

Abstract
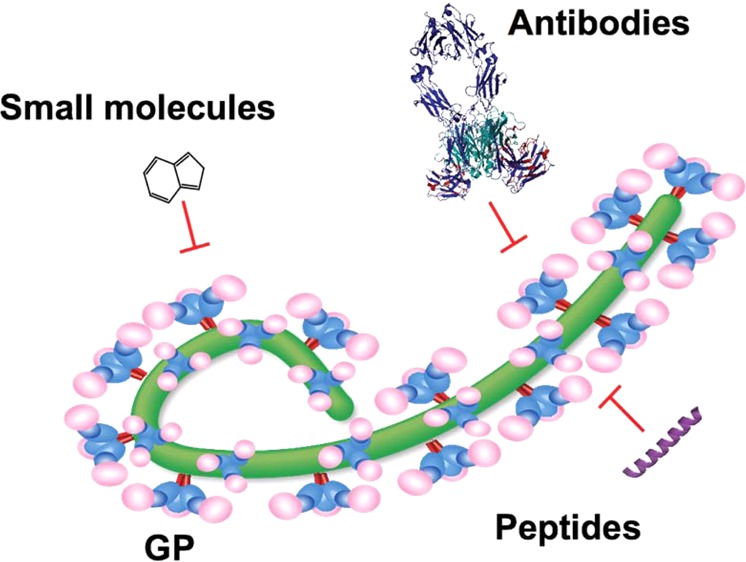
The Ebolaviruses are members of the family Filoviridae (“filoviruses”) and cause severe hemhorragic fever with human case fatality rates as high as 90%. Infection requires attachment of the viral particle to cells and triggering of membrane fusion between the host and viral membranes, a process that occurs in the host endosome and is facilitated by the envelope glycoprotein (GP). One potential strategy for therapeutic intervention is the development of agents (antibodies, peptides, and small molecules) that can interfere with viral entry aspects such as attachment, uptake, priming, or membrane fusion. This paper highlights recent developments in the discovery and evaluation of therapeutic entry inhibitors and identifies opportunities moving forward.
Keywords: Ebola virus, viral hemhorragic fever, viral entry, envelope glycoprotein
The family Filoviridae, the constituents of which are known as filoviruses, includes five species of Ebolaviruses, two species of Marburg virus (MARV), and one species of Cuevavirus.1,2 Infection by pathogenic filovirus species results in severe and rapidly progressing hemorrhagic fever. Each of the Ebolavirus species is named after the region in which it was identified: Zaire (EBOV), Sudan (SUDV), Bundibugyo (BDBV), Tai Forest (TAFV), and Reston (RESTV). Both EBOV and SUDV have been associated with recurring large outbreaks; two isolates of EBOV (Guinea and Sierra Leone) are the cause of the current epidemic in West Africa. The human case fatality rates for each of these Ebolavirus species varies according to outbreak, but in general EBOV has been associated with the highest mortality (up to 90%) followed by SUDV and BDBV.3 Only one case of TAFV has been recorded, and RESTV, although lethal to monkeys, is not known to cause human disease.4 Given this variation in attributes, it is useful to compare the sequences among the envelope glycoproteins (GP), which are critical for viral cell entry and the primary target for antibody responses (elaborated below). Table 1 shows the amino acid similarity and identity among the GPs of the five Ebolavirus species; it is interesting to note that the two most pathogenic species (EBOV and SUDV) are the most distantly related. The prefusion X-ray structures for EBOV GP and SUDV GP have been reported and, although the greater topology of the GP spike remains quite similar, there are subtle variations.5−7 Prefusion GP roughly consists of a chalice-and-bowl morphology, with GP1 comprising the bowl, which sits within the chalice formed by the GP2 subunits.6,7 In this prefusion structure, the GP2 fusion loop wraps around GP1. Ostensibly, the “post-fusion” structures of the fusion subunit (GP2) ectodomain for EBOV and MARV are the same, except for features of specific side chain–side chain interactions that give rise to pH-dependent stability.8
Table 1. Amino Acid Percent Identity and Similarity of the Glycoproteins Compared to GP EBOV for the Five Species of Ebolavirus.
| species (strain) | identity (%) | similarity (%) |
|---|---|---|
| EBOV (Mayinga) | ||
| SUDV (Boniface) | 55 | 69 |
| SUDV (Uganda/Gulu) | 56 | 71 |
| RESTV (Reston) | 58 | 72 |
| TAFV (Cote d’Ivoire) | 65 | 76 |
| BDBV (R4QRCO-9MONO) | 66 | 77 |
The pathology of Ebola and Marburg viral disease (EVD or MVD) arises from the ability of filoviruses to rapidly proliferate in their hosts and overcome the immune response. Although the correlates for survival in humans have not been extensively documented, it appears that survivors of an infection with SUDV continue to produce GP-specific antibodies as late as 12 years postinfection.9 In nonhuman primates (NHPs), protection from viral challenge afforded by convalescent antibody therapy or monoclonal antibody (mAb) cocktails appears to involve seroconversion of the surviving animals and production of endogenous GP-specific IgG in the range of 12 days postviral challenge.56 Ebolaviruses and MARV have broad cellular tropism, but in natural infection macrophages and dendritic cells are the primary targets of infection.10,11 Infected macrophages are unable to stimulate a robust immune response and cause a “cytokine storm” that is proposed to be the primary cause of the blood-clotting abnormalities and vascular leakage characteristic of filovirus hemorrhagic fever.12,13 Damage to other tissues (e.g., liver, kidneys, vascular endothelia) is thought to contribute to hemorrhagic fever symptoms and to late-stage multiorgan failure. Thus, any agent that decreases the spread of the virus in early infection stages has the potential to act as a postexposure therapeutic or pre-exposure prophylactic.
