation-specific polymerase chain reaction; R, reverse.
Figure 1.

Bisulfite (BS) sequencing analysis of the RASSF1A promoter and first exon region in normal liver, diseased liver and non-liver normal tissues. (a) RASSF1A promoter region (GenBank accession no. DQ444319.1) and position of BS sequencing primers for three regions: P1 (nucleotides 357–548), P2 (nucleotides 530–736) and E1 (nucleotides 680–981). Vertical lines represent CpG sites; the transcription start site (TSS) is also indicated. CpG sites designated 1–59 are indicated with arrows. Note, the BS sequencing data for CpG sites 12–14 were not available due to sequencing-related technical issues; thus, they are not listed. (b) The extent of DNA methylation in three regions, P1, P2 and E1. Qualitative index of each CpG site (open boxes, <50% methylation detected; filled boxes, 350% methylation detected) is based on the relative heights of cytosine and thymine peaks within the chromatogram generated by a reconstituted standard as shown in the Figure S1. The non-liver normal tissues are: 1, spleen; 2, lung; 3, breast; 4, stomach; 5, colon; 6, trigeminal ganglion; 7, pancreas; 8, kidney; and 9, fetal liver. (c) The percentage of CpG sites with high (350%) methylation per total number of CpG sites examined in each region, P1, P2 and E1, in each liver tissue type as listed in the table. HCC, hepatocellular carcinoma.
Reference index for data analysis of BS–PCR sequencing
To normalize the primer or sequencing software bias, we established, on the basis of the BS–PCR sequencing data of the reconstituted standards from two reproducible experiments, a reference index for data analysis of each primer set (Fig. S1). Sequencing results were analyzed using chromatograms and comparisons of thymine versus cytosine peaks at each CpG site of the PCR products of reconstituted DNA standards. As shown in Figure S1, the P1 and E1 primer pairs were not able to detect the DNA sample with 10% of methylated DNA, however, they could detect the reconstituted sample with 25% of methylated DNA. The P2 primer set, however, was the least sensitive of the three as it did not detect 25% methylated DNA. Thus, for all three primer sets, the fact that the height of the cytosine peak exceeded that of the thymine peak was an indication of greater than 50% methylation reproducibly. Therefore, we categorized the methylation of each CpG site into two groups: less than 50% methylation (T only and C ≤ T); and 50% or more methylation (C > T and C only).
Quantitative methylation-specific PCR (qMSP) assays
Two qMSP assays, P1 and P2, were conducted in a Roche LightCycler 480 (LC480) system (Roche Applied Science, Mannheim, Germany). PCR conditions for both the P1 and the P2 assays were 95°C for 10 min (95°C for 10 s, 56°C for 15 s, 72°C for 10 s), 45 cycles with 1X LightCycler 480 Probes Master (Roche), 1.0 μM primers and 0.2 μM TaqMan probe. For the E1 assay, PCR conditions were 95°C for 5 min (95°C for 10 s, 56°C for 15 s, 72°C for 10 s) and 45 cycles.
For the short amplicon two-step qMSP targeting the P1 region, the step one reactions were set up at 10 μL with 1× Qiagen PCR buffer, 250 μM deoxyribonucleo-tide triphosphate mix, 1.0 μM primers, Hotstart Taq Polymerase Plus (Qiagen), at 95°C for 5 min (95°C for 30 s for 57°C for 30 s, 72°C for 30 s), and 25 cycles, 72°C for 4 min. The step one PCR product was diluted 1:10 and 1 μL of diluted PCR product was added as the template for the second PCR, which was set up in LC480 with 1× LightCycler 480 SYBR Green I Master (Roche) and 1.0 μM step two primers. The PCR conditions were 95°C for 5 min (95°C for 10 s, 58°C for 30 s, 72°C for 10 s) and 40 cycles. All experiments were performed in duplicate to ensure reproducibility. All MSP primer information is tabulated in Table 5.
Statistical analysis
Distribution of age and sex across the HCC and non-HCC groups was evaluated using Student's t-test and Fisher's exact test, respectively. For analysis of the results, appropriate statistical tests, as mentioned in the text, were performed using Graph Pad software (La Jolla, CA, USA). Receiver–operator curves (ROC)for all assays, parallel charts and distribution graphs were constructed using SPSS Statistics 20 (IBM, Armonk, NY, USA). ROC were compared using the StAR Tool48 available online.
RESULTS
Comparison of the extent of DNA methylation on P1, P2 and E1 regions of the RASSF1A gene in hepatocarcinogenesis by BS–PCR sequencing
To test our hypothesis that the extent of DNA methylation of a particular promoter loci of the RASSF1A gene varies during the process of hepatocarcinogenesis, our strategy was first to comprehensively examine every CpG site in the region of interest by BS–PCR sequencing in a small cohort of tissue DNA and then validate the findings in a larger tissue DNA cohort using MSP, which is a higher throughput assay.
Figure 1(a) shows CpG sites (vertical bars) in the promoter and first exon regions of the RASSF1A gene, known as the P1, P2 and E1 regions,35 along with locations of the BS–PCR sequencing primers used in this study. CpG sites within the 626-bp region studied were numbered 1–59 in the 5′-to-3′ direction.
We first established a reference index for sensitivity of each primer set for detecting methylation as detailed in Methods and in Figure S1. We then used this index to estimate the extent of DNA methylation of the three regions, P1, P2 and E1, on DNA isolated from tissues of various liver diseases. The data is summarized in Figure 1(b). To compare the methylation status of these three regions across the various tissue types, we pooled total CpG sites from all samples and assigned them to one of two categories: “low methylation” (<50% methylation) and “high methylation” (350% methylation). We then calculated the percent of CpG sites in the high-methylation category (Fig. 1c).
Hepatocellular carcinoma tissue samples had the highest level of methylation in all three regions. Although more than 85% of the CpG sites studied in each region were significantly methylated (350%) in the HCC samples, the methylation of the P1 region was significantly less in non-cancerous, diseased liver tissue (25.5% for hepatitis and 23.6% for cirrhosis), compared with E1 and P2 (57.9% and 38.6% for E1 and 77.5% and 67.5% for P2, for hepatitis and cirrhosis, respectively) as compared by Fisher's exact test (P < 0.0001) (Fig. 1c), suggesting that P1 is the region most specifically methylated in HCC.
