

Abstract
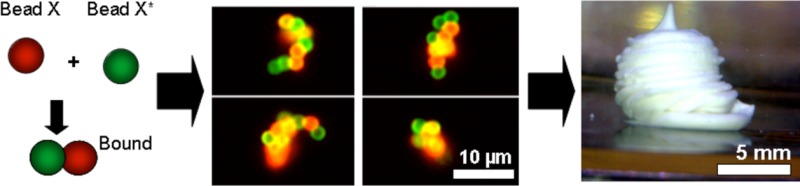
By relying on specific DNA:DNA interactions as a “smart glue”, we have assembled microparticles into a colloidal gel that can hold its shape. This gel can be extruded with a 3D printer to generate centimeter size objects. We show four aspects of this material: (1) The colloidal gel material holds its shape after extrusion. (2) The connectivity among the particles is controlled by the binding behavior between the surface DNA and this mediates some control over the microscale structure. (3) The use of DNA-coated microparticles dramatically reduces the cost of DNA-mediated assembly relative to conventional DNA nanotechnologies and makes this material accessible for macroscale applications. (4) This material can be assembled under biofriendly conditions and can host growing cells within its matrix. The DNA-based control over organization should provide a new means of engineering bioprinted tissues.
Keywords: DNA, hybridization, self-assembly, microparticles, colloidal gel, 3D printing
Nucleic acid hybridization has previously been used for the programmed assembly of materials.1 However, the objects that have been constructed are frequently restricted to the nanoscale, with very few examples of nucleic acid hybridization leading to the production of visible materials.2,3 Moreover, even when visible objects are generated, the cost of using DNA for their generation is exorbitant; for example millimeter crystals were generated from gold nanoparticles measuring 7.2 nm in diameter that were functionalized with ∼30 molecules of 10 nm long DNA.2 If these nanoparticles had been assembled into a colloidal gel with a density of 1% v/v, it would have cost on the order of $2500 per liter (excluding the price of the gold). By using 2.3 μm polystyrene microparticles functionalized with an equivalent DNA density, the DNA price could potentially be reduced to a more reasonable $60 per liter. This price difference will likely be a critical determination for applications of DNA as an adhesive for the production of human scale objects.
To this end, we have developed methods to assemble DNA-functionalized microparticles into a colloidal gel, and to extrude this gel with a 3D printer at centimeter size scales. This process produces materials with several unique properties, which we demonstrate: (1) Unlike conventional 3D printed objects, the extruded semisolids are assembled solely by DNA:DNA interactions that are strong enough to support the object at the macroscale. (2) These objects have internal, microscale properties that are programmed by the nanoscale DNA interactions; by controlling the assembly of materials from the molecular to the macroscale, one of the challenges for self-assembling materials has been realized. (3) The size of these objects can be large—up to centimeters—and the cost for this material is reasonable as the bulk of its volume is an inexpensive polymer rather than expensive DNA. (4) This bulk material is assembled under conditions in which cells can survive and grow. This material can be “seeded” with cells during extrusion and these cells will proliferate within the colloidal gel matrix.
Unlike conventional 3D printed objects, the extruded materials have internal, microscale properties that are programmed by the microparticles and their nanoscale DNA interactions. By controlling the assembly of materials from the molecular to the micron to the macroscale, one of the long-standing challenges for self-assembling materials has been realized. Using DNA adhesives to leverage functional microparticle assembly should provide great advantages relative to charge-based assembly.4,5 Micro- and nanoparticles have previously been engineered to have a range of important properties from controlled release of growth factors and morphogens6 to cell capture7 by binding to specific cell surface antigens. Any or all of these properties could in principle be integrated into the microparticles that comprise the self-assembled colloidal gel. Particles could release compounds into the surrounding environment, draw specific cells into proximity, or both. The ability to rationally integrate control over material connectivity, material-cell connectivity, and controlled release may prove important in the construction of novel, rationally designed biomaterials.
Results
Microparticle Cluster Assembly by DNA Hybridization
To demonstrate DNA-mediated assembly of objects, we functionalized 2.3 μm polystyrene microparticles with fluorescent oligonucleotides using established EDC coupling chemistry.8 The ability of microparticles bearing two different oligonucleotides to hybridize to one another was analyzed via imaging flow cytometry9 (Figure 1).
Figure 1.
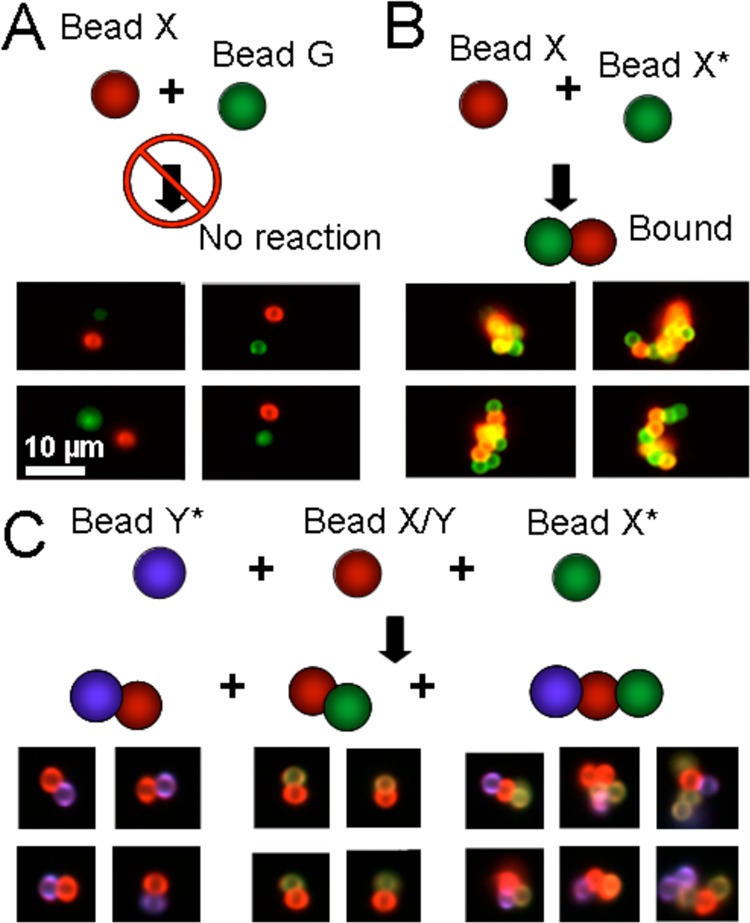
Imaging cytometry of hybridized microparticles. (A) Representative images from imaging flow cytometry show that mixing microparticles bearing noncomplementary DNA yields dispersed, single microparticles. Scale bar is 10 μm throughout. (B) Representative images show self-assembled clusters of microparticles bearing complementary DNA (green X and red X). (C) Schematic shows beads bearing orthogonal specificities; representative images show the specific microparticle clusters.