The filovirus genome contains one gene for the envelope GP. MARV GP is encoded by a single open reading frame (ORF),14 and Ebola virus as well as Cuevavirus GP genes express three proteins from individual partially overlapping ORFs: GP1,2 and two secreted glycoproteins (sGP and ssGP).15−17 sGP is translated as a precursor (pre-sGP), which is cleaved by furin protease at its C-terminus to yield mature sGP and a secreted cleavage product (Δ-peptide). Although high levels of sGP and Δ-peptide circulate in the blood, their respective function during the filovirus host cell entry process remains to be elucidated. GP is the sole protein on the viral surface, is necessary and sufficient for infection, and is the primary target of neutralizing antibodies.14,18,19 The prefusion spike consists of three copies each of the two GP subunits, GP1, which mediates cell recognition and uptake, and GP2, which performs the viral membrane fusion reaction. GP1 and GP2 are disulfide bonded in the prefusion spike and result from furin cleavage of a single GP0 precursor.20−22 A brief overview of GP structure and the filovirus entry process is provided here; readers are referred elsewhere for detailed descriptions.18,19,23,24 Filovirus particles are filamentous and studded on the exterior by GP spike assemblies which, in the prefusion form, consist of three copies each of GP1 and GP2. Viral particles bind to the cell and are taken up via a macropinocytosis-like mechanism.23,25−28 GP mediates viral attachment to cells via multiple cell-surface molecules, including lectins (e.g., L-SIGN and DC-SIGN),29−31 the tyrosine kinase receptor Axl,32 and human T cell mucins.33 However, recent studies indicate that the latter two enhance binding and entry of Ebolaviruses into host cells by interacting with phosphatidylserines in the viral membrane rather than through interactions with the GP.34,35 As the host vesicle (containing the viral particle) matures toward an endolysosome, there are at least three critical aspects that are required for viral membrane fusion (Figure 1). The first is cleavage of the prefusion GP spike by host endosomal cysteine proteases cathepsins L and B (CatL/CatB), reducing the 130 kDa GP1 subunit to ∼17 kDa; this processing removes major glycosylated and highly variable regions and exposes a receptor binding domain.36−38 Second, an interaction between the remaining GP1 fragment and a critical endosomal host receptor (or receptors) mediates fusion with the endosomal membrane. Niemann Pick C1 (NPC1) is one critical host factor, and there may be others unidentified; ultimately these trigger GP2 into its active fusogenic conformation.39−41 NPC1, a highly conserved late endosome-residing protein, was identified from a haploid screen and is required for Ebola virus infection in vitro and in vivo. Other host factors involved in the architecture and trafficking of endosomal/lysosomal compartments (cellular GTPases Rab5 and Rab7, and members of the homotypic fusion and vacuole protein-sorting (HOPS) tethering complex) have been shown to contribute to Ebola virus cellular uptake.28,39
Figure 1.

Overview of GP-mediated viral membrane fusion. Upon cell attachment and uptake, the prefusion spike is first processed by CatL/CatB, leaving a 17 kDa fragment of GP1. Interaction of this remaining fragment with NPC1, and potentially other host factors, triggers the membrane fusion cascade. The GP2 fusion loop (FL) inserts into the host cell, creating an extended intermediate conformation that spans both membranes. Collapse of the N- and C-heptad repeat regions (NHR and CHR, respectively) into a six-helix bundle is promoted by low pH and facilitates progression to a hemifusion intermediate. Subsequent events lead to full fusion of both membranes. All of the steps in the fusion pathway, as well as initial cell attachment (not shown here), are susceptible to inhibition by entry inhibitors.
Third, the decreased pH of the maturing endosome is believed to have a direct conformational effect on the fusion subunit, GP2.8,42−44 The primary sequence of GP2 contains an N-terminal fusion loop that has been shown to induce membrane mixing at low pH. By analogy to other class I fusion mechanisms, it is thought that initial triggers result in extension of the fusion loop into the host endosomal membrane, leading to an extended or “pre-hairpin” intermediate.43,45,46 Collapse of this intermediate, by folding of the N- and C-terminal heptad repeat regions (referred to as NHR and CHR, respectively) into a six-helix bundle, is hypothesized to provide the energetic driving force for bringing the two membranes into proximity and promoting initial lipid mixing events. The “post-fusion” ectodomain conformation (exemplified by the six-helix bundle structure) is strongly promoted in low pH for both EBOV and MARV;42,44 this feature likely acts as a method for conformational control so that this late-stage fusion conformation is promoted only in conditions of appropriately matured endosomes and not earlier. After initial lipid mixing events, the precise mechanism of transition to formation of the full fusion pore through which the contents of the viral particle enter the cell has yet to be elucidated.
Virus entry is an attractive target for drug development, because its inhibition hinders viral propagation at an early stage, minimizing the chance of the virus acquiring drug resistance during a later step of the virus life cycle. Entry inhibitors for all viruses can target any critical step of the entry pathway by blocking fusion-associated conformational changes, inhibiting critical protein–protein interactions, or preventing key processing steps. Indeed, entry inhibitors reported for Ebolaviruses and MARV have targeted both host proteins (e.g., NPC1) and viral proteins (e.g., prefusion GP). The goal of this paper is to review efforts within the past five years to develop Ebolavirus entry inhibitors, placing an emphasis on chemical and structural aspects, and to identify opportunities for future development. For simplicity, we have organized this discussion according to class of inhibitor (antibodies, peptides, and small molecules), but we emphasize that the themes for mechanisms of such inhibitors run parallel in many cases. Thus, any particular step could be inhibited by a number of different approaches. Finally, we provide a discussion of opportunities and challenges in this area.
Therapeutic Antibodies
There has been significant recent interest in the use of convalescent antibody therapy or mAbs as postexposure treatments. A large number of antibodies for EBOV have been described.47,48 Here we focus on a handful to illustrate key points. From a systemic perspective, antibody therapies likely work by preventing viral entry (“neutralization”) and/or activation of host complement, antibody-dependent cell-mediated cytotoxicity (ADCC), or other functions related to the antibody Fc region. The extent to which these independent mechanisms contribute is not completely clear, but most antibody cocktails that have been demonstrated to be effective in small or large animals contain at least one mAb with neutralizing activity in vitro. Key components of two first-generation antibody cocktails, namely, MB-003 (MappBio) and ZMab (Defyrus), were recently combined to yield ZMapp, which demonstrates increased efficacy. ZMapp is a cocktail of three mouse-derived mAbs against EBOV GP that were chimerized into human IgG scaffolds (c13C6, c2G4, and c4G7).49−51 This cocktail was provided for compassionate use in several human cases this year and has been demonstrated to reverse the course of EVD 5 days post viral challenge in NHPs.49 Each of the ZMapp mAbs alone is capable of affording partial protection in guinea pigs; two of these mAbs, c2G4 and c4G7, have neutralizing activity in vitro, whereas the third (c13C6) requires complement pathway components.49,52,53 Moreover, recent structural analysis has shown that none of the mutations observed in the currently circulating West Africa EBOV strains occurred within the epitopes of the ZMapp antibodies.54,55 In NHPs, convalescent antibody (derived from distinct GP-vaccinated animals) provides postexposure protection for both EBOV and MARV, up to 48 h postchallenge, and these sera also neutralize in vitro.56 Among the most potent neutralizing antibodies described to date are SUDV-specific mAbs 16F6 (murine) and similar synthetic human mAbs, which can neutralize infection by nearly two logs at midnanomolar concentrations.6,57 Two of the synthetic human antibodies (E10 and F4) as well as 16F6 can provide postexposure protection >80% in an immunocompromised mouse model.57 However, protection of NHPs by any antibody therapy has yet to be demonstrated for SUDV. Postexposure protection of NHPs against any filovirus by a single mAb (as opposed to polyclonal IgG in convalescent antibody therapies or cocktails of mAbs) also has not been shown. An early study indicated that one EBOV-specific mAb, KZ52, did not afford protection from challenge, but it is unknown if altered or increased dosing would have made a difference.48,58
At present, only two antibody–GP X-ray crystal structure complexes have been published (Figure 2),6,7 although others are likely forthcoming, and single-particle electron microscopy (EM) reconstructions of every mAb in the three most efficacious mAb cocktails (MB-003, ZMAb, and ZMapp) were recently made available.55 Combined, these data show that KZ52, a human EBOV-specific mAb, and 16F6, a murine SUDV-specific mAb, as well as the two chimerized antibodies c2G4 and c4G7 bind at similar locations on the overall GP prefusion structure, namely, at the base of the chalice-and-bowl between GP1 and GP2.5,7,59 Structures are not available for the synthetic human mAbs E10 and F4, but competition ELISA suggests they also bind SUDV GP in a similar location.57 Other components of the protective cocktails, c13C6 and c1H3, bind within the glycan cap.55 The KZ52 structural epitope spans both GP1 and GP2, whereas the 16F6 epitope is contained mostly on GP1; the two structural epitopes overlap by 10 residues. These X-ray structures in combination with the structural information on c4G7 and c2G4 suggest that the GP1–GP2 interface of the prefusion spike is particularly susceptible for neutralization.5,7 Thus, a potential strategy for isolating additional neutralizing antibodies, or non-antibody protein therapeutics, would be to target specifically this region on GP.
