

Abstract

An ultrahigh-throughput screen was performed to identify novel small molecule inhibitors of influenza virus replication. The screen employed a recombinant influenza A/WSN/33 virus expressing Renilla luciferase and yielded a hit rate of 0.5%, of which the vast majority showed little cytotoxicity at the inhibitory concentration. One of the top hits from this screen, designated S20, inhibits HA-mediated membrane fusion. S20 shows potent antiviral activity (IC50 = 80 nM) and low toxicity (CC50 = 40 μM), yielding a selectivity index of 500 and functionality against all of the group 1 influenza A viruses tested in this study, including the pandemic H1N1 and avian H5N1 viruses. Mechanism of action studies proved a direct S20–HA interaction and showed that S20 inhibits fusion by stabilizing the prefusion conformation of HA. In silico docking studies were performed, and the predicted binding site in HA2 corresponds with the area where resistance mutations occurred and correlates with the known role of this region in fusion. This high-throughput screen has yielded many promising new lead compounds, including S20, which will potentially shed light on the molecular mechanisms of viral infection and serve as research tools or be developed for clinical use as antivirals.
Keywords: screen, antiviral, fusion, hemagglutinin, influenza, docking
Influenza viruses are enveloped, negative-stranded RNA viruses that are part of the Orthomyxoviridae family. Influenza A and B viruses are a major cause of human respiratory disease and account for up to 5 million cases of severe disease and 500,000 deaths per year worldwide (WHO). Periodic pandemics of antigenically novel influenza A viruses exacerbate these levels of morbidity and mortality due to the lack of pre-existing immunity in the population. The most recent occurrence was the novel swine H1N1 pandemic virus of 2009,1−3 but the greatest example of the destructive potential of a pandemic was that which occurred in 1918 and caused an estimated 50–100 million deaths worldwide.4
The influenza virus membrane contains three proteins: hemagglutinin (HA), neuraminidase (NA), and the proton channel (M2). Early in infection HA is responsible for binding to sialic acid receptors and, after uptake into endosomes, for pH-dependent fusion of virus and host membranes.5 Once in the late endosome the M2 channel allows for protons to pass into the virion, where the low pH facilitates uncoating of the viral particle and release of the genome through the fusion pore. NA acts later in infection and has the enzymatic function of cleaving sialic acid from glycoproteins to allow release of budding particles from the cell surface. NA inhibitors (oseltamivir and zanamivir) target the NA protein, and adamantanes (amantadine and rimantadine) target the transmembrane domain of the M2 protein.
Influenza continues to be a significant threat to public health, and the inadequate production of vaccine for the recent 2009 pandemic underscores the need for antivirals.6 Currently, NA inhibitors are the only drugs recommended for clinical use by the CDC as the circulating strains of both H1N1 and H3N2 influenza A viruses are resistant to the adamantane class of drugs. Also, the H1N1 viruses circulating prior to the 2009 pandemic presented nearly complete resistance to oseltamivir without any loss in fitness,7 and there are sporadic reports of oseltamivir resistance among contemporary H1N1 viruses, too.8−12 Taken together, these facts emphasize the need for new influenza-specific antiviral drugs with novel mechanisms of action to be used in conjunction with current treatments to control infection and reduce incidences of resistance. This strategy would mimic the well-established success of the highly active anti-retroviral therapy (HAART) against HIV13 and the recent advancements in HCV treatment.46
The influenza virus HA protein is a homotrimer, in which each monomer is composed of two disulfide-linked subunits named HA1 and HA2. HA1 forms the head region of the protein, which is primarily involved in the receptor binding function of HA, whereas HA2 is referred to as the stem region and is involved in the virus–host membrane fusion process. Following viral binding and uptake into the endosome, the low-pH environment triggers irreversible conformational changes within the HA. The hydrophobic fusion peptide of the HA is exposed from its buried position and is followed by several structural rearrangements of the HA2, which ends in fusion of the host and viral membranes. This membrane fusion facilitates the release of viral ribonucleoproteins (RNPs) from the virion into the cytoplasm. Upon release they are trafficked to the nucleus, where the genome is replicated and transcribed.45
Several small molecules that inhibit these conformational changes required for membrane fusion by the HA protein have been identified in recent years.14−22 These studies have highlighted the HA-mediated membrane fusion process as a relevant target for antiviral development. The obstacle that is frequent among these fusion inhibitors is that they are all limited by subtype specificity. There are currently 18 known HA subtypes of influenza A virus,23 which can be divided into two broad phylogenetic groups: group 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, and H18) and group 2 (H3, H4, H7, H10, H14, and H15). Most of the inhibitors that target HA are restricted to blocking fusion of either group 1 or group 2 HA proteins. The fact that current seasonal circulating influenza viruses include both group 1 (H1) and group 2 (H3) makes it important that a future clinical inhibitor has pan-subtype efficacy or that both group 1 and group 2 inhibitors are included in a drug cocktail. Discovery of new inhibitors of this class and further understanding of their collective mechanisms are key to developing effective therapies.
In a recent screen of 919,960 compounds we have found several potent influenza-specific inhibitors of viral replication with submicromolar IC50 concentrations. One of these compounds, which we have termed S20, was determined to be an inhibitor of HA-mediated membrane fusion for group 1 influenza A viruses. In this study we explore the mechanism of action of this inhibitor in depth and propose its continued development to expand efficacy against all influenza A viruses.
Results and Discussion
Identification of Potent Influenza Virus Inhibitors Using a Cell-Based High-Throughput Screen Assay
A cell-based assay that allows for multicycle influenza virus replication, and thus the ability to capture inhibitors of all steps in the viral life cycle, was developed for the purposes of a high-throughput screen (HTS). This assay used a recombinant influenza A/WSN/33 virus that had been engineered to express Renilla luciferase27 (Figure 1a). Briefly, the open-reading frame for the viral HA gene was replaced with that of Renilla luciferase. The packaging signals of the HA segment were maintained to ensure that the recombinant segment is incorporated into progeny virions. As this WSN-Renilla virus lacks the ability to express HA, it can undergo multicycle replication only in an HA complementing cell line (MDCK-HA). For the HTS assay MDCK-HA cells were plated in solid white 1536-well plates and incubated overnight at 37 °C (Figure 1b). Two hours prior to infection, media containing library or control compounds (2 μM in DMSO) were added to each well. Cells were then infected with the WSN-Renilla virus at a multiplicity of infection (MOI) of 0.05 and infection was allowed to proceed for an additional 30 h. Renilla luciferase activity was determined using a luminescent readout, which served as a measurement of viral replication. The luminescent signal from the wells containing library compounds was compared to those containing positive controls (ribavirin and oseltamivir) and DMSO as the negative control. As an NA inhibitor, oseltamivir affects the very last stage of the viral life cycle, the release step. Thus, inhibition by oseltamivir can be seen only in the context of multicycle infections, so its inclusion and effectiveness ensured that multicycle replication was occurring in our assay and that inhibitors of the release step, as well as all prior steps, could be detected. Ribavirin, which is a broad-spectrum inhibitor of RNA viruses and targets polymerase function, showed an even larger window of inhibition because it targets an earlier step in the life cycle. The screen assay was determined to have a Z′ factor between 0.4 and 0.6, a CV ranging from 10 to 20%, and a signal window of 5–7-fold, indicating that it was robust and would yield relevant hits.
Figure 1.

High-throughput screen design and execution: recombinant influenza virus and high-throughput screen design and results. (a) Schematic of the recombinant influenza A/WSN/33 virus expressing Renilla luciferase. The Renilla luciferase open reading frame was inserted in the reverse orientation and complementary sense between the 3′ and 5′ packaging sequences of the HA segment, which contains the viral promoter and ensures correct packaging of the recombinant segment. Due to the lack of HA ORF, this WSN-Renilla virus must be grown in an HA-complementing cell line. Upon infection, the influenza virus polymerase recognizes the promoter and the reporter gene is transcribed and expressed. (b) MDCK-HA cells were plated in 1536-well plates and infected with WSN-Renilla virus (MOI = 0.05). Compounds were added 120 min prior to infection, and expression of Renilla luciferase was assayed 30 h later. A 50% reduction in luminescence signal was employed as a cutoff. (c) Results from the HTS of 919,960 compounds indicating the number of primary hits, the hits confirmed in dose–response, and the selection of hits for revalidation.