When the oligonucleotides were noncomplementary, only approximately 0.03% of all detection events contained aggregates with both types of microparticles. However, of these, virtually all were false positives; the red and green microparticles shared the same image, but were not bound to one another (Figure 1A). In contrast, when the oligonucleotides were complementary, 33% of the imaged events were bound clusters. To examine higher-order clustering, three types of microparticles were generated using combinations of two different DNA oligonucleotides bearing fluorescent labels: X/Y-red microparticles, X*-green microparticles and Y*-purple microparticles (where * indicates a complement; false colored for the figure). These three microparticles types were mixed at a 1:1:1 ratio (Figure 1C), and again analyzed by imaging flow cytometry. No X*-Y* clusters were found but the suspension contained representatives of all other cluster types (X/Y bound to Y*, X/Y bound to X*, and higher-order clusters containing all three microparticle types). Assembly appeared to be efficient, as some 50% of all measured objects were self-assembled clusters; in noncomplementary controls, only about 1% of events represented apparent clusters.
Critical Parameters for Large-Scale Aggregation via Hybridization
At high concentrations of complementary microparticles, amorphous aggregates form and fall out of suspension (even with mild shaking). This observation suggested that we could use optical density as a simple assay for oligonucleotide mediated aggregation. At low concentrations, both complementary and noncomplementary microparticles disperse into suspension and yield visible light scattering that can be easily measured with a densitometer (Figure 2A). At a critical concentration, complementary microparticles form an aggregate that sinks to the bottom of the cuvette and the optical density falls to zero (Figure 2B). The critical concentration at which the trend toward increasing optical density reverses is thus a metric for the strength of aggregation. In the case of short, surface immobilized DNA (40 base pairs), aggregates formed virtually instantaneously at between 20 and 40% w/v.
Figure 2.
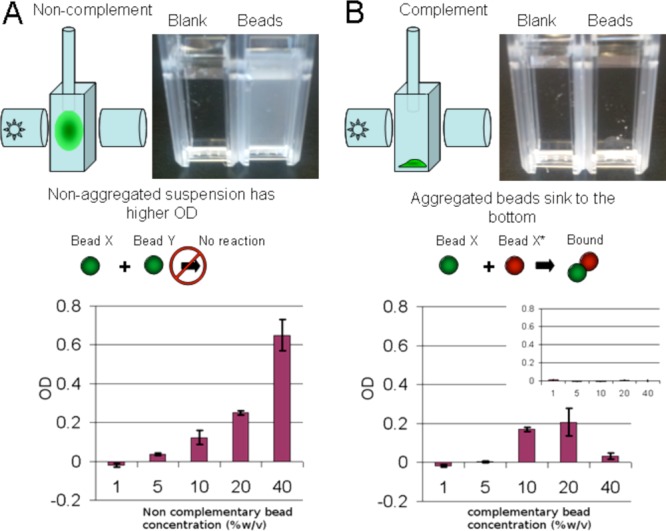
Microparticle aggregation assay. (A) Left, a diagram shows how a dispersed cloud of microparticles can be detected by optical density measurements. Right, a photograph of a blank cuvette and experimental cuvette shows the change in optical density when noncomplementary microparticles are added. Bottom, a graph shows the optical density (OD) as a function of noncomplementary microparticle concentration. (B) Left, a diagram shows how microparticles bearing complementary DNA assemble and fall to the bottom of the cuvette. Right, a photograph of a blank cuvette and experimental cuvette shows that the assembled clusters do not increase the optical density. Bottom, a graph shows OD as a function of complementary microparticle concentration; the OD decreases sharply at a critical concentration when assembly is rapid and complete. An inset shows a graph of these results repeated with microparticles bearing long poly-A and poly-T DNA.
The length and base composition of the surface DNA has an impact on cluster formation9 and also on the critical concentration at which large scale aggregation occurs. To generate longer, surface bound DNA molecules, immobilized oligonucleotides were used as primers for untemplated extension by deoxynucleotidyl terminal transferase (dNTT), which can produce poly-A or poly-T with lengths of 100+ nucleotides.10 Microparticles bearing long poly-A and poly-T tracts were produced by extending surface-bound oligonucleotides with terminal transferase and dATP or dTTP. The microparticles formed aggregates at 5% w/v concentrations or lower (Figure 2B, inset).
3D Printed Objects from Hybridized Colloidal Gels
The fact that microparticle aggregates precipitated from solution indicated that it might be possible to form solid objects based on DNA:DNA interactions. To this end, we attempted to extrude complementary microparticles at high concentrations with a 3D printer. We show in Figure 3 that an amorphous colloidal gel is formed by the particle suspension and that this material can be printed so as to form different morphologies. The Replicator 3D printer (Makerbot Industries) was originally developed for printing thermoplastics. For this study, it was modified for printing microparticle-based gels by directing the print head to extrude a suspension provided via a programmable syringe pump. By actuating the print head while controlling the dispense rate of the syringe pump, the 3D printer directed the extrusion of the colloidal gel into 3D shapes (Figure 3C–E). While there are clearly limitations on the precision of the printed shape, these are in large measure the result of the rheology of the material itself. The printed object holds its form against gravity, and as additional contacts between the particles are put in place, either with DNA or with other materials, it seems likely that the overall form of the soft object will become even more defined.
Figure 3.
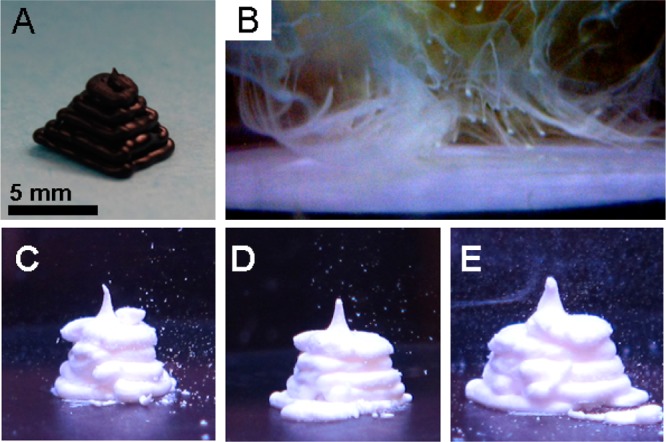
3D printing of DNA-derivatized microparticles. (A) Digital photograph shows an ABS thermoplastic pyramid printed by the 3D printer to show the desired output pattern. Scale bar is 5 mm throughout. (B) Digital micrograph shows the output of the 3D printer when it attempts to extrude microparticles bearing noncomplementary DNA. (C–E) Digital micrographs show DNA cross-linked colloidal gel printed into the pyramidal shape with extrusion rates of 1.3, 1.7, and 2.1 μL/s, respectively. The 3D printer head motion pattern was identical in all three cases.