Figure 2.
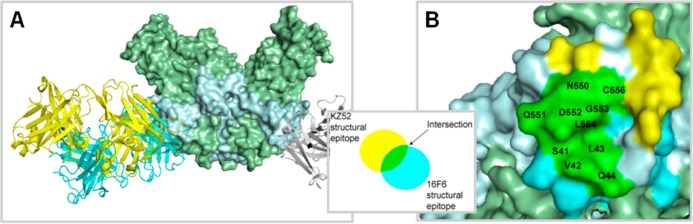
Structure of prefusion GP bound to antibodies. (A) Overlay of complexes for GP (spacefill) bound to KZ52 Fab (yellow, which targets GPEBOV, PDB ID 3CSY, ref (7)) and 16F6 Fab (cyan, which targets GPSUDV, PDB ID 3S88, ref (6)). GP1 is colored green, and GP2 is colored light gray. There is partial overlap in the structural epitopes. (B) Surface representation of GPSUDV with residues that correspond to 16F6, KZ52, or shared structural epitopes color-coded according to the scheme shown in the inset. The two structural epitopes overlap at 10 residues, labeled on the surface representation.
One paradoxical attribute of the SUDV mAbs 16F6, E10, and F4 and the EBOV mAb KZ52 is that, whereas KZ52 binds with a low nanomolar dissociation constant, it leaves ∼10% of virus unneutralized at high antibody concentrations (>130 nM)6,60,61 in both pseudotype and authentic viral entry assays. In contrast, 16F6, E10, and F4 are all much more potent against their respective GPs, neutralizing 2 logs or ∼99% at much lower antibody concentrations. These mAbs bind with affinities in the micromolar range and 16F6 has a much smaller structural epitope than KZ52.5,6 Within the synthetic antibody panel from which E10 and F4 were discovered (all highly related in terms of sequence), there was reasonable correlation between neutralization potency and binding affinity.57 However, at a broader level it appears that binding affinity is not predictive of neutralization potency, nor is neutralization predictive of protection in animals. There are likely other regions in GP that contain neutralizing epitopes that could be mined to some extent by naïve library phage panning or mouse immunizations. Furthermore, GP from EBOV is known to undergo some subtle conformational changes upon exposure to low pH or mild denaturants.62,63 It is possible that cryptic epitopes exist and are accessible on the prefusion structure that can be targeted using a variety of approaches.60 Antibodies against the glycan cap and mucin-like domains (e.g., 13F6 and 14G7) provide in vivo protection, but usually appear non-neutralizing in vitro, possibly due to the removal of their epitopes through cathepsin cleavage in the late endosome.50,53,55,64
From the structural and binding data, it is tempting to assume that the mechanism of antibody neutralization is prevention of fusion-associated conformational changes, but there is little direct evidence for this. Initial cell attachment of filovirus particles is mediated through glycans of the heavily glycosylated mucin-like domain, far away from the sites of binding for KZ52, 16F6, E10, and F4, and therefore inhibition of cell attachment is not likely part of their mechanism.31,65,66 Additionally, Dias et al. found that 16F6 and KZ52 did not block cellular attachment or inhibit cathepsin L cleavage.6 However, because triggering of the viral membrane fusion pathway is dependent upon endosomal factors such as CatL/B, NPC1, and low pH, a model whereby neutralizing antibodies function by blocking fusion-associated conformational changes would necessitate the antibody–GP complex survives with some reasonable lifetime in the endosome. A potential mechanism for KZ52 is to act as a “staple”, locking the two subunits together in the prefusion form, because its structural epitope spans both subunits.7 However, this mechanism would posit that KZ52 must remain bound to GP throughout maturation of the endosome, presumably to the lysosome where the viral particle is degraded before it has a chance to infect. A similar mechanism can be proposed for 16F6 and, by extension, E10 and F4, but the 16F6 structural epitope is contained mostly on GP1; thus it is less clear how 16F6 binding can occlude GP2 conformational rearrangements as efficiently as KZ52.5 A structural and mechanistic dissection of these potential neutralization mechanisms may lend itself to the design of new, more potent antibodies or protein therapeutics. Moreover, efficient antibody-mediated protection likely requires a mixture of antibodies that recognize both the GP1–GP2 interface and the outer regions of the GP. In regard to this, KZ52, which failed to provide protection as a single mAb in NHP studies,58 might be effective in an adequate antibody cocktail.
Peptide Entry Inhibitors
Peptides present an alternative to antibodies for targeting proteins, owing to their ease of synthesis in small scale and potential for chemical modification to improve features such as structure and stability. Precisely folded peptides can be utilized to engage in numerous energetically favorable contacts over a relatively large surface. In addition, peptides serve as excellent tools to identify key pharmacophores and can act as templates on which small molecules that mimic those interactions can be developed. One FDA-approved peptide drug is the human immunodeficiency virus type 1 (HIV-1) fusion inhibitor Fuzeon (enfurvitide/T-20), which corresponds to the CHR of the gp41 fusion subunit.67 It is thought that Fuzeon and related peptides (“C-peptides”) work by binding the NHR segments during the extended/pre-hairpin intermediate and preventing formation of the six-helix bundle that is critical for membrane fusion. This approach has been applied to several class I viruses, and similar concepts have been applied to class II viruses.68,69
Early attempts to develop C-peptides against filoviruses yielded mixed results; one report showed that a peptide corresponding to the CHR could reduce the infectivity of pseudotyped virus, albeit at high concentrations, but another study suggested similar peptides had little activity.70,71 One potential issue with development of a filovirus C-peptide is accessibility of the putative target, the transient extended intermediate. It is presumed that the initial membrane fusion events are not triggered until the virus is deep within the endocytic pathway, because early steps require host endosomal resident proteins (CatL/CatB and NPC1).36,40 Therefore, any therapeutic seeking to engage mid- or late-stage fusion intermediates, such as the GP2 extended intermediate, would have to be present in endosomal compartments.