DNA methylation of the RASSF1A gene varies among different tissue types
Next, we compared the DNA methylation pattern of normal liver tissue to that of other normal non-liver tissues. Although the methylation profile of the RASSF1A gene does not seem to exert a liver-specific pattern because a similar degree of DNA methylation was found in kidney and pancreatic tissue, most of the other normal tissues examined did not contain detectable levels of mRASSF1A when analyzed by BS–PCR sequencing. Interestingly, CpG sites 30 in the P2 region and 59 in the E1 region were consistently methylated across all tissue types examined (Fig. 1b).
Methylation of the P1 region is most specific for distinguishing HCC from hepatitis and cirrhosis of the three regions studied
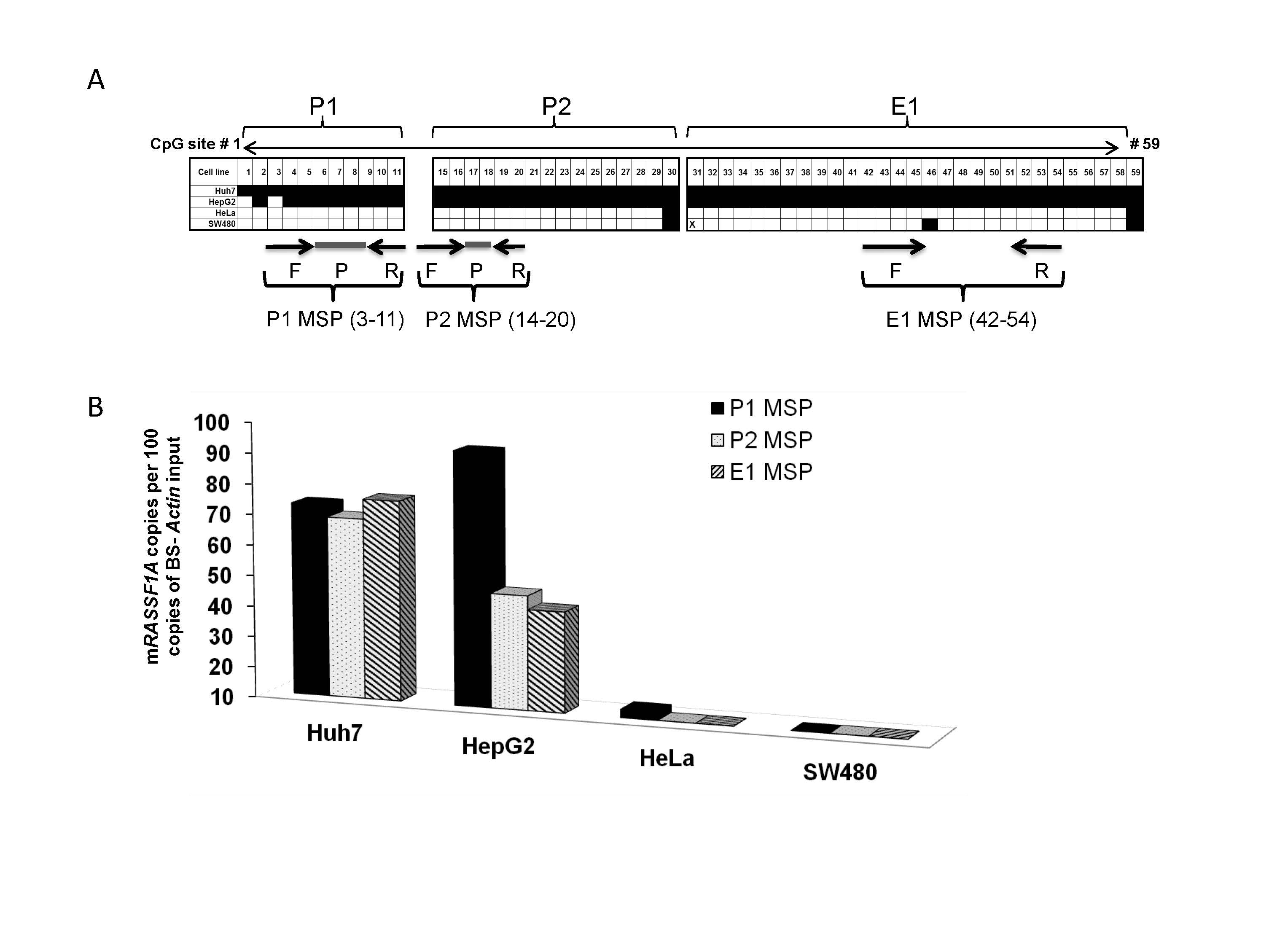
In order to determine whether methylation of the P1 region is most specific for distinguishing HCC from hepatitis and cirrhosis in a larger sample size, higher throughput, qMSP assays for P1, P2 and E1, were developed, as shown in Figure 2(a). The performance of the MSP assays were first compared with BS–PCR sequencing data, using a panel of four cell lines: two hepatoma cell lines (Huh7 and HepG2) with both undetectable RASSF1A expression and methylated promoters22,24 and two non-hepatoma carcinoma cell lines (HeLa, human cervical adenocarcinoma, and SW480, colorectal adeno-carcinoma) with both high RASSF1A expression and unmethylated promoters.11,49 The data is summarized in Figure S2(a) for BS–PCR sequencing and in Figure S2(b) for the MSP assays. The methylation analysis obtained by these two different assay platforms is comparable, although only the MSP assays are quantitative (Fig. S2). Of interest, the overall methylation of the RASSF1A promoter region, quantified by the qMSP assays, was significantly greater in the “RASSF1A-low” cell lines Huh7 and HepG2 than in the “RASSF1A-high” cell lines HeLa and SW480 (P < 0.01 by Student's t-test). Subsequently P1, P2 and E1 qMSP were performed to evaluate the finding from the BS–PCR sequencing in a larger sample size (120 HCC, 35 cirrhosis and 35 hepatitis tissues), as described in Table 3.
Figure 2.