In this study, 919,960 compounds from the academic library at the Genomics Institute of the Novartis Research Foundation (GNF) were screened. The GNF Academic Screening Collection consists of a collection of compounds that were selected after applying proprietary algorithms designed to select for optimal compound properties and eliminate undesirable functional groups The screen yielded 4582 hits (0.5% hit rate) that met the cutoff of 50% inhibition (Figure 1c). These hits where then tested in an 8-point dose response confirmation screen in 96-well format in the presence of reporter virus or in the absence of virus for a cytotoxicity counter-screen. Seven hundred and forty-four compounds were confirmed with an IC50 < 2 μM and CC50 > 2 μM. Structural analysis of the validated hit compounds revealed that they could be clustered into 420 groups based on distinct structural scaffolds. Of these groups, 360 were composed of a single compound with no structural relatives within the screen hits, whereas the other 60 groups contained between 2 and 31 related small molecules. The most potent compound from each scaffold was then reordered from commercial vendors and revalidated with the reporter virus assay. Of the 420 compounds, 250 were verified for activity and these were next tested in an 8-point dose response assay against wild type influenza A/WSN/33 virus in non-HA-expressing MDCK cells. Viral titers were assessed initially through hemagglutination assay and then by plaque assay. Compounds that showed significant antiviral effects were then reassessed for cytotoxicity to compile a set of compounds with IC50 values <1 μM, selective indices (SI = CC50/IC50) >40, and maximum viral titer reductions of ≥1 log.
Whereas most previous screens have relied on indirect readouts such as cytopathic effect or have utilized mini-replicon systems that exclude the entry and egress stages of the viral life cycle, our luciferase reporter virus, in combination with MDCK cells stably expressing the HA protein, provides a direct readout of viral gene expression in the context of the entire replication cycle and therefore is capable of identifying compounds such as the egress inhibitor oseltamivir. Using this system we identified 21 compounds that inhibit influenza A/WSN/33 replication as well as or better than oseltamivir in cell culture.
S20 Is a Specific Inhibitor of Influenza A Virus
One of the most potent compounds to emerge from our secondary screening and validation assays was designated S20 (structure indicated in Figure 2a). Compound S20 potently inhibited viral replication of the wild type WSN and WSN-Renilla viruses in MDCK or MDCK-HA cells, respectively. When tested against wild type WSN in the physiologically more relevant A549 (human lung epithelial) cells, even greater inhibition of viral titers was detected with an IC50 value of 80 nM and a maximum reduction in viral titer of >3 logs (Figure 2b). Cytotoxicity remained low in A549 cells with a CC50 value of 40 μM, yielding an impressive SI of 500. S20 also showed activity against other influenza A viruses such as A/California/04/09 (H1N1) and A/Vietnam/1203/04 (H5N1), but viruses of the H3N2 subtype appear to be naturally resistant to the inhibitory effects of S20 (Table 1). S20 also failed to impact the replication of influenza B virus and vesicular stomatitis virus, indicating that it is a specific inhibitor of influenza A viruses, particularly the H1N1 and H5N1 subtypes.
Figure 2.

S20 has potent anti-influenza activity: compound S20 and its antiviral activity against influenza A virus. (a) Chemical structure of compound S20 and its molecular weight (MW). (b) A549 cells were infected with influenza A/WSN/33 virus (MOI = 0.01) in the presence of increasing concentrations of compound S20. Viral titers were determined at 24 h postinfection (left-hand scale, solid line) and the IC50 and IC90 calculated. Cell viability was determined independently for a 24 h incubation period (right-hand scale, dashed line), and the CC50 and CC10 were calculated. Means of three replicates ± SD are shown.
Table 1. Breadth of S20 Antiviral Activitya.
| virus strain | IC50 (μM) | 95% CIb | IC90 (μM) | 95% CIb |
|---|---|---|---|---|
| A/WSN/1933 (H1N1) | 0.08 | 0.02–0.16 | 0.2 | 0.01–0.8 |
| A/Puerto Rico/8/1934 (H1N1) | 0.8 | 0.18–3.35 | 2.5 | 1.38–3.9 |
| A/California/04/2009 (H1N1) | 0.15 | 0.02–0.37 | 1.8 | 0.93–2.76 |
| A/Vietnam/1203/2004 (H5N1) | 0.08 | 0.01–0.24 | 4 | 1.7–6.1 |
| A/Hong Kong/1/1968 (H3N2) | >10 | NA | >10 | NA |
| A/Panama/2007/1999 (H3N2) | >10 | NA | >10 | NA |
| A/Wyoming/03/2003 (H3N2) | >10 | NA | >10 | NA |
| A/Victoria/361/2011 (H3N2) | >10 | NA | >10 | NA |
| A/Perth/16/2009 (H3N2) | >10 | NA | >10 | NA |
| B/Yamagata/16/1988 | >20 | NA | >20 | NA |
| vesicular stomatitis virus | >20 | NA | >20 | NA |
Inhibitory potencies of S20 against a range of influenza A virus subtypes, influenza B virus, and VSV. All infections were performed in A549 cells, with the exception of influenza B virus infection, which was performed in MDCK cells.
95% confidence intervals reported as (lower limit – upper limit).
S20 Inhibits Influenza Entry and Targets the HA Protein
To determine whether S20 acts at an early or a late stage of the viral life cycle, time of addition studies were conducted at an MOI of 1 with S20 treatments at −2, 0, 2, 4, 6, and 8 h relative to infection. S20 inhibited virus growth only when added prior to infection, indicating that the compound is likely acting during one of the early entry steps in the life cycle (Figure 3a). This correlates well with the lack of activity observed for S20 in the influenza A mini-genome system, which reconstitutes only influenza transcription/replication and would not detect inhibitors of viral entry or egress (data not shown). Next, we employed a pseudotyped particle assay to determine if S20 directly inhibits HA-mediated entry. This system utilizes a replication deficient HIV provirus expressing gaussia luciferase that was pseudotyped with either influenza virus HA and NA or the VSV G protein as a control.27 As indicated in Figure 3b, S20 potently inhibited entry of the particles pseudotyped with HA/NA while having no effect on the VSV control. As expected, the replication inhibitor A3, which targets the cellular protein DHODH,30 had no effect on the pseudotyped particle entry, indicating that this was a specific effect of S20.
Figure 3.

S20 inhibits at early times during infection and is effective against HA-mediated viral entry. (a) Time-of-addition assay for inhibition of influenza A/WSN/33 virus by S20. A549 cells were infected with influenza virus A/WSN/33 (MOI = 1). Compound S20 was present in the culture medium 2 h before infection or added to the medium at the indicated time points postinfection at its CC10 concentration. Viral titers were determined 24 h postinfection by plaque assay. The assay was performed in triplicate; results are presented as the mean ± SD. (b) Inhibitory activity of S20 at the indicated concentrations on infection of A549 cells with luciferase-expressing lentiviruses pseudotyped with VSV-G or WSN HA and NA. The non-entry inhibitor A3 has no effect on the pseudotyped particle system and served as a negative control. The samples were tested in triplicate, and the data are presented as the mean ± SD.