To overcome these challenges, targeted EBOV C-peptides have been developed in which CHR segments are localized to endosomal compartments or membranes by conjugation to cell penetrating peptide (CPP) domains or cholesterol. One study demonstrated that the potency of C-peptide inhibitors can be substantially enhanced by conjugation to the HIV-1 Tat arginine-rich sequence (referred to as Tat-Ebo).72 The antiviral activity of these endosome-targeting peptides is dependent on both the Tat sequence and the native EBOV CHR sequence. Mechanistic studies suggested that the peptide acts by blocking a membrane fusion intermediate, although this was not unequivocally demonstrated, but defects were evident in late-stage entry steps. However, the concentrations of peptide required for substantial reduction in viral infection are likely too high for the C-peptide to be of clinical relevance. Subsequent efforts focused on the generation and characterization of C-peptides conjugated to cholesterol and Tat-Ebo analogues containing covalent side chain–side chain cross-links to promote α-helical conformation.73 Although the mechanism of inhibition appears to be unspecific (the peptides also showed activity against the vesicular stomatitis virus glycoprotein G), the cholesterol-conjugated C-peptides were potent inhibitors of EBOV glycoprotein-mediated cell entry. Interestingly, it was found that cholesterol conjugation enhances α-helical structure in a concentration-independent manner. These results highlight that caution must be exercised in considering cholesterol conjugated C-peptides as a general therapeutic approach to improve the antiviral activity of C-peptide analogues of class I fusion proteins. In the specific case of EBOV, conjugation to cholesterol enhanced potency but at the cost of specificity. Moreover, side chain–side chain cross-linking enhanced α-helical stability of the Tat-Ebo variants only at neutral pH. However, one side chain–side chain cross-linked peptide with moderately higher activity than the parent compound Tat-Ebo was identified. Recently, a mirror image phage display strategy was utilized to develop reverse chirality peptides (d-peptides) that bind to mimics of the EBOV extended intermediate.74
Although MARV does not produce a Δ-peptide (C-terminal cleavage product of sGP secreted by Ebola virus-infected cells), Fc-tagged versions of EBOV, SUDV, and TAFV Δ-peptides specifically inhibited entry by Ebolaviruses and Marburg virus, indicating that they interfere with a pathway used by all filoviruses to gain entry into their target cells.75 It seems likely that other aspects of GP-mediated viral entry could be targeted with peptides, perhaps acting as starting points for the development of small molecules or themselves acting as potential therapeutic candidates. For example, cyanovirin N (CVN) is a small 10 kDa glycan-binding protein that binds mannose-containing oligosaccharides and, thus, inhibits viral entry of diverse enveloped viruses that possess glycoproteins with a high density of oligomannose glycosylation sites, including HIV-1, hepatitis C, measles, and EBOV, presumably by preventing initial virus glycan–cell interactions.76−78 CVN was also able to delay death due to EBOV challenge in a murine model, although this required multiple high doses.76 Given this, it is conceivable that other peptide inhibitors that can prevent virus–cell attachment may hold promise as entry inhibitors and would have the added advantage that they need not function in the endosome. Later points in the entry pathway might also be susceptible to inhibition by peptides including virus–receptor interactions.
Small Molecules
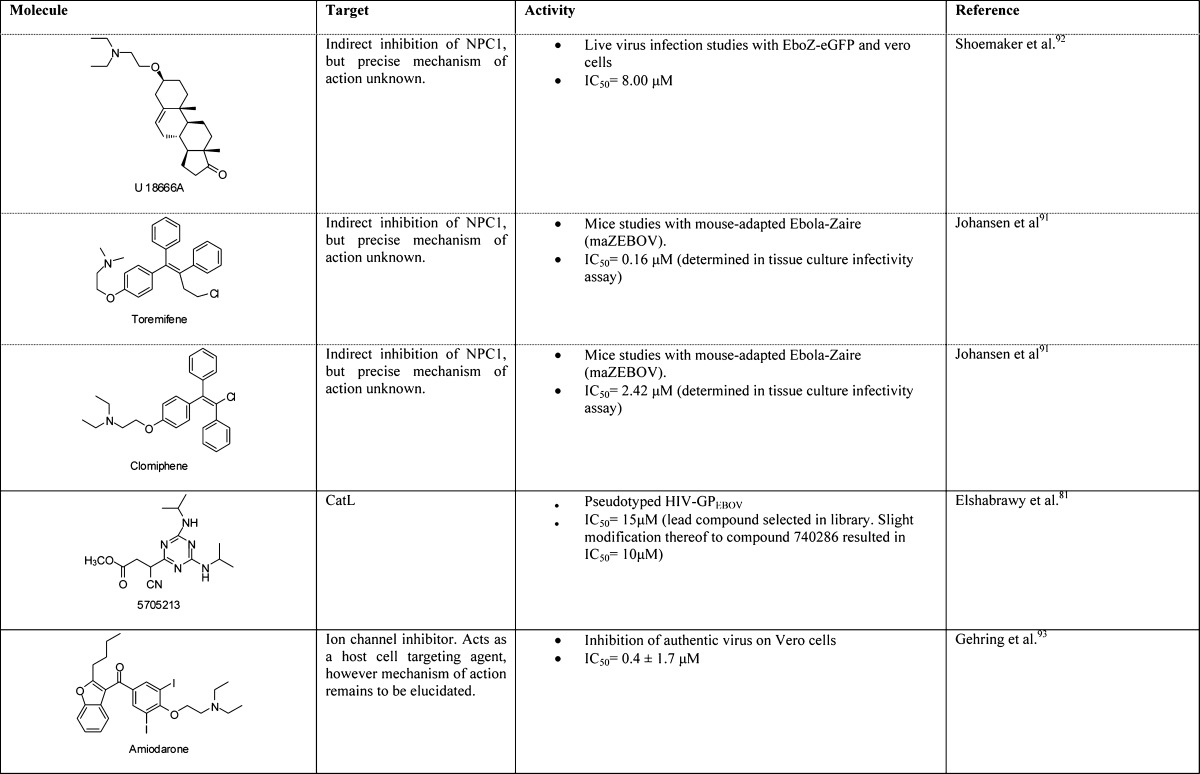
A unique advantage that small molecules possess, particularly in the context of emergency prophylactic or postexposure therapeutics, is that they are orally available and would likely not require refrigeration. Numerous small molecule therapeutics against the filoviruses have been described, some with demonstrated efficacy in nonhuman primates. These compounds fall into a broad range of mechanistic classes, perhaps the most advanced being polymerase inhibitors;79 we focus here on entry inhibitors. An overview of lead compounds and their targets is shown in Table 2. Roughly speaking, these small molecules can be categorized as follows: (i) broad-spectrum viral entry inhibitors, targeting host proteases or the viral membrane; (ii) inhibitors that target specifically elements of filovirus entry, such as GP or NPC1; and (iii) FDA-approved therapeutics that have been repurposed for filoviruses but for which the mechanisms of action are unclear (cationic amphiphiles).