In these instances, the colloidal gel was held together by 40 base pairs of complementarity between two microparticle types (see Materials and Methods). Like other self-assembled colloidal gels, we suspect that the colloids behave as shear thinning fluids.5 The colloidal gel does not clog the tubing and settles into a more solid form in the absence of shear.
The extruded gels held their form with slump heights (height without any visible gravitational collapse11) of greater than 6 mm under high salt conditions (see Figure 3C–E). Under physiological salt conditions, the printed object holds its shape up to a slump height of >2 mm. Noncomplementary microparticles disperse into suspension and do not form a colloidal gel (Figure 3B).
These results are proof of concept that DNA connectors can mediate complex assembly of a macro scale object with internal properties defined by the nanoscale interactions. Our initial studies with HEK 293T cells expressing GFP indicated that these aggregates are tissue culture compatible. Because the aggregates are highly opaque, individual cells were obscured within the material; nonetheless, the presence of cells within the material and their growth and proliferation were evident from a positive and increasing GFP signal over many days (see Figure S1).
Generating Substructure in Three-Dimensional Colloidal Gel
A number of other biocompatible materials have been printed in 3D.12 The advantage of DNA as a “smart glue” for such materials is that substructures can be programmed within the overall printed object, often under physiological conditions. As a first example of this programmed substructure, two binary pairs of complementary microparticles assembled to form two specific cluster types within the three-dimensional matrix. This could not be accomplished with, for instance, charge–charge attraction of colloidal microparticles.4
Two pairs of microparticle types were generated bearing complementary DNA. One pair was coupled to fluorescein modified DNA and the other pair to Cy5 modified DNA as shown in Figure 4A. When these four microparticle types were mixed at a total concentration of 40% w/v, they formed a colloidal gel consistent with other results with a single pair of complementary microparticle types. Upon examining the margins of this structure (Figure 4B) with confocal microscopy, it was clear that the two assembly processes had produced independent clusters of red and green microparticles (Figure 4C).
Figure 4.
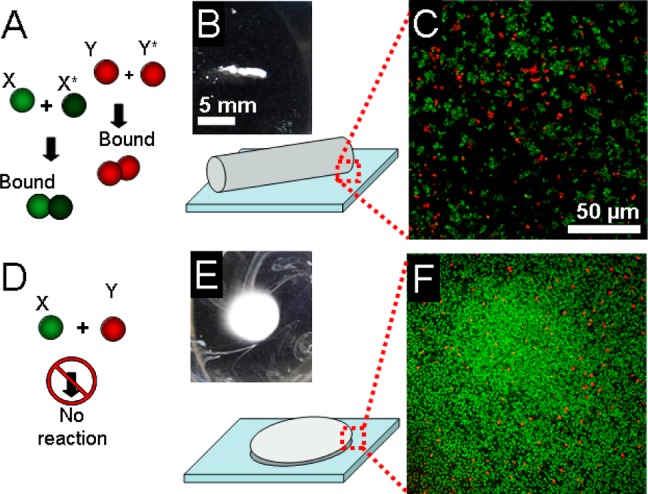
Internal organization of printed materials. (A) Schematic shows how two pairs of microparticle types bearing DNA complementary to each other should form a binary mixture of red and green clusters. (B) Micrograph shows a millimeter sized assembled aggregate generated from the complementary microparticle mixture; the diagram indicates how the outer margin of the object was imaged. Scale bar is 5 mm. (C) Confocal micrograph of the edge of the aggregate shows red and green clusters. Scale bar is 50 μm. (D–F) Corresponding experiments with noncomplementary microparticles show no assembly. Scales are identical to those in A–C.
As a second method for generating controlled substructures within the self-assembled aggregate the stoichiometry of large and small complementary microparticles was varied. An overabundance of small particles relative to large particles would effectively block the large particles from assembling into aggregates, with concomitant impacts on the overall morphology and granularity of the structure. The plans for both simulation and experiment are shown in schematic in Figure 5A. In the simulation, particle types of diameters 2.3 and 1 μm were mixed at various proportions and the number of independent nucleation events was recorded. The details of the simulation are given as MATLAB source code available as Supporting Information. In brief, particles were iteratively generated and paired in a random order on a 2D simulated surface. If a newly generated particle overlapped with a particle already in place, it was moved to a random location and could serve as a new center for binding newly generated particles. The results of the simulations are shown in Figure 5B. As the fraction of small microparticles increases and prevents larger particle interactions, there were more new nucleation, creating a new cluster and leading to greater granularity overall.
Figure 5.
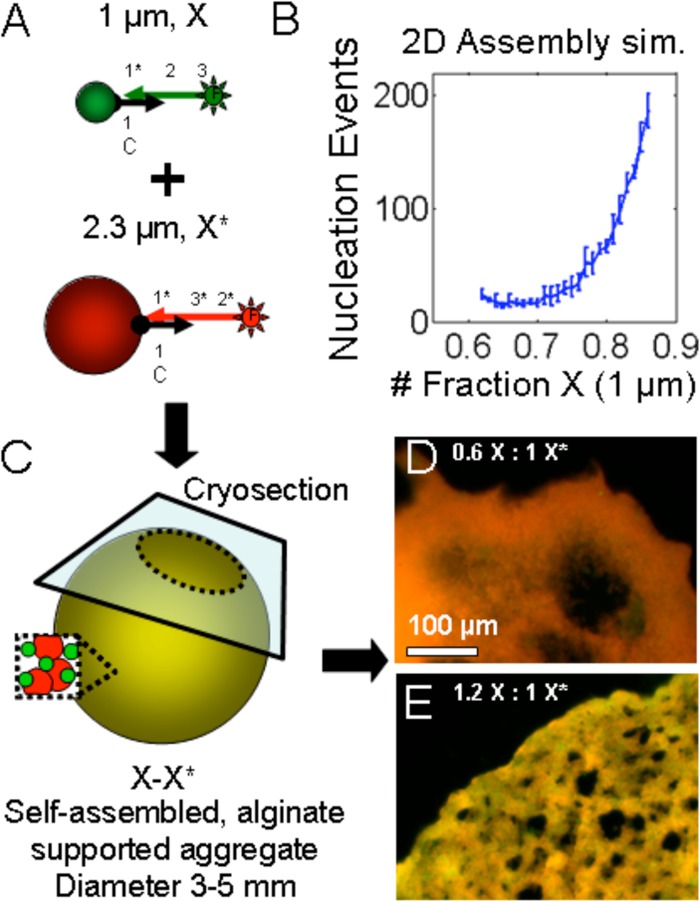
Microparticle stoichiometry affects granularity of printed objects. (A) Schematic shows how small, green microparticles were mixed with large, red microparticles bearing complementary DNA. (B) Simulation of nucleation events as a function of the number fraction of small microparticles. (C) Schematic shows large and small microparticles assembled within a carrier gel and sectioned for fluorescence microscopy. (D, E) Fluorescence micrographs show the structures resulting from different fractions of small microparticles. Scale bar is 100 μm.