Performance of the methylated P1, P2 and E1 regions of the RASSF1A gene as a biomarker for distinguishing hepato-cellular carcinoma (HCC) tissue from other liver disease tissues by quantitative methylation-specific polymerase chain reaction (qMSP) assays. (a) RASSF1A promoter. Vertical lines represent CpG sites. Forward (F) and reverse (R) primers and probes (P) are indicated for the three qMSP assays in their respective regions. The transcription start site (TSS) is also indicated. (b) Receiver– operator curves (ROC) for the RASSF1A gene as a marker to discriminate HCC (n = 120) from non-HCC liver tissues including hepatitis (n = 35) and cirrhosis (n = 35), generated by the qMSP assays of the P1, P2 and E1 regions, respectively. The quantity of methylated DNA was the average of two duplicate qMSP assays as detailed in Methods. Each area under the ROC and the specificity and sensitivity determined by the cut-off of 10 copies per input of 300 copies of DNA is shown in the inserted table.
Table 3.
Clinicopathological characterization of the liver tissues analyzed by MSP assays
| Characteristic | Hepatitis (n = 35) | Cirrhosis (n = 35) | HCC and adjacent non-HCC (n = 120) | P |
|---|---|---|---|---|
| Mean age ± SD | 55 ± 11.62 | 56 ± 13.8 | 60 ± 11.3 | 0.03† |
| Male/female | 17/18 | 23/12 | 81/39 | 0.16† |
| HBV/HCV/non-viral or unknown (n) | 3/22/9/1 | 6/16/0/13 | 59/29/4/28 | – |
| Stage 1/2/3/4/unknown (n) | – | – | 48/48/16/4/4 | – |
| Grade 1/2/3/unknown (n) | – | – | 18/74/23/5 | – |
| Mean size of tumor ± SD | – | – | 5.31 ± 3.69 cm | – |
| AFP levels ≤20 ng/mL/>20 ng/mL/unknown | – | – | 62/53/5 | – |
Across all subjects (n = 190), age was analyzed by Student's t-test and sex by Fisher's exact test.
AFP, α-fetoprotein; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; MSP, methylation-specific polymerase chain reaction; SD, standard deviation.
A ROC was constructed, and the area under the ROC (AUROC) was calculated for each region to evaluate the performance of methylation of these three regions as a biomarker to distinguish HCC from other liver diseases (cirrhosis and hepatitis) (Fig. 2b). The AUROC of the P1 (0.90) is statistically better than both E1 (0.84) and P2 (0.72) (P1 vs E1, P = 0.0256; P1 vs P2, P < 0.0001). Note, a sample size of 120 from the positive group (HCC) and 70 from the negative group (cirrhosis and hepatitis) achieves 85% power to detect a difference of 0.10 between the two diagnostic tests, with an AUROC of 0.90, and another diagnostic test with an AUROC of 0.80, using a two-sided z-test at a significance level of 0.05.50,51 Thus, this sample size is statistically powered to make a conclusion for the comparison of AUROC analysis. To compare the specificity, the sensitivity was fixed at 90%, 75%, 50% and 25%, as listed in the insert table in Figure 2(c); at 90% sensitivity, the P1 assay had a specificity of 72.9%, almost twice as specific as the E1 assay, which was only 38.6% specific, and far better than the P2 assay, which had a specificity of a mere 27.1% (P < 0.0001 by Fisher's exact two-tailed test when comparing the specificity of methylation for P1 vs P2 and P1 vs E1). A similar trend was observed at all other fixed values of sensitivity.
Methylation of the P1 region of the RASSF1A promoter during hepatocarcinogenesis
Having identified the P1 region as the most specifically methylated location in the RASSF1A gene in HCC, we studied its methylation distribution during hepatocarcinogenesis by analyzing the quantitative P1 qMSP data. We compared the methylation of hepatitis, cirrhosis, adjacent non-HCC and HCC tissues to each other by generating a 2-D dot plot (Fig. 3). Each open circle represents one case and the horizontal dark line indicates the median value.
Figure 3.

Analysis of the methylated RASSF1A in the liver tissues during hepatocarcinogenesis. 2-D dot plot illustrating the distribution of mRASSF1A in the hepatitis (n = 35), cirrhosis (n = 35), adjacent nonhepatocellular carcinoma (HCC) (n = 120) and HCC (n = 120) liver tissues. The Y-axis indicates the number of mRASSF1A copies detected by the P1 quantitative methylation-specific polymerase chain reaction (qMSP) assay per 300 copies of bisulfite (BS)-actin input. ***P < 0.001 by Kruskal–Wallis test.
As indicated in Figure 3, mRASSF1A increases gradually during the process of hepatocarcinogenesis. The amount of mRASSF1A is significantly greater in: (i) cirrhosis as compared with hepatitis; and (ii) HCC tissues as compared with hepatitis, cirrhosis and adjacent non-HCC tissues. There is no significant difference between the mRASSF1A between cirrhosis and the adjacent non-HCC tissues. To evaluate the performance of mRASSF1A in tissue as a marker for hepatocarcinogenesis, we constructed ROC curves (Fig. S3) and obtained AUROC values for distinguishing: (i) cirrhosis from hepatitis (AUROC = 0.697, 95% confidence interval [CI] = 0.563–0.831]; and (ii) HCC and cirrhosis from hepatitis (AUROC = 0.881, 95% CI = 0.827– 0.935).
Correlation of the age and methylation of the P1, P2 and E1 region of the RASSF1A gene
Previous studies have reported an age-dependent methylation of the RASSF1A promoter regions P2 and E1.11,31,52 To dissect the impact of age from that of hepatocarcinogenesis on the mRASSF1A, two analyses were performed. Spearman's rank correlation coefficient test between age and the level of methylation revealed that the age-dependent effect was only statistically significant in the hepatitis and cirrhosis group for the E1 and P1 regions (P < 0.05), and insignificant in adjacent non-HCC and HCC tissue (P > 0.05) (Table 6). To eliminate any influence of age-related methylation, we performed the Wilcoxon rank sum test between the HCC and the matched adjacent non-HCC tissue for each individual. All three regions had significantly higher methylation in the HCC tissue as compared with the matched adjacent non-HCC tissue (P < 0.001). Thus, the elevated mRASSF1A gene detected in HCC tissue resulted mainly from liver carcinogenesis.
Table 6.