In an attempt to determine whether S20 is targeting a viral protein, we selected for S20 resistant influenza viruses. Briefly, influenza A/WSN/33 virus was passaged in A549 cells at an MOI of 0.01 in the presence of the maximum concentration (0.5 μM) of S20 that yielded enough virus for subsequent passages at the same MOI. Following four passages under these conditions, resistant viruses were isolated in seven independent experiments, with three virus plaques purified from each and submitted for complete sequencing of the viral genome. Fourteen of the 21 sequenced viruses contained mutations, revealing 10 single amino acid changes (Figure 4a). All of these mutations were in the HA gene, indicating that this is the likely target of S20. Eight of 10 of these mutations occurred either within or surrounding the large “B” loop structure, which connects the large and small α-helices of the HA2 subunit. This B loop in HA2 is thought to be the “spring” in the proposed spring-loaded mechanism of HA-mediated membrane fusion due to its high propensity for helical conformation.31 We found that these resistance-associated residues were conserved among HAs from group 1, but not group 2 (Figure 4b), offering a possible explanation for the group 1 specificity of S20. Within the loop itself there were three mutations (A65 V, T61I, and M59I, using WSN HA2 amino acid numbering), the large α-helix had one mutation (K75R) adjacent to the top of the loop, and two mutations were found in the small α-helix (V52I and N50S) near the bottom of the loop. Interestingly, two of the mutations occurred within the HA1 subunit (K321T and M330 V). Although distant within the primary sequence, these mutations in HA1 align perfectly with the loop region of HA2, further highlighting this region as likely important for S20 inhibition. Two other mutations occurred within HA2, but outside this loop region (N114S and V122A). These mutations within the stalk that are not localized near the B loop could possibly cause resistance to S20 by decreasing the stability of the prefusion conformation of HA, therefore allowing fusion to be triggered more easily and bypassing S20 inhibition. Indeed, a mutation at position 114 of HA2 has previously been shown to allow fusion to occur at higher pH,35 suggesting destabilization of the native HA conformation. To confirm resistance, the M59I mutation was introduced into a recombinant influenza A/WSN/33 virus using reverse genetics.28 The virus rWSN-M59I was not impaired for growth compared to a wild type recombinant virus under multicycle conditions (data not shown), but did show resistance to S20 inhibition with a 20-fold higher IC50 value compared with WT rWSN (Figure 4c), confirming the importance of this residue for S20 antiviral activity. A compound structurally related to S20, MBX2546, was recently reported to inhibit fusion in a similar manner to what we propose for S20 in this work, but no resistance mutation was identified in that study.21 In Figure 4d, we show that the M59I mutation, which falls within the region predicted to bind to MBX2546,21 also causes resistance to that compound. When the M59I mutation was analyzed within the pseudotyped particle entry assay, a similar resistance phenotype was observed (Figure 4e).
Figure 4.

Mutations within the HA protein cause resistance to the inhibitory effects of S20. (a) Crystal structure of A/PR/8/34 HA (PDB 1RU7) protein showing the HA1 (green) and HA2 (silver) subunits. Residue positions where S20 escape mutations occurred are indicated in orange (HA1) and yellow (HA2). (b) Sequence alignment of group 1 and 2 HA proteins within the region of highest escape mutation density. Positions of escape mutations are shaded. WSN amino acid numbering is used in both panels a and b. Virus titers from A549 cells infected with either rWSN-WT or rWSN-HA/M59I viruses (MOI = 0.01) were treated with increasing concentrations of (c) S20 or (d) MBX2546 for 24 h. Curves represent means of triplicate values ± SD. IC50 and IC90 values are indicated. (e) S20 inhibition of VSV-G, WSN HA/NA, or WSN HA-M59I/NA containing pseudotyped particles in A549 cells. The non-entry inhibitor A3 was used as a negative control. The samples were tested in triplicate, and the data are presented as the mean ± SD.
S20 Stabilizes the HA Prefusion Conformation and Inhibits HA-Mediated Membrane Fusion
The resistance mutations within the HA stalk indicated that HA-mediated fusion may be affected by S20. The B loop structure and the surrounding region within HA2 in which the S20 resistance mutations occur have been extensively studied. A conformational change from a loop to a helical structure is a critical step in HA-mediated membrane fusion,32,33 facilitating fusion of the viral and endosomal membranes during influenza virus entry. To determine whether S20 was inhibiting the HA membrane fusion machinery, we employed a hemolysis assay, which measures the ability of influenza virus to induce membrane fusion and lysis of red blood cells under low-pH conditions. In this assay S20 displayed potent inhibition of fusion induced at a pH of 5 (Figure 5a), whereas the rWSN-M59I virus was resistant to this effect and appeared similar to the negative control. Previously, we had observed S20 to have subtype specificity for influenza A viruses with group 1 HAs (H1N1 and H5N1) while having no appreciable effect on viruses with group 2 HAs (H3N2). To confirm this specificity, the ability of S20 to inhibit the fusion of a virus with a group 2 HA was tested. No effect of S20 on influenza A/Perth/16/2009 (H3N2) fusion was observed in this assay at a maximum concentration of 50 μM (data not shown).
Figure 5.

S20 blocks HA-mediated membrane fusion in a pH-dependent manner. After mixing a suspension of chicken erythrocytes with (a, b) rWSN-WT virus or (c) rWSN-HA/M59I virus on ice, S20 was added at the indicated concentrations. The mixture was then acidified with a pH of either (a) 5 or (b, c) a range from 4.8 to 6. The suspension was then incubated at 37 °C for 30 min and assayed at 340 nM for NADH released from the erythrocytes, as a measurement of fusion. Data are expressed as percentage relative to the DMSO control and means of triplicates ± SD are shown. (d, e) Trypsin sensitivity assay showing S20 protection of purified (d) WT HA but not (e) HA-M59I from trypsin digestion in a pH-dependent manner. Purified A/WSN/33 HA was incubated with DMSO (D) or S20 at the CC10 concentration for 15 min at 31 °C prior to acidification to the indicated pH. The mixture was neutralized to a final pH of 7.4 and treated with trypsin for 30 min at 37 °C. The extent of trypsin cleavage was analyzed on a gradient SDS-PAGE gel and visualized with Coomassie staining. Trypsin digestion of HA at neutral pH (pH 7.4) was used as a control and is shown in the first lane.
We were then interested in testing the pH dependence of S20 fusion inhibition, so we varied the pH of fusion activation from 6 to 4.8 in the presence of DMSO or 20 μM S20. Under these conditions we observed S20 inhibition of fusion from pH 6 down to 5. When the pH was lowered to 4.8, inhibition by S20 was lost and fusion was identical to that of the DMSO control (Figure 5b). Of note, the rWSN-M59I virus showed no difference in the optimal pH for fusion whether in the presence of DMSO or S20 (Figure 5c). To investigate whether S20 was inhibiting fusion by directly blocking the low-pH conformational change of HA, a trypsin digestion assay was used, which detects exposure of cryptic trypsin cleavage sites in HA during the prefusion to postfusion conformational change. As indicated in Figure 5d, the purified baculovirus expressed HA was protected from trypsin digestion at pH 5 when in the presence of S20, but this effect was lost at lower pH, similar to observations in the fusion experiments. The M59I mutant HA showed minimal protection by S20 at a pH of 5, again providing evidence for the central role of this residue in S20 activity. Of the possible modes of inhibiting HA-mediated membrane fusion, the most common is prevention of HA refolding into its postfusion conformation. Alternatively, a fusion inhibitor can also function via premature triggering of the fusion machinery and therefore irreversibly block the process. The fact that S20 inhibition can be overcome at pH 4.8 indicates that it is stabilizing the prefusion conformation of HA, and this is supported by the results of a tryptic digestion assay, where S20 protected HA from conversion to its trypsin-sensitive conformation under low-pH conditions.