Corresponding experiments in which the ratios of particle sizes were varied were evaluated visually for granularity or smoothness. The internal morphology of printed aggregates was observed with fluorescence microscopy. The total concentration of microparticles was 4% w/v; this was the maximum density of microparticles that could be reliably imaged with optical methods. Because the colloidal assemblies were too fragile to be manipulated, they were reinforced (after assembly) with alginate carrier gel. Alginic acid (1%) was present in the initial slurry and after particle–particle assembly was gelled by the addition of calcium chloride (Figure 5C).
The colloidal gel within its carrier gel was sectioned using a cryostat as shown in the diagram in Figure 5C. As predicted by simulation, the internal structure was found to depend on the ratio of small to large microparticles in the suspension. A high proportion of large microparticles produced smooth, nongranular material with some large pores (Figure 5D). A low proportion of large microparticles produced a much more granular appearance with many small micropores (∼10 μm, Figure 5E). The assembly of micrometer-scale particles into aggregates has an effect on the structure of the colloidal gel at the multimicrometer scale. The choice of particle shape could be used to control the structure at the 1–20 μm scale as has been demonstrated by McGinley et al.13 Additionally, several groups have shown that emergent structure can be created by assembling crystals using DNA:DNA interactions including the Merkin group14 and the Crocker group.13 Such work previously demonstrated that structure can be more precisely controlled by colloidal crystallization, but for smaller overall objects (∼100 μm). The structural control presented here is stochastic rather than deterministic: the average porosity (for example) is a function of the microparticle size and stoichiometry rather than precise crystalline order.
Creation of DNA-Coated, Acrylamide-Based Colloidal Gel
As noted above, polystyrene-based colloidal gels scatter light very efficiently and are highly opaque to light microscopy. Preliminary experiments indicated that mammalian tissue culture could be performed within these self-assembled structures, but we were unable to make microscopic observations within the mass. These results were therefore recreated in a colloidal gel generated from hydrogel microparticles because hydrogels have an index of refraction much closer to that of water.
Polyacrylamide microparticles were generated by dispersion polymerization. Briefly, acrylamide monomers in an aqueous solution were mixed with a DNA complex bearing three modifications: a labile double bond (acrydite), a fluorophore, and a cholesterol modification. This DNA/acrylamide mixture was mixed with radical initiators and rapidly dispersed into mineral oil. Polymerization takes several minutes. During this time, the cholesterol modified DNA migrates to the oil–water interface, concentrating at the margins of the polymerizing microdroplets. Once polymerized, the microparticles are decorated at their surface with DNA. This arrangement is optimal for self-assembly (Figure 6A). Without the cholesterol modification, the DNA is evenly distributed throughout the microparticle volume and a smaller fraction is available for bridging between adjacent microparticles (see Materials and Methods).
Figure 6.
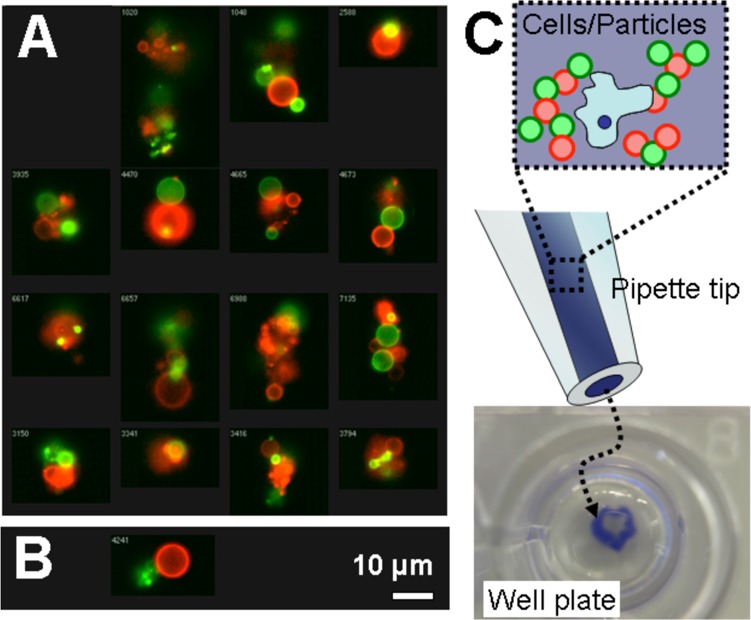
Visualization of hybridized hydrogel microparticles. (A) Representative images from imaging flow cytometry show self-assembled clusters of polyacrylamide hydrogel microparticles bearing complementary DNA (green X and red X*). (B) Single micrograph of the single false-positive event from imaging flow cytometry with noncomplementary microparticles. (C) Schematic drawing shows how blue dye, cells, and hydrogel microparticles bearing complementary DNA were mixed and loaded into a pipet tip. At bottom, a digital micrograph shows the stable blue aggregate in a well.
These DNA-coated polyacrylamide microparticles were washed extensively over the course of several days to remove any acrylamide monomer. Their self-assembly behavior was observed with an imaging flow cytometer. DNA mediated self-assembly occurred with complementary DNA as shown by representative imaging flow cytometry images in Figure 6A. In the complementary case, many events (12.4%) were clusters of large, assembled aggregates. In the noncomplementary case, only a single false positive event (shown in Figure 6B) shared the same highly intense red and green fluorescence as the complementary aggregates pictured. Only 1.3% of the events detected in the noncomplementary showed any aggregation.
Complementary acrylamide microparticles can form the same type of colloidal gel demonstrated with polystyrene microparticles. Complementary polyacrylamide microparticles at 40% weight:volume were cosuspended with cells and then extruded from a pipet tip into a well plate. The extruded object held its shape (Figure 6C; blue dye added to enhance contrast).
Cell Behavior within DNA-Mediated Self-Assembled Polyacrylamide Colloidal Gel
Mammalian cells (A431 human epithelial carcinoma) were coextruded with complementary or noncomplementary polyacrylamide microparticles into a well plate of tissue culture media and allowed to grow for 4 days. Because the hydrogel microparticles did not scatter light as effectively as the polystyrene microparticles, the cells within the self-assembled mass could be readily observed (Figure 7A). Cells growing within the mass of DNA-assembled colloidal gels showed changes relative to cells grown either without any microparticles or with noncomplementary microparticles in two ways: first, many spherical bodies could be seen inside and outside of the plane of focus (Figure 7B). These bodies were spherical colonies of A431 cells within the colloidal gel (Figure 7C). Second, the activation of fluorescein diacetate, a marker for cell integrity and metabolism, was much higher than was the case for mere surface-bound cells. Overall, the cells within the colloidal gel were apparently more healthy and active than those on the polystyrene surface. The same cells grown without the microenvironment of the self-assembled colloidal gel did not form colonies. Moreover, because the noncomplementary microparticles did not aggregate, they were largely washed away from the cells during staining (despite efforts to wash extremely gently). This can be seen in Figure 7D in the form of a shadow in the upper left. All observed cells grown in the presence of noncomplementary particles adhered to the polystyrene floor of the well plate and grew into a confluent lawn. Fluorescein diacetate staining of these negative control cells indicated that only the outermost edge cells actively hydrolyzed the fluorogenic substrate (Figure 7E).