Correlation between age and methylation levels of the P1, P2 and E1 regions of the RASSF1A gene in hepatitis and cirrhosis, adjacent non-HCC tissue and HCC tissues
| Spearman's rank correlation coefficient | Age correlation with methylation, Spearman's rho (P-values) |
||
|---|---|---|---|
| P1 | P2 | E1 | |
| Hepatitis and cirrhosis | 0.20 (0.045†) | 0.09 (0.221) | 0.23 (0.025†) |
| Adjacent non-HCC | –0.006 (0.474) | –0.08 (0.455) | 0.01 (0.455) |
| HCC | –0.09 (0.17l) | –0.14 (0.064) | –0.01 (0.453) |
Correlation is significant at the 0.05 level (one-tailed).
HCC, hepatocellular carcinoma.
Detection of mRASSF1A in the cell-free urine DNA of patients with hepatitis, cirrhosis and HCC by qMSP assay
We, and others, have shown that urine contains DNA from the circulation36,41,53,54 and that circulation-derived DNA in urine is fragmented into segments of length fewer than 300 bp that can be used to detect cancer-derived genetic modifications if the tumor is present. A short-amplicon, two-step qMSP assay targeting a 49-bp P1 region (including CpG sites 3–9, as referred to in the numbering in Fig. 1a) was developed for detecting cell-free circulating mRASSF1A in the urine to explore whether mRASSF1A (P1 region) can be a urine-based biomarker for HCC development.
Urine samples from 45 hepatitis, 50 cirrhosis and 78 HCC subjects were tested for the presence of mRASSF1A DNA using the P1 short-amplicon qMSP assays. Total urine DNA was isolated and the DNA less than 1 kb, designated as LMW urine DNA, was obtained with the purpose of enriching circulation-derived DNA. BS-converted LMW urine DNA derived from 0.2 mL urine was subjected to the P1 short-amplicon qMSP assays.
As indicated in Figure 4(a), the amount of mRASSF1A DNA detected was significantly higher in the urine samples from patients with cirrhosis and HCC as compared with the urine samples from patients with hepatitis (Fig. 4b, Mann–Whitney U-test, P < 0.0001, for hepatitis vs cirrhosis and hepatitis vs HCC). Also, mRASSF1A DNA was significantly higher in the HCC group as compared with the non-HCC group (hepatitis + cirrhosis) (P < 0.0001). However, there was no significant difference between the cirrhosis group versus the HCC group (P = 0.069), suggesting that mRASSF1A appears to be an early event in the hepatocarcinogenesis.
Figure 4.

Analysis of methylated RASSF1A in the urine of patients with hepatitis, cirrhosis and hepatocellular carcinoma (HCC). (a) Box plot showing the distribution of mRASSF1A copies in the urine of patients with hepatitis, cirrhosis and HCC. The median value is indicated by a line within each box and the diamond symbols (◇,  ) indicate outliers in the respective group. (b) Performance of mRASSF1A in urine as a biomarker for severe liver diseases. Area under the receiver–operator curve values for mRASSF1A in urine as a marker to discriminate severe liver diseases, HCC and cirrhosis from hepatitis and P-values of each comparison by Mann– Whitney U-test are listed in the inserted table.
) indicate outliers in the respective group. (b) Performance of mRASSF1A in urine as a biomarker for severe liver diseases. Area under the receiver–operator curve values for mRASSF1A in urine as a marker to discriminate severe liver diseases, HCC and cirrhosis from hepatitis and P-values of each comparison by Mann– Whitney U-test are listed in the inserted table.
To evaluate the performance of mRASSF1A in urine as a biomarker for severe liver diseases or hepatocarcinogenesis, we generated ROC curves for distinguishing: (i) HCC from hepatitis; (ii) HCC from cirrhosis; (iii) HCC from cirrhosis and hepatitis; and (iv) HCC and cirrhosis from hepatitis. The AUROC was calculated for each comparison and listed in Figure 4(b). As expected, the detection of mRASSF1A in urine as a biomarker for severe liver diseases (cirrhosis and HCC) has an AUROC of 0.846 but an AUROC of 0.595 to distinguish HCC from cirrhosis. It was of interest to determine whether mRASSF1A would have the potential as a biomarker to detect α-fetoprotein (AFP) negative HCC, because this is the type of HCC that would be undetected by the most widely used biomarker, serum AFP level. We thus analyzed the incidence of mRASSF1A in the urine of HCC patients who were negative for serum AFP (<20 ng/mL). Encouragingly, the mRASSF1A DNA was detected in 36 of 44 (81.8%) urine samples (data not shown).
DISCUSSION
This study semonstrated that location of CpG dinucleotide methylation affects the specificity of mRASSF1A to distinguish HCC tissue from other liver diseases, such as cirrhosis and hepatitis. In addition, this study identified that among the three regions studied (P1, P2 and E1), methylation of the P1 region exerts the most specificity to HCC. To our knowledge, this is the first report examining the methylation of the P1 region in association with liver diseases, although there have been more than 30 publications studying the association of RASSF1A methylation and HCC carcinogenesis. We further showed that the aberrantly methylated RASSF1A gene could be detected in urine samples and is significantly higher in patients with cirrhosis and HCC, which suggests that mRASSF1A in urine can be utilized as a potential biomarker for liver carcinogenesis.
By comparing the methylation status of the three regions (P1, P2 and E1) by qMSP assays, we found that the promoter P1 region was far more specific than those examined in previous studies (P2 and E1).4,11,14,22,31,55–57 We also found that methylation of the RASSF1A promoter is an early event and increases progressively during the process of HCC pathogenesis. Wild-type Ras proteins have been described as tumor suppressors, namely, negative regulators of mitosis and the cell cycle, as well as activators of cell death; thus, it is likely that their malfunction (e.g. via aberrant hypermethylation) has great potential to trigger carcinogenesis.18
The impact on methylation by only liver carcinogen-esis was demonstrated by comparing the level of methylation between adjacent non-HCC and HCC for each individual and by showing a significant elevation of methylation in all three regions. To our knowledge, this is the first report comparing the level of methylation between adjacent non-HCC and HCC by individual subjects to eliminate possible effects of age or other individual variations. It is an important comparison to distinguish HCC-related methylation from age-related methylation, particularly in this study, as the average age in the HCC group is significantly higher than that of the hepatitis and cirrhosis groups (P < 0.05, Table 3). Although age-related methylation was revealed in hepatitis and cirrhosis, it was significantly lower than HCC-related methylation.