Table 2. Small Molecule Entry Inhibitors for Ebolaviruses.
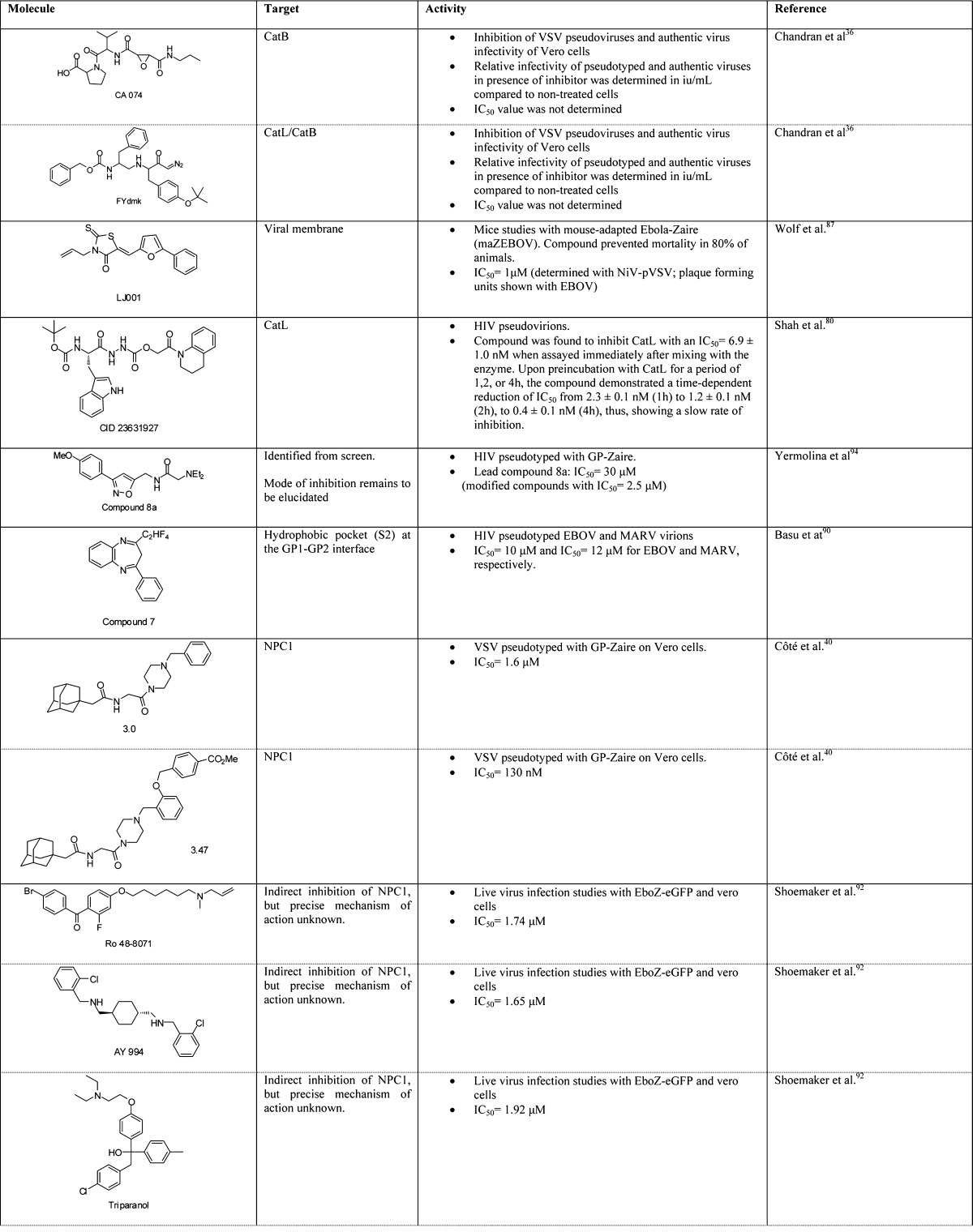
Within the first category are numerous cysteine cathepsin inhibitors. Cysteine cathepsin proteases are susceptible to mechanism-based suicide inhibitors such as epoxide-containing or diazomethane-containing compounds.36 Using the selective CatB inhibitor CA074 and the CatL/CatB inhibitor FYdmk, an essential role for CatB and an accessory role for CatL in EBOV GP-dependent target cell entry could be identified. More recent efforts for targeting CatL include oxocarbazate80 or more diverse structures isolated from a library screen.81 In all cases, inhibition was observed with pseudotyped virus cell-based entry assays and with authentic viruses in some cases. In vitro, there appears to be a strong but not absolute dependence on CatB for GP-mediated entry by some filoviruses (EBOV, TAFV, BDBV), whereas CatL is less critical.36,37,82 Other filoviruses (SUDV and MARV) do not require CatB.82 In vivo, CatB and CatL knockout mice are both still susceptible to EBOV infection and death, suggesting that redundant proteolytic mechanisms may exist.83 Moreover, EBOV can readily adapt to use non-CatB cysteine cathepsins, at least in tissue culture, and a similar situation may exist in animals. Thus, a likely requirement for filovirus entry inhibitors is potent, broad-spectrum inhibition of cysteine proteases residing in the endocytic pathway of all cells. Targeting a general mechanism such as cathepsin cleavage may prove advantageous in that such agents could be used for other endosomal viruses that also require these proteases (such as severe acute respiratory syndrome coronavirus; SARS-CoV);81 however, a potential disadvantage may be toxicity due to loss of host protease function. A critical consideration for these compounds may be whether systemic toxicity (if any) can be tolerated within the required treatment window for postexposure therapy. Several of the epoxide-based cathepsin inhibitors are well-tolerated in mice upon prolonged administration, an important factor in the consideration of compounds with this mechanism of action.84−86
An alternative to targeting a general host factor is to target the viral membrane, a task that has been accomplished recently with the aryl methyldiene rhodanine derivative LJ001.87 The proposed mechanism of this compound and its derivates is specific intercalation into membranes. Unlike biogenic mammalian cells, virions lack the ability to repair membrane damage. Thus, while being capable of binding both membrane types, LJ001 inhibits only virus–cell but not cell–cell fusion, because the intercalation probably causes defects in membrane architecture, and the activation of a thioxo functionality leads to local membrane damage. LJ001 had potent in vitro antiviral activity against EBOV and a number of other enveloped viruses, including influenza A, flaviviruses, poxviruses, and HIV-1. Although 80% of mice pretreated with LJ001 survived a lethal dose of mouse-adapted EBOV challenge, LJ001 did not show efficacy in postexposure mouse model experiments due to a suboptimal pharmacokinetic profile. Nonetheless, these results provide proof-of-concept that a general membrane-targeted reagent could serve as a broad-spectrum antiviral.