S20 Binds to the Influenza HA Protein
The S20 resistance mutations that were selected within the influenza HA protein are suggestive of a direct interaction between the compound and protein. To examine this further, we used biolayer interferometry (BLI) to detect binding of S20 to the WSN HA protein. BLI is a technique that detects real-time binding of small molecules to an immobilized protein through shifts in interference patterns of white light passed through the biolayer containing the protein.34 We found that S20 binds to baculovirus-expressed and purified WSN HA in a dose-dependent manner with a calculated Kd of 5.29 μM (Figure 6a). As a negative control, ribavirin was tested at 5-fold higher concentrations, and no binding to HA was observed (data not shown). When binding to purified WSN HA containing the M59I mutation was tested, we found that S20 had a 6-fold lower affinity with a Kd of 30.9 μM, suggesting that this loop region of the stalk is the site of interaction. Whereas the loss of binding was small with the mutant HA (6-fold), it is in line with the finding that the HA/M59I recombinant virus showed only a moderate resistant phenotype with 20-fold lower sensitivity to S20. Mutations within this region that cause a greater loss in binding activity would likely yield more impressive resistance profiles, but this may also be accompanied by a decrease in viral fitness. When tested against the Perth/09 HA, an H3 that is resistant to S20 inhibition, no binding of S20 was observed. As a positive control, we used the compound tert-butylhydroquinone (TBHQ), previously described in the literature to bind to and inhibit group 2 HAs.20 TBHQ bound to Perth/09 HA with a Kd of 5.3 μM, indicating the protein was of proper quality for binding experiments (data not shown). The binding data best fit to a 2:1 binding model, suggesting two binding modes for S20 on HA. The secondary association event that was detected through BLI probably reflects nonspecific binding, which is unrelated to the antiviral activity of S20. We believe this to be true because the affinity of the secondary binding is 2 orders of magnitude lower than that of the primary interaction (Figure 6a). Also, this secondary binding is unchanged in the presence of the S20 resistance mutation M59I, further suggesting that it is unrelated to the antiviral activity of S20.
Figure 6.
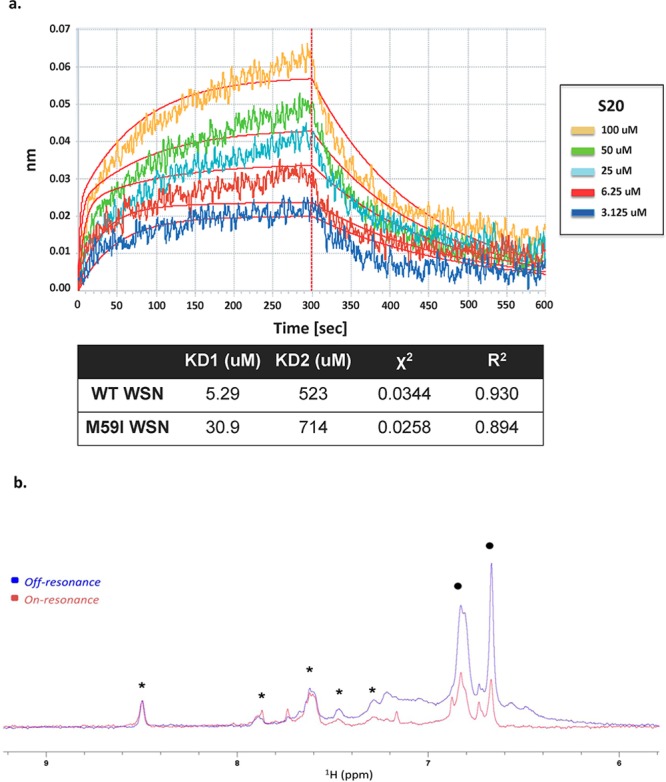
S20 directly binds to the influenza virus hemagglutinin protein. (a) Biolayer interferometry was used to assay the binding of the small molecule S20 to purified WSN HA protein. The association and dissociation curves of increasing concentrations of S20 binding to WT WSN HA are shown. A negative control of buffer (PBS, 0.1% Tween-20, 10% DMSO) containing no HA was run for each experiment. The affinity of S20 for HA–WT and HA–M59I was calculated and represented as two separate dissociation constants for the high-affinity specific interaction (KD1) and the low-affinity nonspecific binding (KD2). A χ2 of 0.0344 and an R2 of 0.93 indicate that the binding data fit to this 2:1 binding model (i.e., two binding events on HA). (b) NMR binding of S20 to WT WSN HA via saturation transfer difference (STD). Overlay of the aromatic region of on-resonance (0.5 ppm, red) and off-resonance (20 ppm, blue) 1H NMR spectra of 75 μM compound S20 (resonances between 6.6 and 7.0 ppm, black dots) and 75 μM of a unrelated small molecule used as negative control (resonances between 8.5 and 7.25 ppm, asterisks) in PBS, pH 7.4 (+10% D2O) in the presence of 7.5 μM WT WSN HA. Selective attenuation (saturation) of S20 protons (indicated with dots) is evident, whereas the resonances of the negative control compound (indicated with asterisks) appear unperturbed.
To further confirm direct interactions between S20 and HA, we performed NMR saturation transfer difference experiments (STD).48,49 In this experiment, selective irradiation of the aliphatic hydrogen nuclei is achieved by a train of selective pulses centered at 0.5 ppm (see Methods). This selective irradiation causes the saturation of surrounding protein hydrogen nuclei via spin diffusion. Ultimately, by the same principle, the saturation is transferred to the hydrogen nuclei of the bound ligand, and the effect manifests itself in the attenuation of the signal intensity of the compound resonances. The STD NMR spectra of S20 in the presence of wild type WSN HA are reported in Figure 6b, where the attenuation of the S20 signals in the irradiated protein spectrum is evident compared to a negative control molecule. Binding was also observed to the M59I mutant WSN HA in this assay, in agreement with our BLI data (data not shown). Taken together, this suggests that S20 blocks fusion via a specific physical interaction with group 1 influenza HA proteins and that resistance mutations decrease the affinity of this interaction.
In Silico Docking of S20 to the HA Crystal Structure
The crystal structure of the PR8 HA protein was loaded into MOE (Molecular Operating Environment, version 2013.08). Docking simulations of S20 were performed with the MOE-dock system and allowed the entire HA trimer to be considered for possible binding sites. Induced fit modeling was employed to allow movement of the protein during refinement to simulate the dynamic molecular environment. The affinity scoring function London dG was used to assess and rank the receptor–ligand complexes. The top-ranked docking score is shown in Figure 7. The predicted binding pocket is formed by the large α-helix and loop of HA2 and a loop consisting of amino acids 300–310 of HA1. This correlates well with our S20 escape mutants (residues indicated in Figure 7a), which highlighted the HA2 loop region as being important for S20 inhibition. Two of the residues, from both HA1 (K321) and HA2 (A65), which when mutated cause S20 resistance, form part of the surface of this predicted binding pocket (Figure 7b). S20 is predicted to form a hydrogen bond of 1.66 Å with residue Q62 of the HA2 loop (see alignment in Figure 4B), which is stabilized by a hydrogen bond that forms between Q62 and K321 in HA1. Furthermore, in silico mutagenesis studies indicate that the K321T and A65 V mutations, which directly form the predicted binding pocket, would cause significant losses (Δaffinity = +1.41 and 0.95, respectively) in the affinity of S20 for the pocket. The mutation M59I caused a smaller loss in affinity (Δaffinity = +0.20) in line with the small loss of affinity observed via our BLI experiments. All other resistance mutations caused no predicted loss of affinity for S20, indicating that they are not involved in the binding pocket, but more likely causing decreases in stability of the prefusion conformation of HA. Finally, docking of S20 with the compound-sensitive HA from A/Vietnam/1203/04 (H5N1) (PDB 2FK0) protein yielded a similar binding pocket within the HA2 loop structure, whereas attempts to model the compound with the S20-resistant HA from A/Aichi/2/68 (H3N2) (PDB 2YPG) yielded no predicted binding pockets within this region. Therefore, the presence of this predicted binding pocket correlates with virus sensitivity to S20 inhibition.
Figure 7.

In silico docking and predicted binding pocket of S20 within the HA protein structure. (a) Location of the S20 docking site in the HA trimer in the simulated three-dimensional structure of the A/PR/8/34 HA (PDB 1RU7) with HA1 in green and HA2 in silver. Residues where S20 escape mutations occurred are indicated in orange (HA1) and yellow (HA2). The Q62 residue, which forms a predicted hydrogen bond with S20, is highlighted in light green. (b) The surface of the docking pocket is presented with blue indicating positive charge and red indicating negative charge; escape mutation residues, which help form the pocket, are represented by their corresponding color.