Figure 7.
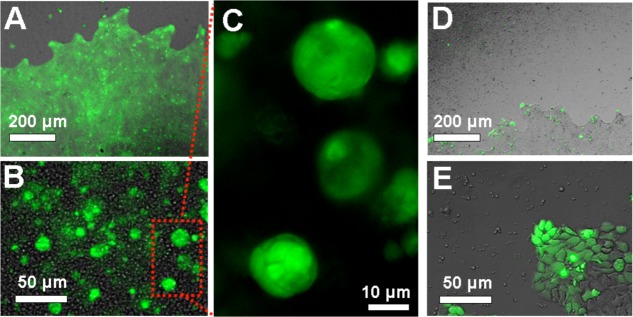
Microscopy of living cells within colloidal gels composed of hydrogel particles. (A) Low-magnification brightfield/fluorescence micrograph of complementary microparticles and A431 cells. False color green overlay is fluorescence from hydrolyzed fluorescein diacetate. Scale bar is 200 μm. (B) High-magnification bright-field/fluorescence overlay image focused within the self-assembled mass. Scale bar is 50 μm. (C) Zoomed fluorescence micrograph of multicellular colonies formed within the mass. Scale bar is 10 μm. (D) Low-magnification brightfield image of cells extruded with noncomplementary microparticles. Scale bar is 200 μm. (E) High-magnification bright-field/fluorescence overlay micrograph of the edge of the confluent lawn of cells. Scale bar is 50 μm.
Conclusions
We have for the first time printed a macroscale object that is held together solely by DNA interactions. Such printed colloidal gels held together by DNA adhesives are programmable at three distinct scales: (i) at the nanometer scale, specific DNA interactions mediate individual microparticle:microparticle interactions. (ii) At the micrometer scale, microparticle clusters form DNA dependent substructures. This microscale topology can be further controlled by using different sizes and stoichiometries of oligonucleotide derivatized microparticles.9 The length of the DNA linker also impacts the morphology of substructures, because long linkers produce larger, stronger clusters. (iii) Finally, the shape of the object can be patterned at the centimeter scale by 3D printing and its material properties, such as porosity, can be altered through control over the composition of the colloidal mixtures.
This work demonstrates that by using multiple binary pairs of complementary DNA types to assemble microparticles, microparticle:microparticle and ultimately material assembly can be programmed in a rational manner (e.g., as opposed to charge–charge assembly). Given the progress that has been made in the DNA nanotechnology community,15−18 such programmability may provide an interesting avenue for creating new materials with programmed structure at the microscale. DNA circuits could mediate the conditional or algorithmic assembly of DNA-modified subunits into higher-order morphologies. Such processes have previously been demonstrated at the nanoscale with subunits such as DNA origami tiles, and our methods provide a potential for further scaling.19 For example, specific microparticle types could be added such that clusters are formed and then bridged (a hierarchical assembly process). Logical conditional operators, such as AND gates based on conformation switching,20 could be used to locally determine if assembly is appropriate and permitted. More advanced DNA computation might also allow us to algorithmically determine the assembly process at the nano- to microscales (as has been demonstrated for tile-based assembly19). We have also previously demonstrated that light can be used to modify the behavior of DNA reaction networks and form two-dimensional patterns;21 such techniques could also be used to selectively alter the morphology of three-dimensional colloidal gels.22
Nucleic acid programmability should prove to be generalizable to a wide variety of materials. Polystyrene microparticles with highly monodisperse size distributions were readily acquired commercially; this made them attractive substrates for developing new methods. These methods can now be applied to other materials. For example, hydrogels were a particularly attractive substrate because they are biocompatible and can release encapsulated compounds into the surrounding environment.23 By creating hydrogel microparticles adorned with DNA, it was proved that the principles developed with polystyrene microparticles could be immediately applied on a completely different platform. Polyacrylamide-based microparticles were generated with a similar index of refraction to water. This allowed for microscopy that probes deep into assembled aggregates.
DNA-mediated, self-assembled colloidal gels made from other substrates may eventually find application as a prototype tissue scaffold material. Polyacrylamide may not be the ideal scaffold material because it is not approved in the United States for use as an injectable polymer; however, it has been used in Europe as filler material in cosmetic surgery with a low incidence of side effects.24 Microparticles of the appropriate size and surface composition for DNA mediated assembly have also been generated from both gelatin6 and PLGA/HLA,25 and our methods could also be applied to collagen or hyaluronic acid biomaterials, both of which have been approved by the FDA for in vivo use in the US.26
It is clear that self-assembly affects the behavior of cells trapped within the self-assembled matrix. Mammalian cancer cells in self-assembled colloidal gels were found to form spheroid colonies, very different behavior from their adherent spreading morphology on a polystyrene plate. This was not an effect of the polyacrylamide substrate as the nonassembled acrylamide microparticles had no apparent impact. We hope to use this paradigm to upgrade an inexpensive biological hydrogel to a tailored biomimetic environment for tissue growth. Previously, porous biomaterials created by electrospinning27 or using inverted colloidal crystals28 have approximated the physical environment found in decellularized tissue (already evaluated in human clinical trials29,30), but had to be seeded with cells postsynthesis. Our biomimetic scaffolds can be coprinted with cells and this may lead to the generation of more specific microscale morphologies and improved performance of bioprinted tissue.
Future work will focus on controlling the self-assembly process using the properties of both DNA hybridization and DNA circuitry in order to test the effects of different self-assembly processes. It will be determined how microscale structure affects cells seeded within it. The ability to control the macroscale shape, the microscale topology by DNA computation-mediated self-assembly, and the ability to choose the chemistry of the “dumb” substrate material is a unique combination of features for tissue engineering.
Materials and Methods
Preparation of DNA-Modified Polystyrene Microparticles
HPLC-purified DNA was purchased from IDT (Coralville, IA) and used without further purification. Sequences: (X), CTTCTATTA CTGAATAAG ACGAGAATA CTAAACCTC CCTCGTCAG TGAGCTAGG TTAGATGTCG; (X*), GAGGGAGGT TTAGTATTC TCGTCTTAT TCAGTAATA GAAGGTCAG TGAGCTAGG TTAGATGTCG; (Y), GTTAATGGC ACAAAGTTT TAGGAGGGA GGTCAGTGA GCTAGGTTA GATGTCG; (Y*), CTCCCTCCT AAAACTTTG TGGTCAGTG AGCTAGGTT AGATGTCG; (C), 5′ Amine - CGACATCTA ACCTAGCTC ACTGAC. Amine-modified sequence C acted as a common linker for coupling to bead surfaces. Sequence C is complementary to a region at the 3′ end of sequences X, X*, Y, and Y*. When fluorophores are noted in the text, they appear at the 5′ end of X, X*, Y, or Y*.