Upon cleavage, the protein–protein interaction between cleaved GP1 and NPC1 is critical for triggering the membrane fusion machinery, although it remains to be determined if other factors are involved.39,40 Both genetic and chemical ablations of this interaction have been shown to reduce infectivity in vitro and, in NPC1 heterozygotic (NPC1+/− mice), there is a substantial resistance to both EBOV and MARV lethal challenge. Protein–protein interactions have been a challenging class of targets for antagonism with small molecules, because interfacial surfaces are usually large, dynamic, and lack deep binding pockets into which a small molecule could bind and efficiently occlude the interaction.88 Nonetheless, an adamantane class of compounds (e.g., 3.47) appears to function by preventing GP binding to NPC1.40 A parent compound, 3.0, was identified from a small molecule screen for inhibition of pseudotyped virus infection, specific to EBOV GP. Subsequent structure–activity relationship (SAR) analysis and pull-down experiments suggested this class interacts directly with NPC1 and inhibits a GP–NPC1 interaction.89 However, neither a direct KD measurement nor structural evidence of the small molecule–NPC1 interaction has been reported. A separate small molecule screen identified a benzodiazepine derivative (compound 7), which likely targets a hydrophobic pocket (S2) at the GP1–GP2 interface in the prefusion conformation, as evidenced by ablation of activity upon mutation of specific GP residues.90 Whereas it remains to be determined whether compound 7 inhibits GP–NPC1 interactions or prevents GP-associated conformational changes, these studies suggest that the S2 hydrophobic pocket can serve as a well-defined small molecule drug target for the inhibition of EBOV infection.
A third mechanistic class includes cationic amphiphiles, which have been isolated from multiple screens of FDA-approved drugs. For example, the selective estrogen receptor modulators clomiphene, used as an infertility treatment to induce ovulation, and toremifiene, used as a breast cancer chemotherapeutic, both inhibit EBOV infection in vitro at a late-stage entry step.91 A broader panel of cationic amphiphiles has recently been shown to have similar activities,92 and an independent study identified amiodarone,93 used to treat cardiac arrhythmias, and other ion channel inhibitors with similar properties. The precise mechanism by which cationic amphiphiles inhibit viral entry is not completely understood, but it appears that they are distinct from that of 3.47 in that they do not directly inhibit the GP–NPC1 interaction. Alternative proposed mechanisms include allosteric modulation of NPC1, disruption of NPC1 by general disfunction of the endolysosomal membrane, and interaction with a yet-unidentified additional required host factor.91,92 Although the prospect of repurposing FDA-approved drugs for use as antifilovirus drugs is appealing from the standpoint that these agents have already passed safety studies, it is important to emphasize that dosing will likely be a critical issue. Such compounds likely have a very specific treatment window for their intended indications, and their use as antifilovirus drugs may require increased amounts to combat the high viral titers that may go beyond the approved therapeutic window. Nonetheless, the in vitro data for these compounds are compelling enough to warrant further evaluation in animals.
In addition to the classes listed above, Yermolina et al. reported a newly developed antifiloviral screening system based on EBOV-GP pseudotyped HIV particles to identify compound 8a, which selectively inhibited EBOV and MARV infection in human cells.94 Although subsequent modifications improved the antiviral activity of this lead compound, its mode of entry inhibition remains to be elucidated.
Present Challenges and Opportunities
Among the three classes of entry inhibitors discussed here, antibodies are the most advanced in terms of development. Although the efficacy of cocktails, such as ZMapp, for EBOV in nonhuman primates is clear, a challenge moving forward will be to identify similar treatments for other filoviruses. The convalescent serum protection studies with MARV suggest that it can also be targeted with appropriate antibody therapies. Furthermore, analysis of sera in NHPs treated with one broad vaccine candidate, a mixture of vesicular stomatitis virus particles containing GPs from EBOV, SUDV, and BDBV, suggests that induction of neutralizing antibodies is a significant component of vaccine-induced protection.95 It therefore seems likely that protection from other filovirus species with mAb cocktails is possible and that cross-protection with appropriately engineered cross-neutralizing cocktails or broadly neutralizing antibodies merits investigation. In addition, further molecular studies on the mechanism of neutralization may yield new insight into how to develop more potent and effective mAbs for future development.
Peptide inhibitors of viral membrane fusion have yielded important mechanistic insights into infection, but a critical challenge for use of these compounds will be their in vivo half-life and whether the extended intermediate is accessible enough to be a viable therapeutic target. One HIV-1 C-peptide, fuzeon/enfurvirtide/T-20, has provided some clinical utility; however, filovirus C-peptides have the added complication that they must be delivered to endosomes of susceptible cell types at high enough levels to induce a therapeutic effect. Whereas conjugation to various targeting agents (CPP or cholesterol) was an interesting and straightforward strategy, this approach may also divert the peptides to a broad number of cell types and therefore dilute antiviral activity. Furthermore, cationic peptides in general suffer from high systemic toxicity. Nonetheless, such agents could likely serve as valuable research reagents, and new peptides could be developed to target other aspects of the entry pathway.
Small molecule entry inhibitors that target the host or virus may prove to be clinically useful, especially when considered as part of a cocktail with compounds that inhibit viral replication (e.g., polymerase chain terminators). A critical challenge moving forward will be identifying compounds that exhibit therapeutic effects in animal models within reasonable treatment windows. Evaluation of these compounds in mouse protection models will be critical to prioritizing them for potential therapeutic efficacy. As with antibodies, additional mechanistic information specifically for the cationic amphiphiles will be beneficial from both the basic and applied perspectives. Finally, one unexplored area is the potential for combination of antibody and small molecules as antibody–drug conjugates (ADCs). This approach is widely popular for cancer therapies, and proof-of-concept has been demonstrated for HIV-1.96,97 Such an approach could leverage the specificity of monoclonal antibodies with the in vitro potency of small molecule inhibitors.
Filovirus entry is a complicated process requiring multiple endosomal components. The inhibitors discussed herein are thought to function at a variety of steps including cell attachment (CVN), proteolytic processing (cathepsin inhibitors), host receptor interaction (3.47), and other aspects of membrane fusion (C-peptides, cationic amphiphiles, and antibodies). Which of these steps is most susceptible in vivo remains to be determined, but those agents that target the early prefusion intermediates of GP have the added advantage that they do not require delivery to the endosome for function. Nonetheless, strategies that seek to disrupt these specific steps in the pathway have all been validated in vitro, and some in vivo, thereby paving the way for other structural and chemical approaches to the development of filovirus entry inhibitors.
Acknowledgments
We thank Kartik Chandran and Jayne F. Koellhoffer for assistance with figure preparation and helpful discussions.