This loop in the prefusion HA structure undergoes a radical conformational change during low-pH treatment to the final postfusion conformation in which it assumes an α-helical structure to act as the “spring” that allows insertion of the fusion peptide into opposing membranes.31 This conformational restructuring is critical for both the extension of the fusion peptide into the opposing membrane and the eventual collapse of HA, via a coiled-coil structure, to bring the two membranes within proximity for fusion to occur. S20 association with this region could stabilize the prefusion conformation and prevent these necessary structural changes. The region of HA1 close to the HA2 loop structure is believed to act as a “clamp” to prevent the triggering of the “spring” in the spring-loaded mechanism of HA-mediated membrane fusion.36,37 Therefore, another possibility would be that the interaction of S20 with residues from both HA1 and HA2 may prevent the HA1 “clamp” from moving away from the stalk region, which is a requirement for the early steps of HA-mediated fusion. We were unable to model S20 into the H3 structure; however, HA fusion inhibitors that are specific for group 2 HAs have been reported and are predicted to bind to a pocket near the same loop region in the group 2 HA structure. Mutations in the small α-helix adjacent to the loop of an H3 HA (E57K) render the virus resistant to the fusion inhibitor 4c,19 and a similar compound, TBHQ,20 was shown to interact with E57 through a cocrystal structure. Interestingly, several other structurally unrelated group 1-specific fusion inhibitors select for resistance mutations in both the small and large α-helices surrounding the stalk loop region.14,15,17 During preparation of this paper a group 1-specific compound, MBX2546, was published that has a chemical structure similar to that of S20.21 This compound was found to compete for binding to HA with an antibody that is known to associate with the small α-helix and loop region of the stalk.38 Resistance mutations were not well-defined in that study, but here we report that mutations that confer S20 resistance also confer resistance to MBX2546 and in silico docking places MBX2546 in the same pocket as for S20.
Conclusion
Subtype specificity appears to be a hallmark (and a major hurdle for development) of small molecule fusion inhibitors that target this region of the HA protein. Further investigation of S20, and other group 1-specific inhibitors in conjunction with the group 2-specific compounds, could lead to a greater understanding of this inhibition and future development of pan-subtype small molecule fusion inhibitors. It should be noted that the region identified as the binding site for S20, and other HA fusion inhibitors, overlaps with the epitopes of broadly neutralizing HA antibodies39−42 that, like the small molecules, are either group 1- or group 2-specific. Nevertheless, there are massive efforts underway to design vaccines that elicit such antibodies with the goal of developing a “universal” influenza vaccine,43,44 which would protect over many years as opposed to the current seasonal vaccine. With antiviral drugs, it is likely that future influenza therapies will use a combination of virus-specific compounds to increase efficacy and decrease incidents of viral resistance, much like HAART for treatment of HIV infections. Therefore, it is possible that two subtype specific fusion inhibitors could be paired with a drug targeting a disparate stage of the viral life cycle, such as oseltamivir, to produce an effective triple therapy.
In conclusion, we have found a group 1 influenza A virus specific inhibitor that targets HA-mediated viral-host membrane fusion. This inhibition is most likely due to S20 directly binding to the stalk loop region of the HA protein and stabilizing it from the acid-induced conformational change required to undergo fusion. Future studies aimed at obtaining a cocrystal of the S20–HA complex will add to the understanding of structural differences between group 1 versus group 2 fusion inhibitors and perhaps lead to the design of more effective drug-like candidates in the future.
Methods
Cell Culture and Reagents
Madin–Darby canine kidney (MDCK) epithelial cells, human alveolar epithelial (A549) cells, and human embryonic kidney 293T (293T) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Production of the MDCK cell line stably expressing the WSN HA protein was previously described.24 MDCK, A549, and 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, South Logan, UT, USA) and 1% penicillin–streptomycin (P/S) (Gibco). All cells were grown at 37 °C and 5% CO2. Transfection of DNA was performed in Opti-MEM I-reduced serum medium (Opti-MEM) (Gibco) with Lipofectamine LTX (Invitrogen) in A549 cells according to the manufacturer’s specifications. For measurement of luciferase production in reporter assays, the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) was used. The Renilla-Glo Luciferase Assay System was used in the high-throughput primary screen and confirmation screen (Promega).
Expression Plasmids and Cloning
The pRL-TK (Promega) reporter contains a Renilla luciferase gene under the regulation of the herpes simplex virus thymidine kinase promoter. The influenza A virus minigenome reporter (pPolI NP_Luc) was generated as previously described.25 The influenza virus rescue plasmid pPolI-HA M59I was generated by exchanging one nucleotide in the parental plasmid pPolI-HA using the QuickChange site-directed mutagenesis kit (Agilent Technologies, Wilmington, DE, USA) using specific primers (forward 5′-ctctgttatcgagaaaataaacactcaattcacagctgtgg-3′; reverse: 5′-ccacagctgtgaattgagtgtttattttctcgataacagag-3′). The presence of the mutation was confirmed by sequencing (Macrogen, Rockville, MD, USA). The mammalian expression vector pCAGGS containing a chicken β-actin promoter has been previously described.26 The expression plasmid encoding the HA M59I mutant was generated by subcloning from pPolI HA M59I into pCAGGS-HA using ClaI and NheI enzymes (New England Biolabs, Ipswich, MA, USA). Proper insertion and presence of the mutation were confirmed by sequencing (Macrogen).
Viruses
The WSN-Renilla virus construction was previously described.27 The influenza A/WSN/1933 (H1N1) virus (WSN) was propagated in MDCK cells for 2 days at 37 °C. Influenza A/California/04/2009 (H1N1) virus was propagated in MDCK cells for 3 days at 35 °C. Influenza viruses A/Puerto Rico/8/1934 (H1N1) (PR8), A/Hong Kong/1/1968 (H3N2), A/Panama/2007/1999 (H3N2), A/Perth/16/2009 (H3N2), A/Victoria/361/2011 (H3N2), A/Wyoming/03/2003 (H3N2), and A/Vietnam/1203/2004 (H5N1) bearing a mutated polybasic cleavage site in the HA segment (HAlo) were propagated in 10-day-old embryonated chicken eggs for 2 days at 37 °C. Influenza B/Yamagata/16/1988 virus was propagated in 8-day-old embryonated chicken eggs for 3 days at 33 °C. All influenza viruses were titered by standard plaque assay in MDCK cells.19 Vesicular stomatitis virus was grown and titered by plaque assay in VERO cells. Recombinant influenza viruses were generated using the influenza virus rescue protocol as previously described.28 Briefly, 293T cells were transfected with eight pPolI constructs expressing the PB1, PB2, PA, NP, HA (or HA M59I), NA, M, and NS genomic segments as well as pCAGGS expression plasmids encoding the PB1, PB2, PA, and NP proteins. Twenty-four hours post-transfection, MDCK cells were cocultured with the transfected 293Ts for an additional 24–48 h, until cytopathic effects were observed. Newly generated viruses were collected and plaque-purified, and the presence of the mutation was confirmed by sequencing.
Small Molecular Weight Compounds
Compounds for the high-throughput screen were from the academic library at the Genomics Institute of the Novartis Research Foundation (GNF) (San Diego, CA, USA). For secondary analyses, hit compounds were purchased from the vendors indicated through eMolecules (La Jolla, CA, USA) and dissolved in 100% DMSO. The final concentration of DMSO in the culture medium did not exceed 0.5%.