DNA-conjugated microparticles were prepared from carboxylate-modified microparticles (Bangs Laboratories, Fishers, IN). To prepare microparticles bearing DNA, the following method was used (Figure 8). An oligonucleotide (e.g., X) was annealed to C by making a 100 μM solution of X and C in MES buffer. This solution was heated to 80 °C for 3 min then cooled to room temperature at a rate of 0.1 °C per sec. Carboxylate microparticles were washed three times by centrifugation and resuspension in 100 mM MES buffer pH 4.5 (Sigma-Aldrich). 500 pMol of the X-C complex was then added to 50 μL of washed 10% w/v polystyrene microparticles (2.3 μm). EDC, N-(3-(Dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (Sigma-Aldrich, St Louis, MO), was added to a final concentration of 100 mM slowly with shaking. The reaction proceeded for 12–18 h. The microparticles were then washed two times by centrifugation and resuspended in 0.4 M tetraethylammonium bicarbonate buffer (TEAB, Sigma) with 0.1% Tween (Sigma) and 10 μM sodium azide (Life Technologies, Grand Island, NY).
Figure 8.
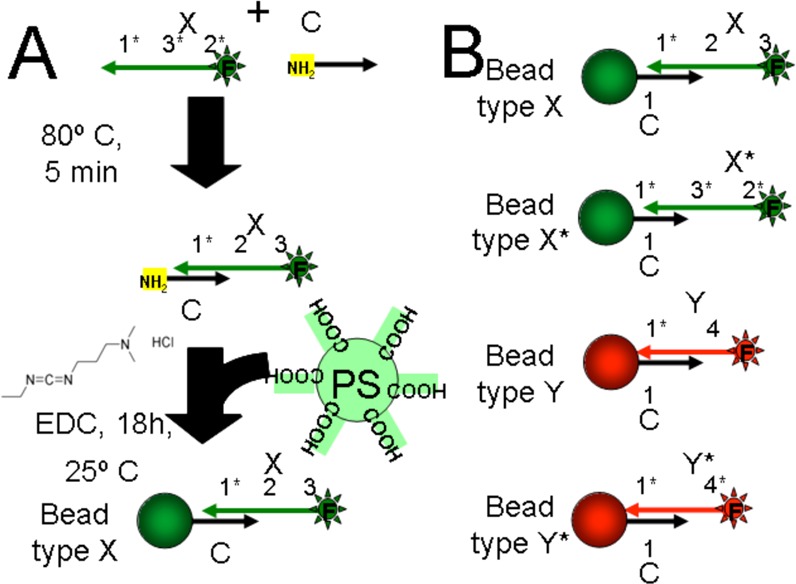
Conjugating DNA to microparticle surfaces. (A) Schematic shows surface functionalization protocol. (B) Schematic drawings show the different microparticle types generated by coupling different sequences.
Imaging Flow Cytometry of DNA-Assembled Clusters
DNA-coated microparticles (as described above) were mixed at a one-to-one ratio. Microparticles of type X display single-stranded domains 2 and 3 (Figure 8). Microparticles of type X* display the domains 3* and 2* (where * indicates reverse complementarity). The microparticle types X and X* were mixed and allowed to assemble at modest concentrations (1%) for approximately 1 min, and then diluted to a final concentration (0.01%) in PBS (phosphate buffered saline, Thermo Fisher Scientific, Waltham, MA). This final sample was analyzed on the imaging flow cytometer (Imagestream X, Amnis, Seattle).
Measurement of Critical Volume Percentage for Aggregation
A cuvette with 500 μL of PBS was placed into a shaker set to 750 rpm. Microparticles bearing complementary or noncomplementary DNA were mixed at the appropriate concentration. Microparticles were then slowly pipetted into the cuvette and shaking was immediately initiated. Microparticles were allowed to shake for 3 min and the optical density was measured. The concentration of microparticles at which the optical density decreased rather than increased was noted.
Terminal Deoxynucleotidyl Transferase Extension of DNA
2.3 μm microparticles were conjugated to DNA C as described above. This DNA was then extended with Terminal Deoxynucleotidyl Transferase (DNTT, Thermo Fisher) according to manufacturer’s specifications. Briefly, the microparticles and enzyme were suspended in the provided reaction buffer along with 5 mM ATP or TTP. The reaction was allowed to progress at 37° overnight with rotation.
Microscopic Imaging of the Internal Structure of Aggregates
Microparticle slurry containing complementary DNA coated microparticles were prepared at 4% w/v in a 1% w/v solution of alginic acid in PBS. To this was added 0.1% bromophenol blue as a contrast agent for sectioning. Various ratios of the two microparticle types were pipetted and mixed by gentle vortexing. A hanging drop of ∼2 μL of the resulting aggregate was dropped into excess 1 M calcium chloride. After gelling, the excess calcium chloride was aspirated away and replaced with tissue freezing medium (TFM). The gel microparticles embedded in TFM were frozen at −20 °C and sectioned with a cryostat (Microm HM 550, Thermo Fisher) then mounted and imaged on an inverted fluorescence microscope (IX51, Olympus, Tokyo).
3D printing of DNA-assembled aggregates
A computer-controlled syringe pump (SIAlab.com, Seattle, WA) was connected to a polyethylene tube. The tube was connected to the 3D printer head. The polyethylene tube was purged thoroughly with PBS. Microparticle slurry containing complementary (X and X*) or noncomplementary (X and Y) DNA coated microparticles was aspirated into the polyethylene tubing (500 μL at 40% w/v). The microparticles were then extruded at a carefully controlled rate tuned to match the rate printer head motion. Print head motion and extrusion were initiated simultaneously.
Confocal Microscopy of DNA-Assembled Aggregates
A sample of microparticles (complementary X/X* or noncomplementary X/Y, 40% w/v) was slowly pipetted into a coverslip-bottom chamber slide containing approximately 100 μL of PBS. Complementary microparticles produced a millimeter sized aggregate at the bottom of the well. Noncomplementary microparticles produced a thin layer of microparticles at the bottom of the well. The slide was then loaded onto a confocal microscope (SP2 AOBS, Leica, Germany). Images were acquired at the margins of the aggregate (the high scattering of the assembled microparticles did not permit us to see deeply into the aggregate). Images were also acquired of the noncomplementary particles sitting on the surface of the well plate.