Glossary
Abbreviations
- EBOV
Zaire Ebola virus
- SUDV
Sudan Ebola virus
- BDBV
Bundibugyo Ebola virus
- TAFV
Tai Forest Ebola virus
- RESTV
Reston Ebola virus
- MARV
Marburg virus
- EVD
Ebola viral disease
- MVD
Marburg viral disease
- NHP
nonhuman primates
- mAb
monoclonal antibody
- GP
envelope glycoprotein
- ORF
open reading frame
- CatB
cathepsin B
- CatL
cathepsin L
- NPC1
Niemann Pick C1 receptor
- NHR
N-terminal heptad repeat
- HOPS
homotypic fusion and vacuole protein-sorting
- CHR
C-terminal heptad repeat
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ELISA
enzyme-linked immunosorbent assay
- FDA
U.S. Food and Drug Administration
- HIV-1
human immunodeficiency virus type 1
- CCP
cell penetrating peptide
- CVN
cyanovirin N
- SARS-CoV
severe acute respiratory syndrome coronavirus
- SAR
structure–activity relationship
- ADC
antibody–drug conjugate
Work in our laboratory is currently funded by the National Institutes of Health (AI090249 and U19AI109762).
The authors declare the following competing financial interest(s): The Albert Einstein College of Medicine has filed two patent applications on the humanized mAbs E10 and F4 discussed herein (U.S. Provisional Patent Application No. 14/291,608 and U.S. Utility Application No. 14/291,608) entitled “Therapy for Filovirus Infection” with J.C.F. and J.R.L. as two of the co-inventors.
References
- Kuhn J. H.; Bao Y.; Bavari S.; Becker S.; Bradfute S.; Brister J. R.; Bukreyev A. A.; Chandran K.; Davey R. A.; Dolnik O. (2013) Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Arch. Virol. 158(1), 301–311 10.1007/s00705-012-1454-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn J. H.; Becker S.; Ebihara H.; Geisbert T. W.; Johnson K. M.; Kawaoka Y.; Lipkin W. I.; Negredo A. I.; Netesov S. V.; Nichol S. T. (2010) Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 155(12), 2083–2103 10.1007/s00705-010-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H.; Geisbert T. W. (2011) Ebola haemorrhagic fever. Lancet 377(9768), 849–862 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M. E. G.; Miranda N. L. J. (2011) Reston ebolavirus in humans and animals in the Philippines: a review. J. Infect. Dis. 204(Suppl. 3), S757–S760 10.1093/infdis/jir296. [DOI] [PubMed] [Google Scholar]
- Bale S.; Dias J. M.; Fusco M. L.; Hashiguchi T.; Wong A. C.; Liu T.; Keuhne A. I.; Li S.; Woods V. L. Jr.; Chandran K. (2012) Structural basis for differential neutralization of ebolaviruses. Viruses 4(4), 447–470 10.3390/v4040447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias J. M.; Kuehne A. I.; Abelson D. M.; Bale S.; Wong A. C.; Halfmann P.; Muhammad M. A.; Fusco M. L.; Zak S. E.; Kang E.; Kawaoka Y.; Chandran K.; Dye J. M.; Saphire E. O. (2011) A shared structural solution for neutralizing ebolaviruses. Nat. Struct. Mol. Biol. 18(12), 1424–1427 10.1038/nsmb.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E.; Fusco M. L.; Hessell A. J.; Oswald W. B.; Burton D. R.; Saphire E. O. (2008) Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454(7201), 177–182 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koellhoffer J. F.; Malashkevich V. N.; Harrison J. S.; Toro R.; Bhosle R. C.; Chandran K.; Almo S. C.; Lai J. R. (2012) Crystal structure of the Marburg virus GP2 core domain in its postfusion conformation. Biochemistry 51(39), 7665–7675 10.1021/bi300976m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobarzo A.; Ochayon D. E.; Lutwama J. J.; Balinandi S.; Guttman O.; Marks R. S.; Kuehne A. I.; Dye J. M.; Yavelsky V.; Lewis E. C.; Lobel L. (2013) Persistent immune responses after Ebola virus infection. New Engl. J. Med. 369(5), 492–493 10.1056/NEJMc1300266. [DOI] [PubMed] [Google Scholar]
- Schnittler H.-J.; Feldmann H. (1998) Marburg and Ebola hemorrhagic fevers: does the primary course of infection depend on the accessibility of organ-specific macrophages?. Clin. Infect. Dis. 27(2), 404–406 10.1086/517704. [DOI] [PubMed] [Google Scholar]
- Bray M.; Geisbert T. W. (2005) Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 37(8), 1560–1566 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Hoenen T.; Groseth A.; Falzarano D.; Feldmann H. (2006) Ebola virus: unravelling pathogenesis to combat a deadly disease. Trends Mol. Med. 12(5), 206–215 10.1016/j.molmed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Takada A.; Kawaoka Y. (2001) The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol. 9(10), 506–511 10.1016/S0966-842X(01)02201-6. [DOI] [PubMed] [Google Scholar]
- Feldmann H.; Volchkov V. E.; Volchkova V. A.; Ströher U.; Klenk H.-D. (2001) Biosynthesis and role of filoviral glycoproteins. J. Gen. Virol. 82(12), 2839–2848. [DOI] [PubMed] [Google Scholar]
- Mehedi M.; Falzarano D.; Seebach J.; Hu X.; Carpenter M. S.; Schnittler H.-J.; Feldmann H. (2011) A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J. Virol. 85(11), 5406–5414 10.1128/JVI.02190-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A.; Trappier S. G.; Mahy B. W.; Peters C. J.; Nichol S. T. (1996) The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U.S.A. 93(8), 3602–3607 10.1073/pnas.93.8.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchkov V. E.; Becker S.; Volchkova V. A.; Ternovoj V. A.; Kotov A. N.; Netesov S. V.; Klenk H.-D. (1995) GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and Vaccinia virus polymerases. Virology 214(2), 421–430 10.1006/viro.1995.0052. [DOI] [PubMed] [Google Scholar]
- Lee J. E.; Saphire E. O. (2009) Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 4(6), 621–635 10.2217/fvl.09.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada A.; Robison C.; Goto H.; Sanchez A.; Murti K. G.; Whitt M. A.; Kawaoka Y. (1997) A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 94(26), 14764–14769 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers S. A.; Sanders D. A.; Sanchez A. (2002) Covalent modifications of the Ebola virus glycoprotein. J. Virol. 76(24), 12463–12472 10.1128/JVI.76.24.12463-12472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A.; Yang Z.-Y.; Xu L.; Nabel G. J.; Crews T.; Peters C. J. (1998) Biochemical analysis of the secreted and virion glycoproteins of Ebola virus. J. Virol. 72(8), 6442–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchkov V. E.; Feldmann H.; Volchkova V. A.; Klenk H.-D. (1998) Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. U.S.A. 95(10), 5762–5767 10.1073/pnas.95.10.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt C. L.; Lennemann N. J.; Maury W. (2012) Filovirus entry: a novelty in the viral fusion world. Viruses 4(2), 258–275 10.3390/v4020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. H.; Chandran K. (2012) Filovirus entry into cells - new insights. Curr. Opin. Virol. 2(2), 206–214 10.1016/j.coviro.