Our data suggest that the P1 region outperforms the E1 region in liver tissues as a biomarker for distinguishing HCC from both cirrhosis and hepatitis. The methylated E1 region of the RASSF1A gene was previously detected in the serum of patients with HCC.10,57–60 Thus, it was of interest to explore the potential of the methylated P1 region of the RASSF1A gene as a screening biomarker by developing a short-amplicon qMSP assay for detection in urine. Encouragingly, similar to our tissue study (Fig. 3), mRASSF1A performed robustly as a biomarker with an AUROC of 0.831 for HCC when compared with hepatitis in urine (Fig. 4b). However, unlike the results obtained in the tissue study (Fig. 3), mRASSF1A was unable to discriminate between HCC and cirrhosis urine (AUROC = 0.595, Fig. 4). This inconsistency of the hypermethylation frequency of the RASSF1A gene between the tissue and periphery (in this case urine) has been previously reported in plasma and serum samples10,58,60 and has been attributed to mRASSF1A being an early common event in chronic liver diseases and the process of hepatocarcinogenesis. In spite of our efforts to map the HCC-specific methylated P1 region in the RASSF1A promoter to boost its specificity as a biomarker for HCC, its performance for distinguishing HCC from cirrhosis was worse in urine than in liver tissues. One possibility is that there were undetectable cancerous cells in the cirrhotic liver that contributed to the mRASSF1A DNA in urine. Another possibility is that, similar to the observation in circulating DNA studies, methylation of the RASSF1A gene is an early event during liver carcinogenesis, which occurs either in cirrhosis or in the transition between cirrhosis to HCC.
We, and others, have suggested that circulation-derived urine DNA is mostly from DNA released by apoptotic cells. Malignant and preneoplastic cells often proliferate at abnormal rates, which is accompanied by an increase in apoptotic cell death,61,62 and this DNA may accumulate in the urine, which is essentially a collection of the body fluid. The statistically insignificant distinction of mRASSF1A observed in urine could also be the consequence of the dilution of the target DNA proportions between HCC and cirrhosis with the DNA coming from the rest of the body. Nevertheless, the amount of mRASSF1A DNA in the urine from patients with hepatitis was significantly less than that from patients with cirrhosis and HCC (P < 0.001, Fig. 4a), suggesting that mRASSF1A can serve as a screening marker for distinguishing HCC or cirrhosis from hepatitis (Fig. 4b) to bring patients more sophisticated imaging tests for early detection of HCC.
Unexpectedly, we observed hypermethylation of the P1 region in normal kidney tissue (Fig. 1b), and it is known that chronic hepatitis B virus/hepatitis C virus are both associated with glomerular disease.63,64 So, it is possible that the methylated P1 DNA detected in urine could be partially coming from the kidney, even though we only used LMW urine DNA as the substrate. In this study, the status of the existing glomerular disease for each patient was not clear but will be important to consider in the further development of this marker.
Overall, these results establish a locus-dependent CpG-site methylation pattern in the RASSF1A gene for the first time in liver cancer and demonstrate a progressive augmentation of mRASSF1A during hepatocarcinogenesis in the liver tissue. In addition, this study also demonstrated for the first time that methylation of the P1 region is associated with hepatocarcinogenesis and is the most specific region of methylation for HCC, as compared with methylation of the P2 and E1 regions. Most importantly, we also demonstrate the feasibility of using circulation-derived urine DNA as a viable approach for non-invasive early indication of hepatocarcinogenesis. We are currently exploring the prospect of combining mRASSF1A with other HCC-specific urine DNA markers, as suggested previously,8,52,53,55 to develop a sensitive and specific urine test for the identification of patients with a “high risk” of developing HCC, thereby greatly improving the prognosis of the disease.
Supplementary Material
Figure S1 Reference index for data analysis of bisulfite polymerase chain reaction (BS–PCR) sequencing. Representative chromatograms of BS–PCR sequencing of the reconstituted standards for the P1, P2 and E1 regions: 0% methylated + 100% unmethylated DNA (0%); 10% methylated DNA + 90% unmethylated DNA (10%); 25% methylated DNA + 75% unmethylated DNA (25%); 50% methylated DNA + 50% unmethylated DNA (50%); and 100% methylated DNA (100%). Boxed areas are the areas of the examples showing the relative “C” and “T” peaks in the chromatogram from each sample of the reconstituted standards by each primer set as indicated. Below is the reference index for P1, P2 and E1 showing percentage of methylation based on relative peak heights of cytosine and thymine at CpG sites obtained from BS sequencing data.
{kind=link}
Figure S3 Biomarker analysis of methylated RASSF1A in the liver tissues during hepatocarcinogenesis. Receiver–operator curve (ROC) for mRASSF1A in tissue as a marker to discriminate cirrhosis from hepatitis and combined cirrhosis and HCC from hepatitis.
{kind=link}
Figure S2 Methylation analysis of the RASSF1A promoter comparing bisulfite polymerase chain reaction (BS–PCR) sequencing and quantitative methylationspecific polymerase chain reaction (qMSP) assays. (a) BS–PCR sequencing analysis of the RASSF1A promoter (P1 and P2) and first exon (E1) regions in DNA isolated from Huh7, HepG2, HeLa and SW480. Each box represents a CpG site and its estimated methylation based on the index in Figure S1. Forward (F) and reverse (R) primers and probes (P) for three qMSP assays with the corresponding CpG sites are indicated. (b) The extent of DNA methylation of the RASSF1A gene as determined by three qMSP assays. The bar chart illustrates the mRASSF1A copies detected by the P1, P2 and E1 qMSP assays as normalized to 100 copies of BS-actin. The limit of detection was 10 copies for all three assays.
Table 2.
Subject information for non-liver tissues and fetal liver
| Tissue | Sex | Age (years) | Pathological profile |
|---|---|---|---|
| Spleen | F | 83 | Normal |
| Lung | F | 50 | Normal |
| Breast | F | 78 | Normal |
| Stomach | M | 27 | Normal |
| Colon | F | 85 | Normal |
| Trigeminal ganglion | M | 75 | Normal |
| Pancreas | M | 27 | Normal |
| Kidney | F | 63 | Normal |
| Fetal liver | M | 29 weeks | Normal |
F, female; M, male.