High-Throughput Small Molecule Screen Assay
The high-throughput small molecule screen was performed in 1536-well microplate format. MDCK cells stably expressing hemagglutinin (MDCK-HA) were cultured to 80–90% confluency, washed with phosphate-buffered saline (PBS, Life Technologies), trypsinized with 0.05% Trypsin-EDTA (Life Technologies), and resuspended in 1× DMEM (Life Technologies) supplemented with 10% FBS (Life Technologies), 1% penicillin–streptomycin/glutamine (P/S/G), and 0.15% sodium bicarbonate (Life Technologies). The cells were then pelleted and resuspended in DMEM containing 1% FBS, 0.3% bovine albumin (Sigma), 20 mM HEPES, and 1% P/S/G to a cell density of 5 × 105 cells/mL. Using an automated high-throughput screening system (GNF Systems), 4 μL of the diluted MDCK-HA cells was dispensed into 1536-well plates (2000 cells/well) and incubated at 37 °C and 5% CO2 overnight. Compounds were then added to each well (10 nL) using a pintool-equipped automated transfer system (GNF Systems) to a final concentration of 2 μM and a final DMSO concentration of 0.2% and incubated at 37 °C and 5% CO2 for 2 h. Next, the cells were infected with 1 μL of recombinant WSN-Renilla luciferase virus (WSN-Ren) at an MOI of 0.05 and incubated at 37 °C and 5% CO2. Thirty hours postinfection, 2 μL of Renilla-Glo (Promega) was added to each well, and Renilla luciferase activity was measured with a ViewLux uHTS Microplate Imager (PerkinElmer). For data analysis, on each plate, the last four columns were reserved for positive (ribavirin, oseltamivir) and negative (DMSO) controls. For each plate, original signal readings were divided by plate median; that is, signals from positive controls were around zero and plate medians were one after normalization. Wells with activity below a cutoff value 0.5 corresponded to >50% inhibition and were designated as hits.
Cell Viability Assay
The CellTiterGlo Cell Viability Assay (Promega) was used to detect ATP levels as a function of cell viability, according to the manufacturer’s specifications. A549 cells were seeded into 96-well plates (1250 cells/well) and incubated at 37 °C and 5% CO2 for 24 h. Culture medium was then replaced with 100 μL of fresh medium containing compound (serially diluted), and this was further incubated for 24 h. Cell viability was measured by adding 50 μL of CellTiterGlo reagent to each well, and the luminescence signal was read using a plate reader (Beckman Coulter, Brea, CA, USA).
Viral Growth Assays in the Presence of Inhibitors
A549 cells (100,000) were seeded into 24-well plates and incubated for 24 h at 37 °C and 5% CO2. Two hours before infection, the medium was replaced with DMEM containing the compound of interest at the indicated concentrations. Compounds were absent during the 1 h virus incubation but were present in the DMEM postinfection medium. Infections were performed at a low MOI (0.01–0.1) for 24 or 48 h, depending on virus used. For infections with influenza viruses, postinfection medium also contained 1 ug/mL TPCK-treated trypsin (Sigma-Aldrich, St. Louis, MO, USA). The infected cells were incubated at 37 °C with the exception of influenza B virus-infected cells, which were incubated at 33 °C. Viral titers were determined by standard plaque assay in MDCK cells.
Selection of S20-Resistant Influenza Viruses
The concentration of S20 required for maximum virus inhibition (3 logs), while maintaining enough virus production for subsequent passages, was determined (0.5 μM S20). A549 cells were infected with WSN at an MOI of 0.01 for 24 h at 37 °C under S20 treatment. The supernatant was then collected and titered by plaque assay. If the recovered S20-treated virus did not show increased viral titer similar to that of the DMSO-treated control, the virus was passaged again by using the same method. Once increased titers in the presence of S20 were detected for two consecutive passages, the viruses were plaque purified. Following plaque purification, all eight genome segments were sequenced and compared to DMSO-treated control virus to detect escape mutations.
Influenza Virus Mini-genome Assays
For influenza A virus mini-genome reporter assays, A549 cells were transfected with lipofectamine LTX (Invitrogen). Transfections were done in 12-well plates at a lipid/DNA ratio of 3:1 (μL/μg). Seventy-five nanograms of WSN pPolI NP-LUC reporter, 50 ng of pRL-TK reporter, 50 ng of WSN PB1, WSN PB2, and WSN PA expression plasmids, and 100 ng of WSN NP expression plasmid (or empty vector for negative control) were cotransfected in 100 μL of Opti-MEM. Incubation of lipid and DNA was done at room temperature for 30 min prior to addition of the transfection complex directly to cells containing DMEM supplemented with S20 or DMSO. Twenty-four hours post-transfection, cells were lysed and luciferase production was measured with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s specifications.
Hemolysis Inhibition Assay
Fresh chicken erythrocytes (RBC) were washed twice with PBS and resuspended to make a 2% (v/v) suspension in PBS that was stored at 4 °C until use. One hundred microliters of RBCs in PBS was mixed with an equal volume of virus stock. After incubation of the virus–RBC mixture on ice for 30 min, compound was added at the indicated concentrations, and the sample was incubated for an additional 10 min on ice. To trigger hemolysis, the sample was centrifuged at 3000 rpm for 5 min and resuspended in 200 μL of PBS at the indicated pH values and mixed well. The mixture was incubated at 37 °C for 30 min to allow for the HA acidification and hemolysis to take place. To separate nonlysed erythrocytes, samples were centrifuged again at 3000 rpm for 5 min. One hundred microliters of supernatant was transferred to a flat-bottom 96-well plate, and absorbance at 340 nm was read on a microtiter plate reader.
HA Purification and Trypsin Protection Assay
Baculovirus-expressed HA was purified as previously described.29 To determine if a compound stabilizes HA native structure in an acidic environment, the sensitivity of HA to trypsin digestion was determined in which only conformationally changed HA resulting from a low-pH treatment is cleavable by trypsin. Purified HA (4–6 mg) was incubated with test compound or controls at 31 °C for 15 min. The mixture was adjusted with 0.25 M citrate (pH 4.2) to a final pH of 5.0 and incubated for another 15 min at 31 °C. The pH was then neutralized with 0.25 M Tris-HCl, pH 9.0, to the final pH of 7.5. Two milligrams of trypsin was added to each reaction, and digestion was carried out for 30 min at 37 °C. Trypsin-mediated HA cleavage was visualized on a 10% SDS-PAGE gel that was stained with Coomassie blue G-250.
S20 Biolayer Interferometry Binding Assay
Real-time binding assays between S20 and purified influenza A/WSN/33 virus HA protein were performed using biolayer interferometry on an Octet Red system (Fortebio, Menlo Park, CA, USA). This system monitors interference of light reflected from the surface of a fiber optic sensor to measure the thickness of molecules bound to the sensor surface. Purified baculovirus expressed HA protein was produced as previously described.29 Purified HA was randomly biotinylated with a 1:1 molar ratio using the EZ-Link Micro-PEO4-Biotinylation kit (Pierce/ThermoFisher, Rockford, IL, USA). Excess biotinylation reagent was removed using Zeba Spin Desalting Columns (Pierce/ThermoFisher). Biotinylated HA was coupled to kinetics grade Super Streptavidin high binding biosensors (Fortebio). Sensors coated with HA were allowed to bind to S20 in PBS with 0.1% (v/v) Tween-20 and 10% DMSO at increasing concentrations. Binding kinetics were calculated using the Octet Red software package, which determined the best fit for the observed binding curves and calculated the association rate constants. S20 was allowed to dissociate by incubation of the sensors in PBS with 0.1% Tween-20 and 10% DMSO. Best fit dissociation curves were determined, and the dissociation rate constants were calculated. Binding affinities were calculated as the kinetic dissociation rate constant divided by the kinetic association rate constant.
NMR Spectroscopy
NMR spectra were acquired on a 600 MHz Bruker Avance spectrometer equipped with a 5 mm TCI cryoprobe. All NMR experiments were performed at 298 K, and data were processed and analyzed using TOPSPIN 2.1 (Bruker Biospin Corp., Billerica, MA, USA). Saturation transfer difference (STD) experiments were acquired with a 2 s presaturation time obtained with a train of selective 5 ms IBURP pulses centered at 0.5 ppm or at 20 ppm in the off-resonance experiment.47 The experiments were acquired with 4096 scans, 512 data points, and a spectral window of 12 ppm.