Generation of DNA-Coated Microparticles from Acrylamide by Suspension Polymerization
In suspension polymerization monomer microdroplets are formed and then polymerized (as opposed to emulsion polymerization31 in which micro- or nanoparticles nucleate outside of the parent microdroplets). Suspension polymerized microparticles are of similar size and morphology to the parent microdroplet. Prior to polymerization, cholesterol can direct the segregation of the DNA to the edges of the microdroplet and this distribution can be maintained in the final microparticle. We followed a protocol similar to that developed by Yamazaki et al.32 Our procedure is illustrated in schematic in Figure 9.
Figure 9.
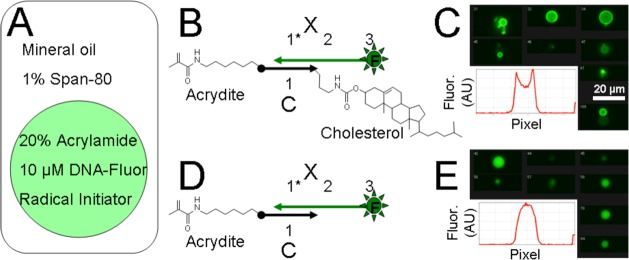
Hydrogel-based microparticle generation. (A) Schematic shows how acrylamide- and acrydite-modified DNA was encapsulated within microbubbles within a water/oil dispersion and then polymerized. (B) Schematic of the cholesterol-modified DNA complex with acrydite and fluorophore. (C) Imaging cytometry data showing the distribution of fluorescent DNA to the outer edge of the final microparticle. Scale bar is 20 μm. The inset shows the line intensity profile of fluorescence from a representative image. (D) Schematic of the noncholesterol-modified DNA complex that was polymerized within control emulsion bubbles. (E) Imaging cytometry data showing the distribution of fluorescent DNA throughout the particle. Scale is the same as E. The inset shows the line intensity profile of fluorescence from a representative image.
Polyacrylamide microparticles bearing DNA were generated as follows. A solution of 20% w/v acrylamide monomer (19:1 acrylamide:bis-acrylamide, BIORAD, Hercules, CA) was mixed to a final concentration of 1× PBS and 0.1 M NaCl. DNA complexes were prepared at 100 μM as described above. The annealed complexes were added to the acrylamide mix to a final concentration of 10 μM. The overall mixture was chilled to 4 °C and TEMED (Thermo Fisher) was added to 2% v/v. Some 100 μL of the DNA/acrylamide/TEMED was then rapidly added to 8 μL of 10% ammonium persulfate (Arcos organics, molecular biology grade, Thermo Fisher) in water, rapidly mixed, and rapidly added to prechilled mineral oil with 1% Span-80. This mixture was immediately (within 5 s) homogenized for 4 min at 42 rpm in a Tissuelyzer LT (Qiagen, Valencia, CA). Once homogenized, the headspace above the dispersion was purged 5 times with a stream of argon. The dispersion was then rotated at RT for ∼20 min. The resulting microparticles were centrifuged, washed 3× with ethanol, and dried under vacuum. The particles were rehydrated in PBS and allowed to soak in PBS for several days before further washing and use in tissue culture.
Incorporation of Tissue Culture Cells into Colloidal Gels
A431 cells were cultured according to standard protocols. Briefly, cells were cultured on Corning T75 flasks in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher) supplemented with FBS (fetal bovine serum) to 5%, 100 μg/mL penicillin and 100 μg/mL streptomycin. Cells were detached with 0.25% trypsin-EDTA, rinsed with PBS, and then mixed with a washed suspension of 40% w/v polyacrylamide microparticles bearing DNA X. Washed suspensions of 40% w/v polyacrylamide microparticles bearing DNA X* were then added to the suspension at a 1:1 volume ratio. Cell:microparticle slurries were slowly pipetted into a microwell filled with media and allowed to incubate at 37 °C with 5% carbon dioxide. Cells within the colloidal aggregate were observed with a fluorescence microscope after incubation for 4 days.
Acknowledgments
This work was funded by the National Institutes of Health (EUREKA, 1-R01-GM094933), The Welch Foundation (F-1654), the NSF (CBET 1159774), and a National Security Science and Engineering Faculty Fellowship (FA9550-10-1-0169). The cover image was designed by Peter Allen, Angel Syrett, and Andrew Ellington.
Supporting Information Available
The following files are available free of charge on the ACS Publications website at DOI: 10.1021/ab500026f.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Seeman N. C. Nanomaterials Based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane R. J.; Lee B.; Jones M. R.; Harris N.; Schatz G. C.; Mirkin C. A. Nanoparticle Superlattice Engineering with DNA. Science 2011, 334, 204–208 10.1126/science.1210493. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Birktoft J. J.; Chen Y.; Wang T.; Sha R.; Constantinou P. E.; Ginell S. L.; Mao C.; Seeman N. C. From Molecular to Macroscopic via the Rational Design of a Self-Assembled 3D DNA Crystal. Nature 2009, 461, 74–77 10.1038/nature08274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Hansen M. B.; Löwik D. W. P. M.; van Hest J. C. M.; Li Y.; Jansen J. A.; Leeuwenburgh S. C. G. Oppositely Charged Gelatin Nanospheres as Building Blocks for Injectable and Biodegradable Gels. Adv. Mater. 2011, 23, H119–H124 10.1002/adma.201003908. [DOI] [PubMed] [Google Scholar]
- Smay J. E.; Cesarano J.; Lewis J. A. Colloidal Inks for Directed Assembly of 3-D Periodic Structures. Langmuir 2002, 18, 5429–5437 10.1021/la0257135. [DOI] [Google Scholar]
- Patel Z. S.; Yamamoto M.; Ueda H.; Tabata Y.; Mikos A. G. Biodegradable Gelatin Microparticles as Delivery Systems for the Controlled Release of Bone Morphogenetic Protein-2. Acta Biomater. 2008, 4, 1126–1138 10.1016/j.actbio.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y.; Liu Y.; Allen P. B.; Asghar W.; Mahmood M. A. I.; Tan J.; Duhon H.; Kim Y.; Ellington A.; Iqbal S. M. Capture, Isolation and Release of Cancer Cells with Aptamer-Functionalized Glass Beads. Lab. Chip 2012, 4693–4701 10.1039/c2lc21251j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh M. K.; Wang X.; Weimer B. C. Optimizing the Immobilization of Single-Stranded DNA onto Glass Beads. J. Biochem. Biophys. Methods 2001, 47, 221–231 10.1016/S0165-022X(00)00146-9. [DOI] [PubMed] [Google Scholar]