ACKNOWLEDGMENTS
This work WAS supported by the National Institutes of Health (no. R01 CA125642 to Y. H. S.); Department of Defense (no. CA093176 to Y. H. S.); National Institutes of Health (no. R43 CA165312 to W. S. and Y. H. S.; no. R44 CA165312 to W. S. and Y. H. S.); and Prevent Cancer Foundation Postdoctoral Fellowship Award (to S. J.). The authors thank Dr Di Chen (ClinPharm) for critical review of the statistical analysis and Ms Pamela Fried, Ms Diana M. Winters and Mr Adam Clemens for assistance in preparation of the manuscript.
Footnotes
Conflict of interest: Wei Song is an employee and shareholder of JBS Science. Surbhi Jain, Lijia Xie and Batbold Boldbaatar are employees of JBS Science. Selena Y. Lin and Ying-Hsiu Su have received funding from JBS Science. All other authors declare that they have no competing interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article at the publisher's website:
REFERENCES
- 1.Jain S, Singhal S, Lee P, Xu R. Molecular genetics of hepatocellular neoplasia. Am J Transl Res. 2010;2(1):105–18. [PMC free article] [PubMed] [Google Scholar]
- 2.Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163(4):1371–8. doi: 10.1016/S0002-9440(10)63495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao W, Kondo Y, Shen L, et al. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis. 2008;29(10):1901–10. doi: 10.1093/carcin/bgn170. [DOI] [PubMed] [Google Scholar]
- 4.Moribe T, Iizuka N, Miura T, et al. Methylation of multiple genes as molecular markers for diagnosis of a small, well-differentiated hepatocellular carcinoma. Int J Cancer. 2009;125:388–97. doi: 10.1002/ijc.24394. [DOI] [PubMed] [Google Scholar]
- 5.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–66. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 6.Mulero-Navarro S, Esteller M. Epigenetic biomarkers for human cancer: the time is now. Crit Rev Oncol Hematol. 2008;68(1):1–11. doi: 10.1016/j.critrevonc.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Eads CA, Danenberg KD, Kawakami K, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:e32, i-e–vi. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Usadel H, Brabender J, Danenberg KD, et al. Quantitative adenomatous polyposis coli promoter methylation analysis in tumor tissue, serum, and plasma DNA of patients with lung cancer. Cancer Res. 2002;62:371–5.. [PubMed] [Google Scholar]
- 9.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan KCA, Lai PBS, Mok TSK, et al. Quantitative analysis of circulating methylated DNA as a biomarker for hepatocellular carcinoma. Clin Chem. 2008;54(9):1528–36. doi: 10.1373/clinchem.2008.104653. [DOI] [PubMed] [Google Scholar]
- 11.Di Gioia S, Bianchi P, Destro A, et al. Quantitative evaluation of RASSF1A methylation in the non-lesional, regenerative and neoplastic liver. BMC Cancer. 2006;6(1):89. doi: 10.1186/1471-2407-6-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeo W, Wong N, Wong W-L, Lai PBS, Zhong S, Johnson PJ. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int. 2005;25:266–72. doi: 10.1111/j.1478-3231.2005.01084.x. [DOI] [PubMed] [Google Scholar]
- 13.Yu J, Ni M, Xu J, et al. Methylation profilinf of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC Cancer. 2002;2:29–43. doi: 10.1186/1471-2407-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez-Vargas H, Lambert M-P, Le Calvez-Kelm F, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS ONE. 2010;5(3):e9749. doi: 10.1371/journal.pone.0009749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Li H, Liu Z, et al. Loss of heterozygosity and methylation of multiple tumor suppressor genes on chromosome 3 in hepatocellular carcinoma. J Gastroenterol. 2013;48(1):132–43. doi: 10.1007/s00535-012-0621-0. [DOI] [PubMed] [Google Scholar]
- 16.Xu B, Di J, Wang Z, et al. Quantitative analysis of RASSF1A promoter methylation in hepatocellular carcinoma and its prognostic implications. Biochem Biophys Res Commun. 2013;438(2):324–8. doi: 10.1016/j.bbrc.2013.07.070. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Tommasi S, Lee D-H, Dammann R, Pfeifer GP. Control of microtubule stability by the RASSF1A tumor suppressor. Oncogene. 2003;22(50):8125–36. doi: 10.1038/sj.onc.1206984. [DOI] [PubMed] [Google Scholar]
- 18.Gordon M, Baksh S. RASSF1A: not a prototypical Ras effector. Small GTPases. 2011;2(3):148–57. doi: 10.4161/sgtp.2.3.16286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song MS, Song SJ, Ayad NG, et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol. 2004;6(2):129–37. doi: 10.1038/ncb1091. [DOI] [PubMed] [Google Scholar]
- 20.Vos MD, Martinez A, Elam C, et al. A role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer Res. 2004;64(12):4244–50. doi: 10.1158/0008-5472.CAN-04-0339. [DOI] [PubMed] [Google Scholar]
- 21.Saelee P, Wongkham S, Chariyalertsak S, Petmitr S, Chuensumran U. RASSF1A promoter hypermethylation as a prognostic marker for hepatocellular carcinoma. Asian Pac J Cancer Prev. 2010;11:1677–81. [PubMed] [Google Scholar]
- 22.Schagdarsurengin U, Wilkens L, Steinemann D, et al. Frequent epigenetic inactivation of the RASSF1A gene in hepatocellular carcinoma. Oncogene. 2003;22(12):1866–71. doi: 10.1038/sj.onc.1206338. [DOI] [PubMed] [Google Scholar]
- 23.Hu L, Chen G, Yu H, Qiu X. Clinicopathological significance of RASSF1A reduced expression and hypermethylation in hepatocellular carcinoma. Hepatol Int. 2010;4(1):423–32. doi: 10.1007/s12072-010-9164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong S, Yeo W, Tang MW, Wong N, Lai PBS, Johnson PJ. Intensive hypermethylation of the CpG island of ras association domain family 1A in hepatitis B virus-associated hepatocellular carcinomas. Clin Cancer Res. 2003;9(9):3376–82. [PubMed] [Google Scholar]