Molecular Docking of S20 to the HA Protein
Docking studies were performed using MOE 2013.08 in the Amber10 force field. The crystal structures of the A/PR/8/34 (PDB 1RU7), A/Hong Kong/68 (PDB ID 2YPG), or A/Vietnam/04 HA (PDB ID 2FK0) proteins were retrieved from the Protein Data Bank (http://www.rcsborg/pdb/home/home.do). To prepare the proteins for docking studies, they were loaded into MOE and all water molecules and heteroatoms were removed. As the protein is a homotrimer, all protein chains were considered for the docking process. The structure was protonated, polar hydrogens were added, and energy minimization was carried out to obtain the stabilized conformation. A docking procedure was followed using the induced-fit protocol implemented in MOE 2013.08. After successful docking, the best energy conformations of receptor–ligand complexes were studied and evaluated to infer the most probable predicted binding site.
Acknowledgments
We thank Dr. Florian Krammer for producing the purified HA proteins and Drs. Thomas Moran and Andrew Duty from the Center for Therapeutic Antibody Development at Mount Sinai for use of the Octet Red system for BLI studies. JChem for Excel was used for structure database management, search, and prediction, JChem for Excel 6.1.1, 2013, ChemAxon (http://www.chemaxon.com). This work was supported in part by National Institutes of Health (NIH) Grants U01 AI1074539, HHSN272200900032C, R21 AI102169, U19 AI106754, U19 AI109946, P01 AI097092, and NIAID CEIRS HHSN272201400008C.
Glossary
Abbreviations
- IC50
half-maximum inhibitory concentration
- CC50
half-maximum cytotoxic concentration
- SI
selective index
- VSV
vesicular stomatitis virus
- DMSO
dimethyl sulfoxide
- DHODH
dihydroorotate dehydrogenase
- BLI
biolayer interferometry
Author Present Address
# (S.S.) Institute of Medical Virology, University of Zurich, 8057 Zurich, Switzerland.
Author Present Address
⊥ (R.K.) Research Group “Host–Pathogen-Interactions,” Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51–59, D-63225 Langen, Germany.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Wang T. T.; Palese P. (2009) Unraveling the mystery of swine influenza virus. Cell 137, 983–985 10.1016/j.cell.2009.05.032. [DOI] [PubMed] [Google Scholar]
- Smith G. J.; Vijaykrishna D.; Bahl J.; Lycett S. J.; Worobey M.; Pybus O. G.; Ma S. K.; Cheung C. L.; Raghwani J.; Bhatt S.; Peiris J. S.; Guan Y.; Rambaut A. (2009) Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- Tang J. W.; Shetty N.; Lam T. T. (2010) Features of the new pandemic influenza A/H1N1/2009 virus: virology, epidemiology, clinical and public health aspects. Curr. Opin Pulm. Med. 16, 235–241 10.1097/MCP.0b013e3283375727. [DOI] [PubMed] [Google Scholar]
- Morens D. M.; Taubenberger J. K.; Harvey H. A.; Memoli M. J. (2010) The 1918 influenza pandemic: lessons for 2009 and the future. Crit. Care Med. 38, e10–20 10.1097/CCM.0b013e3181ceb25b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton B. S.; Whittaker G. R.; Daniel S. (2012) Influenza virus-mediated membrane fusion: determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses 4, 1144–1168 10.3390/v4071144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineberg H. V. (2014) Pandemic preparedness and response – lessons from the H1N1 influenza of 2009. N. Engl. J. Med. 370, 1335–1342 10.1056/NEJMra1208802. [DOI] [PubMed] [Google Scholar]
- Pizzorno A.; Abed Y.; Boivin G. (2011) Influenza drug resistance. Semin. Respir. Crit. Care Med. 32, 409–422 10.1055/s-0031-1283281. [DOI] [PubMed] [Google Scholar]
- Takashita E., Ejima M., Itoh R., Miura M., Ohnishi A., Nishimura H., Odagiri T., and Tashiro M. (2014) A community cluster of influenza A(H1N1)pdm09 virus exhibiting cross-resistance to oseltamivir and peramivir in Japan, November to December 2013 Eurosurveillance 19, article 2, http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20666. [DOI] [PubMed] [Google Scholar]
- Yang J. R.; Huang Y. P.; Chang F. Y.; Hsu L. C.; Huang H. Y.; Pan Y. T.; Lin Y. C.; Wu H. S.; Liu M. T. (2013) Characterization of oseltamivir-resistant influenza A(H1N1)pdm09 viruses in Taiwan in 2009–2011. J. Med. Virol. 85, 379–387 10.1002/jmv.23482. [DOI] [PubMed] [Google Scholar]
- Wang B., Taylor J., Ratnamohan M., McPhie K., Kesson A., Dixit R., Booy R., Hurt A., Saksena N., and Dwyer D. E. (2012) Frequency of oseltamivir resistance in Sydney, during the Newcastle outbreak of community transmitted oseltamivir-resistant influenza A(H1N1)pdm09 virus, Australia, June to August 2011. Eurosurveillance 17, article 4, http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20210. [DOI] [PubMed] [Google Scholar]
- Renaud C.; Kuypers J.; Englund J. A. (2011) Emerging oseltamivir resistance in seasonal and pandemic influenza A/H1N1. J. Clin. Virol. 52, 70–78 10.1016/j.jcv.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Hurt A. C.; Hardie K.; Wilson N. J.; Deng Y. M.; Osbourn M.; Leang S. K.; Lee R. T.; Iannello P.; Gehrig N.; Shaw R.; Wark P.; Caldwell N.; Givney R. C.; Xue L.; Maurer-Stroh S.; Dwyer D. E.; Wang B.; Smith D. W.; Levy A.; Booy R.; Dixit R.; Merritt T.; Kelso A.; Dalton C.; Durrheim D.; Barr I. G. (2012) Characteristics of a widespread community cluster of H275Y oseltamivir-resistant A(H1N1)pdm09 influenza in Australia. J. Infect. Dis. 206, 148–157 10.1093/infdis/jis337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr A.; Amin J. (2009) Efficacy and tolerability of initial antiretroviral therapy: a systematic review. AIDS 23, 343–353(discussion 355–346), 10.1097/QAD.0b013e32831db232. [DOI] [PubMed] [Google Scholar]
- Liu S.; Li R.; Zhang R.; Chan C. C.; Xi B.; Zhu Z.; Yang J.; Poon V. K.; Zhou J.; Chen M.; Munch J.; Kirchhoff F.; Pleschka S.; Haarmann T.; Dietrich U.; Pan C.; Du L.; Jiang S.; Zheng B. (2011) CL-385319 inhibits H5N1 avian influenza A virus infection by blocking viral entry. Eur. J. Pharmacol. 660, 460–467 10.1016/j.ejphar.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Luo G.; Torri A.; Harte W. E.; Danetz S.; Cianci C.; Tiley L.; Day S.; Mullaney D.; Yu K. L.; Ouellet C.; Dextraze P.; Meanwell N.; Colonno R.; Krystal M. (1997) Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J. Virol. 71, 4062–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Li Y.; Li S.; Li H.; Qiu Z.; Lee C.; Lu H.; Lin X.; Zhao R.; Chen L.; Wu J. Z.; Tang G.; Yang W. (2011) Inhibition of influenza A virus (H1N1) fusion by benzenesulfonamide derivatives targeting viral hemagglutinin. PLoS One 6, e29120 10.1371/journal.pone.0029120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi Y.; Igarashi M.; Okamatsu M.; Noshi T.; Sakoda Y.; Yamamoto N.; Ito K.; Yoshida R.; Kida H. (2013) Antiviral activity of stachyflin on influenza A viruses of different hemagglutinin subtypes. Virol. J. 10, 118. 10.1186/1743-422X-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staschke K. A.; Hatch S. D.; Tang J. C.; Hornback W. J.; Munroe J. E.; Colacino J. M.; Muesing M. A. (1998) Inhibition of influenza virus hemagglutinin-mediated membrane fusion by a compound related to podocarpic acid. Virology 248, 264–274 10.1006/viro.1998.9273. [DOI] [PubMed] [Google Scholar]