- Tang H.; Deschner R.; Allen P.; Cho Y.; Sermas P.; Maurer A.; Ellington A. D.; Willson C. G. Analysis of DNA-Guided Self-Assembly of Microspheres Using Imaging Flow Cytometry. J. Am. Chem. Soc. 2012, 15245–15248 10.1021/ja3066896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson A. M.; Orkin S. H. Characterization of the Homopolymer Tailing Reaction Catalyzed by Terminal Deoxynucleotidyl Transferase. Implications for the Cloning of cDNA. J. Biol. Chem. 1982, 257, 14773–14782. [PubMed] [Google Scholar]
- Manley S.; Skotheim J. M.; Mahadevan L.; Weitz D. A. Gravitational Collapse of Colloidal Gels. Phys. Rev. Lett. 2005, 94, 218302. 10.1103/PhysRevLett.94.218302. [DOI] [PubMed] [Google Scholar]
- Landers R.; Hübner U.; Schmelzeisen R.; Mülhaupt R. Rapid Prototyping of Scaffolds Derived from Thermoreversible Hydrogels and Tailored for Applications in Tissue Engineering. Biomaterials 2002, 23, 4437–4447 10.1016/S0142-9612(02)00139-4. [DOI] [PubMed] [Google Scholar]
- McGinley J. T.; Jenkins I.; Sinno T.; Crocker J. C. Assembling Colloidal Clusters Using Crystalline Templates and Reprogrammable DNA Interactions. Soft Matter 2013, 9, 9119–9128 10.1039/c3sm50950h. [DOI] [Google Scholar]
- Mucic R. C.; Storhoff J. J.; Mirkin C. A.; Letsinger R. L. DNA-Directed Synthesis of Binary Nanoparticle Network Materials. J. Am. Chem. Soc. 1998, 120, 12674–12675 10.1021/ja982721s. [DOI] [Google Scholar]
- Zhang G.; Surwade S. P.; Zhou F.; Liu H. DNA Nanostructure Meets Nanofabrication. Chem. Soc. Rev. 2013, 42, 2488–2496 10.1039/c2cs35302d. [DOI] [PubMed] [Google Scholar]
- Wang Z.-G.; Ding B. DNA-Based Self-Assembly for Functional Nanomaterials. Adv. Mater. 2013, 25, 3905–3914 10.1002/adma.201301450. [DOI] [PubMed] [Google Scholar]
- Zahid M.; Kim B.; Hussain R.; Amin R.; Park S. H. DNA Nanotechnology: A Future Perspective. Nanoscale Res. Lett. 2013, 8, 1–13 10.1186/1556-276X-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linko V.; Dietz H. The Enabled State of DNA Nanotechnology. Curr. Opin. Biotechnol. 2013, 24, 555–561 10.1016/j.copbio.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Mao C.; LaBean T. H.; Reif J. H.; Seeman N. C. Logical Computation Using Algorithmic Self-Assembly of DNA Triple-Crossover Molecules. Nature 2000, 407, 493–496 10.1038/35035038. [DOI] [PubMed] [Google Scholar]
- Park K. S.; Seo M. W.; Jung C.; Lee J. Y.; Park H. G. Simple and Universal Platform for Logic Gate Operations Based on Molecular Beacon Probes. Small 2012, 8, 2203–2212 10.1002/smll.201102758. [DOI] [PubMed] [Google Scholar]
- Chirieleison S. M.; Allen P. B.; Simpson Z. B.; Ellington A. D.; Chen X. Pattern Transformation with DNA Circuits. Nat. Chem. 2013, 5, 1000–1005 10.1038/nchem.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D. C.; Yurke B.; Langrana N. A. Mechanical Properties of a Reversible, DNA-Crosslinked Polyacrylamide Hydrogel. J. Biomech. Eng. 2004, 126, 104–110 10.1115/1.1645529 . [DOI] [PubMed] [Google Scholar]
- Kang H.; Liu H.; Zhang X.; Yan J.; Zhu Z.; Peng L.; Yang H.; Kim Y.; Tan W. Photoresponsive DNA-Cross-Linked Hydrogels for Controllable Release and Cancer Therapy. Langmuir 2011, 27, 399–408 10.1021/la1037553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin S. P.; Marmur E. S.; Goldberg D. J. Complications from Injectable Polyacrylamide Gel, a New Nonbiodegradable Soft Tissue Filler. Dermatol. Surg. 2004, 30, 1507–1509 10.1111/j.1524-4725.2004.30551.x. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wei Y. T.; Zu Z. H.; Ju R. K.; Guo M. Y.; Wang X. M.; Xu Q. Y.; Cui F. Z. Combination of Hyaluronic Acid Hydrogel Scaffold and PLGA Microspheres for Supporting Survival of Neural Stem Cells. Pharm. Res. 2011, 28, 1406–1414 10.1007/s11095-011-0452-3. [DOI] [PubMed] [Google Scholar]
- Murray C. A.; Zloty D.; Warshawski L. The Evolution of Soft Tissue Fillers in Clinical Practice. Dermatol. Clin. 2005, 23, 343–363 10.1016/j.det.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Yang F.; Murugan R.; Wang S.; Ramakrishna S. Electrospinning of Nano/micro Scale Poly(l-Lactic Acid) Aligned Fibers and Their Potential in Neural Tissue Engineering. Biomaterials 2005, 26, 2603–2610 10.1016/j.biomaterials.2004.06.051. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Wang S.; Eghtedari M.; Motamedi M.; Kotov N. A. Inverted-Colloidal-Crystal Hydrogel Matrices as Three-Dimensional Cell Scaffolds. Adv. Funct. Mater. 2005, 15, 725–731 10.1002/adfm.200400325. [DOI] [Google Scholar]
- el-Kassaby A.; AbouShwareb T.; Atala A. Randomized Comparative Study between Buccal Mucosal and Acellular Bladder Matrix Grafts in Complex Anterior Urethral Strictures. J. Urol. 2008, 179, 1432–1436 10.1016/j.juro.2007.11.101. [DOI] [PubMed] [Google Scholar]
- Baptista P. M.; Orlando G.; Mirmalek-Sani S.-H.; Siddiqui M.; Atala A.; Soker S.. Whole Organ Decellularization—A Tool for Bioscaffold Fabrication and Organ Bioengineering. In Proceedings of the Annual International Conference of the IEEE Engineering in Medicine and Biology Society;Minneapolis, MN, Sept 3–6, 2009; IEEE: Piscataway, NJ, 2009; pp 6526–6529 10.1109/IEMBS.2009.5333145. [DOI] [PubMed]
- Arshady R. Suspension, Emulsion, and Dispersion Polymerization: A Methodological Survey. Colloid Polym. Sci. 1992, 270, 717–732 10.1007/BF00776142. [DOI] [Google Scholar]
- Yamazaki N.; Naganuma K.; Nagai M.; Ma G.; Omi S. Preparation of W/O (Water-in-Oil) Emulsions Using a PTFE (Polytetrafluoroethylene) Membrane—A New Emulsification Device. J. Dispers. Sci. Technol. 2003, 24, 249–257 10.1081/DIS-120019976. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.