- 25.Ahn EY, Kim JS, Kim GJ, Park YN. RASSF1A-mediated regulation of AREG via the hippo pathway in hepatocellular carcinoma. Mol Cancer Res. 2013;11(7):748–58. doi: 10.1158/1541-7786.MCR-12-0665. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Dai X, Wu B. Study of 5-Aza-CdR on transcription regulation of RASSF1A gene in the BIU87 cell line. Urol Int. 2009;82:108–12. doi: 10.1159/000176036. [DOI] [PubMed] [Google Scholar]
- 27.Xue W-J, Li C, Zhou X-J, et al. RASSF1A expression inhibits the growth of hepatocellular carcinoma from Qidong County. J Gastroenterol Hepatol. 2008;23(9):1448–58. doi: 10.1111/j.1440-1746.2007.05067.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Q-Z, Dou K-F. Methylation of Ras association domain family protein 1, isoform A correlated with proliferation and drug resistance in hepatocellular carcinoma cell line SMMC-7721. J Gastroenterol Hepatol. 2007;22(5):683–9. doi: 10.1111/j.1440-1746.2006.04676.x. [DOI] [PubMed] [Google Scholar]
- 29.Dammann R, Li C, Yoon J-H, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25(3):315–9. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 30.Montenegro MF, Sáez-Ayala M, Piñero-Madrona A, Cabezas-Herrera J, Rodríguez-López JN. Reactivation of the tumour suppressor RASSF1A in breast cancer by simultaneous targeting of DNA and E2F1 methylation. PLoS ONE. 2012;7(12):e52231. doi: 10.1371/journal.pone.0052231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908–18. doi: 10.1002/hep.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Z-H, Fan YC, Yang Y, Wang K. Association between Ras association domain family 1A promoter methylation and hepatocellular carcinoma: a meta-analysis. World J Gastroenterol. 2013;17(41):7189–96. doi: 10.3748/wjg.v19.i41.7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain S, Chang T-T, Hamilton JP, et al. Methylation of the CpG Sites only on the sense strand of the APC gene is specific for hepatocellular carcinoma. PLoS ONE. 2011;6(11):e26799. doi: 10.1371/journal.pone.0026799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain S, Chen S, Chang K-C, et al. Impact of the location of CpG methylation within the GSTP1 gene on its specificity as a DNA marker for hepatocellular carcinoma. PLoS ONE. 2012;7(4):e35789. doi: 10.1371/journal.pone.0035789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan PS, Shi H, Rahmatpanah F, et al. Differential distribution of DNA methylation within the RASSF1A CpG island in breast cancer. Cancer Res. 2003;63(19):6178–86. [PubMed] [Google Scholar]
- 36.Su YH, Wang M, Block TM, et al. Transrenal DNA as a diagnostic tool: important technical notes. Ann N Y Acad Sci. 2004;1022(1):81–9. doi: 10.1196/annals.1318.014. [DOI] [PubMed] [Google Scholar]
- 37.Chan AKC, Chiu RWK, Lo YMD. Cell-free nucleic acids in plasma, serum and urine: a new tool in molecular diagnosis. Ann Clin Biochem. 2003;40:122–30. doi: 10.1258/000456303763046030. [DOI] [PubMed] [Google Scholar]
- 38.Su YH, Wang M, Brenner DE, et al. Human urine contains small, 150 to 250 nucleotide-sized, soluble DNA derived from the circulation and may be useful in the detection of colorectal cancer. J Mol Diagn. 2004;6(2):101–7. doi: 10.1016/S1525-1578(10)60497-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Utting M, Werner W, Dahse R, Schubert J, Junker K. Microsatellite analysis of free tumor DNA in urine, serum, and plasma of patients: a minimally invasive method for the detection of bladder cancer. Clin Cancer Res. 2002;8:35–40. [PubMed] [Google Scholar]
- 40.Serdyuk OI, Botezatu I, Shelepov V, et al. Detection of mutant k-ras sequences in the urine of cancer patients. Bull Exp Biol Med. 2001;131:283–4. doi: 10.1023/a:1017624120807. [DOI] [PubMed] [Google Scholar]
- 41.Botezatu I, Serdyuk O, Potapova G, et al. Genetic analysis of DNA excreted in Urine: a new approach for detecting specific genomic DNA sequences from cells dying in an organism. Clin Chem. 2000;46:1078–84. [PubMed] [Google Scholar]
- 42.Su Y-H, Song J, Wang Z, et al. Removal of high molecular weight DNA by carboxylated magnetic beads enhances the detection of mutated K-ras DNA in urine. Ann N Y Acad Sci. 2008;1137:82–91. doi: 10.1196/annals.1448.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su Y-H, Wang M, Aiamkitsumrit B, Brenner DE, Block TM. Detection of K-ras mutation in urine of patients with colorectal cancer. Cancer Biomark. 2005;1:177–82. doi: 10.3233/cbm-2005-12-305. [DOI] [PubMed] [Google Scholar]
- 44.Su Y-H, Wang M, Norton PA, Brenner DE, Block TM. Detection of mutated K-ras DNA in urine, plasma and serum from patients with colorectal carcinoma or adenomatous polyps. Ann N Y Acad Sci. 2008;1137:197–201. doi: 10.1196/annals.1448.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin SY, Dhillon V, Jain S, et al. A locked nucleic acid clamp-mediated PCR assay for detection of a p53 codon 249 hotspot mutation in urine. J Mol Diagn. 2011;13(5):474–84. doi: 10.1016/j.jmoldx.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren XD, Lin SY, Wang X, Zhou T, Block TM, Su YH. Rapid and sensitive detection of hepatitis B virus 1762T/1764A double mutation from hepatocellular carcinomas using LNA-mediated PCR clamping and hybridization probes. J Virol Methods. 2009;158(1–2):24–9. doi: 10.1016/j.jviromet.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song BP, Jain S, Lin SY, et al. Detection of hypermethylated vimentin in urine of patients with colorectal cancer. J Mol Diagn. 2012;14(2):112–9. doi: 10.1016/j.jmoldx.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vergara I, Norambuena T, Ferrada E, Slater A, Melo F. StAR: a simple tool for the statistical comparison of ROC curves. BMC Bioinformatics. 2008;9(1):265. doi: 10.1186/1471-2105-9-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pijnenborg J, Dam-de Veen G, Kisters N, et al. RASSF1A methylation and K-ras and B-raf mutations and recurrent endometrial cancer. Ann Oncol. 2007;18:491–7. doi: 10.1093/annonc/mdl455. [DOI] [PubMed] [Google Scholar]