- Vanderlinden E.; Goktas F.; Cesur Z.; Froeyen M.; Reed M. L.; Russell C. J.; Cesur N.; Naesens L. (2010) Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J. Virol. 84, 4277–4288 10.1128/JVI.02325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. J.; Kerry P. S.; Stevens D. J.; Steinhauer D. A.; Martin S. R.; Gamblin S. J.; Skehel J. J. (2008) Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 105, 17736–17741 10.1073/pnas.0807142105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A.; Antanasijevic A.; Wang M.; Li B.; Mills D. M.; Ames J. A.; Nash P. J.; Williams J. D.; Peet N. P.; Moir D. T.; Prichard M. N.; Keith K. A.; Barnard D. L.; Caffrey M.; Rong L.; Bowlin T. L. (2014) New small molecule entry inhibitors targeting hemagglutinin-mediated influenza A virus fusion. J. Virol. 88, 1447–1460 10.1128/JVI.01225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlinden E.; Naesens L. (2014) Emerging antiviral strategies to interfere with influenza virus entry. Med. Res. Rev. 34, 301–339 10.1002/med.21289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S.; Zhu X.; Li Y.; Shi M.; Zhang J.; Bourgeois M.; Yang H.; Chen X.; Recuenco S.; Gomez J.; Chen L. M.; Johnson A.; Tao Y.; Dreyfus C.; Yu W.; McBride R.; Carney P. J.; Gilbert A. T.; Chang J.; Guo Z.; Davis C. T.; Paulson J. C.; Stevens J.; Rupprecht C. E.; Holmes E. C.; Wilson I. A.; Donis R. O. (2013) New world bats harbor diverse influenza A viruses. PLoS Pathog. 9, e1003657. 10.1371/journal.ppat.1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh G. A.; Hatami R.; Palese P. (2007) Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J. Virol. 81, 9727–9736 10.1128/JVI.01144-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann H. H.; Palese P.; Shaw M. L. (2008) Modulation of influenza virus replication by alteration of sodium ion transport and protein kinase C activity. Antiviral Res. 80, 124–134 10.1016/j.antiviral.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H.; Yamamura K.; Miyazaki J. (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- Konig R.; Stertz S.; Zhou Y.; Inoue A.; Hoffmann H. H.; Bhattacharyya S.; Alamares J. G.; Tscherne D. M.; Ortigoza M. B.; Liang Y.; Gao Q.; Andrews S. E.; Bandyopadhyay S.; De Jesus P.; Tu B. P.; Pache L.; Shih C.; Orth A.; Bonamy G.; Miraglia L.; Ideker T.; Garcia-Sastre A.; Young J. A.; Palese P.; Shaw M. L.; Chanda S. K. (2010) Human host factors required for influenza virus replication. Nature 463, 813–817 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor E.; Devenish L.; Engelhardt O. G.; Palese P.; Brownlee G. G.; Garcia-Sastre A. (1999) Rescue of influenza A virus from recombinant DNA. J. Virol. 73, 9679–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F.; Margine I.; Tan G. S.; Pica N.; Krause J. C.; Palese P. (2012) A carboxy-terminal trimerization domain stabilizes conformational epitopes on the stalk domain of soluble recombinant hemagglutinin substrates. PLoS One 7, e43603 10.1371/journal.pone.0043603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann H. H.; Kunz A.; Simon V. A.; Palese P.; Shaw M. L. (2011) Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 5777–5782 10.1073/pnas.1101143108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr C. M.; Kim P. S. (1993) A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73, 823–832 10.1016/0092-8674(93)90260-W. [DOI] [PubMed] [Google Scholar]
- Xu R.; Wilson I. A. (2011) Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 85, 5172–5182 10.1128/JVI.02430-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao H.; Pelletier S. L.; Hoffman L.; Hacker J.; Armstrong R. T.; White J. M. (1998) Specific single or double proline substitutions in the “spring-loaded” coiled-coil region of the influenza hemagglutinin impair or abolish membrane fusion activity. J. Cell Biol. 141, 1335–1347 10.1083/jcb.141.6.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartchow C. A.; Podlaski F.; Li S.; Rowan K.; Zhang X.; Mark D.; Huang K. S. (2011) Biosensor-based small molecule fragment screening with biolayer interferometry. J. Comput.-Aided Mol. Des 25, 669–676 10.1007/s10822-011-9439-8. [DOI] [PubMed] [Google Scholar]
- Reed M. L.; Yen H. L.; DuBois R. M.; Bridges O. A.; Salomon R.; Webster R. G.; Russell C. J. (2009) Amino acid residues in the fusion peptide pocket regulate the pH of activation of the H5N1 influenza virus hemagglutinin protein. J. Virol. 83, 3568–3580 10.1128/JVI.02238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godley L.; Pfeifer J.; Steinhauer D.; Ely B.; Shaw G.; Kaufmann R.; Suchanek E.; Pabo C.; Skehel J. J.; Wiley D. C.; et al. (1992) Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell 68, 635–645 10.1016/0092-8674(92)90140-8. [DOI] [PubMed] [Google Scholar]
- Kemble G. W.; Bodian D. L.; Rose J.; Wilson I. A.; White J. M. (1992) Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 66, 4940–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y.; Isegawa Y.; Sasao F.; Ueda S. (1993) A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 67, 2552–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti D.; Voss J.; Gamblin S. J.; Codoni G.; Macagno A.; Jarrossay D.; Vachieri S. G.; Pinna D.; Minola A.; Vanzetta F.; Silacci C.; Fernandez-Rodriguez B. M.; Agatic G.; Bianchi S.; Giacchetto-Sasselli I.; Calder L.; Sallusto F.; Collins P.; Haire L. F.; Temperton N.; Langedijk J. P.; Skehel J. J.; Lanzavecchia A. (2011) A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- Lingwood D.; McTamney P. M.; Yassine H. M.; Whittle J. R.; Guo X.; Boyington J. C.; Wei C. J.; Nabel G. J. (2012) Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 489, 566–570 10.1038/nature11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommakanti G.; Citron M. P.; Hepler R. W.; Callahan C.; Heidecker G. J.; Najar T. A.; Lu X.; Joyce J. G.; Shiver J. W.; Casimiro D. R.; ter Meulen J.; Liang X.; Varadarajan R. (2010) Design of an HA2-based Escherichia coli expressed influenza immunogen that protects mice from pathogenic challenge. Proc. Natl. Acad. Sci. U.S.A. 107, 13701–13706 10.1073/pnas.1007465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert D. C.; Bhabha G.; Elsliger M. A.; Friesen R. H.; Jongeneelen M.; Throsby M.; Goudsmit J.; Wilson I. A. (2009) Antibody recognition of a highly conserved influenza virus epitope. Science 324, 246–251 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F.; Palese P. (2014) Universal influenza virus vaccines: need for clinical trials. Nat. Immunol. 15, 3–5 10.1038/ni.2761. [DOI] [PubMed] [Google Scholar]
- Krammer F.; Palese P. (2013) Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr. Opin. Virol. 3, 521–530 10.1016/j.coviro.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M. L., and Palese P. (2013) Orthomyxoviridae: the viruses and their replication. In Fields Virology (Knipe D. M., and Howley P. M., Eds.), 6th ed., pp 1151–1185, Wolters Kluwer/Lippincott Williams & Wilkins Health, Philadelphia, PA, USA. [Google Scholar]
- Feeney E. R.; Chung R. T. (2014) Antiviral treatment of hepatitis C. BMJ 349, g3308. 10.1136/bmj.g3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellecchia M.; Bertini I.; Cowburn D.; Dalvit C.; Giralt E.; Jahnke W.; James T. L.; Homans S. W.; Kessler H.; Luchinat C.; Meyer B.; Oschkinat H.; Peng J.; Schwalbe H.; Siegal G. (2008) Perspectives on NMR in drug discovery: a technique comes of age. Nat. Rev. Drug Discovery 7, 738–745 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. (1999) Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 38, 1784–1788. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. (2001) Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 123, 6108–6117 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]