- 50.Obuchowski NA, McClish DK. Sample size determination for diagnostic accuracy studies involving binormal roc curve indices. Stat Med. 1997;16(13):1529–42. doi: 10.1002/(sici)1097-0258(19970715)16:13<1529::aid-sim565>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 51.Hanley JA, McNeil BJ. A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology. 1983;148(3):839–43. doi: 10.1148/radiology.148.3.6878708. PMID: 6878708. [DOI] [PubMed] [Google Scholar]
- 52.Lehmann U, Berg-Ribbe I, Wingen LU, et al. Distinct methylation patterns of benign and malignant liver tumors revealed by quantitative methylation profiling. Clin Cancer Res. 2005;11(10):3654–60. doi: 10.1158/1078-0432.CCR-04-2462. [DOI] [PubMed] [Google Scholar]
- 53.Lichtenstein AV, Melkonyan HS, Tomei D, Umansky SR. Circulating nucleic acids and apoptosis. Ann N Y Acad Sci. 2001;945:239–49. doi: 10.1111/j.1749-6632.2001.tb03892.x. [DOI] [PubMed] [Google Scholar]
- 54.Chan KCA, Leung SF, Yeung SW, Chan ATC, Lo YMD. Quantitative analysis of the transrenal excretion of circulating EBV DNA in nasopharyngeal carcinoma patients. Clin Cancer Res. 2008;14(15):4809–13. doi: 10.1158/1078-0432.CCR-08-1112. [DOI] [PubMed] [Google Scholar]
- 55.Oh BK, Kim H, Park HJ. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int J Mol Med. 2007;20(1):65–73. [PubMed] [Google Scholar]
- 56.Park H-J, Yu E, Shim Y-H. DNA methyltransferase expression and DNA hypermethylation in human hepatocellular carcinoma. Cancer Lett. 2006;233(2):271–8.. doi: 10.1016/j.canlet.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 57.Hua D, Hu Y, Wu Y-Y, et al. Quantitative methylation analysis of multiple genes using methylation-sensitive restriction enzyme-based quantitative PCR for the detection of hepatocellular carcinoma. Exp Mol Pathol. 2011;91(1):455–60. doi: 10.1016/j.yexmp.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 58.Huang Z-H, Hu Y, Hua D, Wu Y-Y, Song M-X, Cheng Z-H. Quantitative analysis of multiple methylated genes in plasma for the diagnosis and prognosis of hepatocellular carcinoma. Exp Mol Pathol. 2011;91(3):702–7. doi: 10.1016/j.yexmp.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y-J, Wu H-C, Shen J, et al. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin Cancer Res. 2007;13(8):2378–84. doi: 10.1158/1078-0432.CCR-06-1900. [DOI] [PubMed] [Google Scholar]
- 60.Chang H, Yi B, Li L, et al. Methylation of tumor associated genes in tissue and plasma samples from liver disease patients. Exp Mol Pathol. 2008;85(2):96–100. doi: 10.1016/j.yexmp.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 61.Arai N, Mitomi H, Ohtani Y, Igarashi M, Kakita A, Okayasu I. Enhanced epithelial cell turnover associated with p53 accumulation and high p21WAF1/CIP1 expression in ulcerative colitis. Mod Pathol. 1999;12:604–11. [PubMed] [Google Scholar]
- 62.Schulte-Hermann R, Bursch W, Grasl-Kraupp B, Torok L, Ellinger AML. Role of active cell death (apoptosis) in multi-stage carcinogenesis. Toxicol Lett. 1995;82–83:143–8. doi: 10.1016/0378-4274(95)03550-8. [DOI] [PubMed] [Google Scholar]
- 63.Chan T. Hepatitis B and renal disease. Curr Hepatitis Rep. 2010;9(2):99–105. doi: 10.1007/s11901-010-0042-6. (English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sabry AA, Sobh MA, Irving WL, et al. A comprehensive study of the association between hepatitis C virus and glomerulopathy. Nephrol Dial Transplant. 2002;17(2):239–45. doi: 10.1093/ndt/17.2.239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Reference index for data analysis of bisulfite polymerase chain reaction (BS–PCR) sequencing. Representative chromatograms of BS–PCR sequencing of the reconstituted standards for the P1, P2 and E1 regions: 0% methylated + 100% unmethylated DNA (0%); 10% methylated DNA + 90% unmethylated DNA (10%); 25% methylated DNA + 75% unmethylated DNA (25%); 50% methylated DNA + 50% unmethylated DNA (50%); and 100% methylated DNA (100%). Boxed areas are the areas of the examples showing the relative “C” and “T” peaks in the chromatogram from each sample of the reconstituted standards by each primer set as indicated. Below is the reference index for P1, P2 and E1 showing percentage of methylation based on relative peak heights of cytosine and thymine at CpG sites obtained from BS sequencing data.
Figure S3 Biomarker analysis of methylated RASSF1A in the liver tissues during hepatocarcinogenesis. Receiver–operator curve (ROC) for mRASSF1A in tissue as a marker to discriminate cirrhosis from hepatitis and combined cirrhosis and HCC from hepatitis.
Figure S2 Methylation analysis of the RASSF1A promoter comparing bisulfite polymerase chain reaction (BS–PCR) sequencing and quantitative methylationspecific polymerase chain reaction (qMSP) assays. (a) BS–PCR sequencing analysis of the RASSF1A promoter (P1 and P2) and first exon (E1) regions in DNA isolated from Huh7, HepG2, HeLa and SW480. Each box represents a CpG site and its estimated methylation based on the index in Figure S1. Forward (F) and reverse (R) primers and probes (P) for three qMSP assays with the corresponding CpG sites are indicated. (b) The extent of DNA methylation of the RASSF1A gene as determined by three qMSP assays. The bar chart illustrates the mRASSF1A copies detected by the P1, P2 and E1 qMSP assays as normalized to 100 copies of BS-actin. The limit of detection was 10 copies for all three assays.