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt C. L.; Kolokoltsov A. A.; Davey R. A.; Maury W. (2011) The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 85(1), 334–347 10.1128/JVI.01278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulherkar N.; Raaben M.; de la Torre J. C.; Whelan S. P.; Chandran K. (2011) The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 419(2), 72–83 10.1016/j.virol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbo A.; Imai M.; Watanabe S.; Noda T.; Takahashi K.; Neumann G.; Halfmann P.; Kawaoka Y. (2010) Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathogens 6(9), e1001121 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed M. F.; Kolokoltsov A. A.; Albrecht T.; Davey R. A. (2010) Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathogens 6(9), e1001110 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez C. P.; Lasala F.; Carrillo J.; Muñiz O.; Corbí A. L.; Delgado R. (2002) C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 76(13), 6841–6844 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X.; Olinger G. G.; Aris S.; Chen Y.; Gewurz H.; Spear G. T. (2005) Mannose-binding lectin binds to Ebola and Marburg envelope glycoproteins, resulting in blocking of virus interaction with DC-SIGN and complement-mediated virus neutralization. J. Gen. Virol. 86(9), 2535–2542 10.1099/vir.0.81199-0. [DOI] [PubMed] [Google Scholar]
- Takada A.; Fujioka K.; Tsuiji M.; Morikawa A.; Higashi N.; Ebihara H.; Kobasa D.; Feldmann H.; Irimura T.; Kawaoka Y. (2004) Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Virol. 78(6), 2943–2947 10.1128/JVI.78.6.2943-2947.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima M.; Ikeda Y.; Kawaoka Y. (2007) The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 196(Suppl. 2), S259–S263 10.1086/520594. [DOI] [PubMed] [Google Scholar]
- Kondratowicz A. S.; Lennemann N. J.; Sinn P. L.; Davey R. A.; Hunt C. L.; Moller-Tank S.; Meyerholz D. K.; Rennert P.; Mullins R. F.; Brindley M. (2011) T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. U.S.A. 108(20), 8426–8431 10.1073/pnas.1019030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemielity S.; Wang J. J.; Chan Y. K.; Ahmed A. A.; Li W.; Monahan S.; Bu X.; Farzan M.; Freeman G. J.; Umetsu D. T. (2013) TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathogens 9(3), e1003232 10.1371/journal.ppat.1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller-Tank S.; Kondratowicz A. S.; Davey R. A.; Rennert P. D.; Maury W. (2013) Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J. Virol. 87(15), 8327–8341 10.1128/JVI.01025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K.; Sullivan N. J.; Felbor U.; Whelan S. P.; Cunningham J. M. (2005) Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308(5728), 1643–1645 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schornberg K.; Matsuyama S.; Kabsch K.; Delos S.; Bouton A.; White J. (2006) Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 80(8), 4174–4178 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube D.; Brecher M. B.; Delos S. E.; Rose S. C.; Park E. W.; Schornberg K. L.; Kuhn J. H.; White J. M. (2009) The primed ebolavirus glycoprotein (19-kilodalton GP1, 2): sequence and residues critical for host cell binding. J. Virol. 83(7), 2883–2891 10.1128/JVI.01956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J. E.; Raaben M.; Wong A. C.; Herbert A. S.; Obernosterer G.; Mulherkar N.; Kuehne A. I.; Kranzusch P. J.; Griffin A. M.; Ruthel G. (2011) Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477(7364), 340–343 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M.; Misasi J.; Ren T.; Bruchez A.; Lee K.; Filone C. M.; Hensley L.; Li Q.; Ory D.; Chandran K. (2011) Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477(7364), 344–348 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. H.; Obernosterer G.; Raaben M.; Herbert A. S.; Deffieu M. S.; Krishnan A.; Ndungo E.; Sandesara R. G.; Carette J. E.; Kuehne A. I. (2012) Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 31(8), 1947–1960 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison J. S.; Higgins C. D.; Chandran K.; Lai J. R. (2011) Designed protein mimics of the Ebola virus glycoprotein GP2 α-helical bundle: stability and pH effects. Protein Sci. 20(9), 1587–1596 10.1002/pro.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison J. S.; Higgins C. D.; O’Meara M. J.; Koellhoffer J. F.; Kuhlman B. A.; Lai J. R. (2013) Role of electrostatic repulsion in controlling pH-dependent conformational changes of viral fusion proteins. Structure 21(7), 1085–1096 10.1016/j.str.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison J. S.; Koellhoffer J. F.; Chandran K.; Lai J. R. (2012) Marburg virus glycoprotein GP2: pH-dependent stability of the ectodomain α-helical bundle. Biochemistry 51(12), 2515–2525 10.1021/bi3000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malashkevich V. N.; Schneider B. J.; McNally M. L.; Milhollen M. A.; Pang J. X.; Kim P. S. (1999) Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-Ã... resolution. Proc. Natl. Acad. Sci. U.S.A. 96(6), 2662–2667 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W.; Carfí A.; Lee K.-H.; Skehel J. J.; Wiley D. C. (1998) Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell 2(5), 605–616 10.1016/S1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- Richardson J. S.; Dekker J. D.; Croyle M. A.; Kobinger G. P. (2010) Recent advances in Ebolavirus vaccine development. Human Vaccines 6(6), 439–449 10.4161/hv.6.6.11097. [DOI] [PubMed] [Google Scholar]
- Saphire E. O. (2013) An update on the use of antibodies against the filoviruses. Immunotherapy 5(11), 1221–1233 10.2217/imt.13.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X.; Wong G.; Audet J.; Bello A.; Fernando L.; Alimonti J. B.; Fausther-Bovendo H.; Wei H.; Aviles J.; Hiatt E. (2014) Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 10.1038/nature13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olinger G. G.; Pettitt J.; Kim D.; Working C.; Bohorov O.; Bratcher B.; Hiatt E.; Hume S. D.; Johnson A. K.; Morton J. (2012) Delayed treatment of Ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques. Proc. Natl. Acad. Sci. U.S.A. 109(44), 18030–18035 10.1073/pnas.1213709109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin L.; Pettitt J.; Scully C.; Bohorova N.; Kim D.; Pauly M.; Hiatt A.; Ngo L.; Steinkellner H.; Whaley K. J. (2011) Enhanced potency of a fucose-free monoclonal antibody being developed as an Ebola virus immunoprotectant. Proc. Natl. Acad. Sci. U.S.A. 108(51), 20690–20694 10.1073/pnas.1108360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X.; Fernando L.; Melito P. L.; Audet J.; Feldmann H.; Kobinger G.; Alimonti J. B.; Jones S. M. (2012) Ebola GP-specific monoclonal antibodies protect mice and guinea pigs from lethal Ebola virus infection. PLoS Neglected Trop. Dis. 6(3), e1575 10.1371/journal.pntd.0001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. A.; Hevey M.; Bakken R.; Guest S.; Bray M.; Schmaljohn A. L.; Hart M. K. (2000) Epitopes involved in antibody-mediated protection from Ebola virus. Science 287(5458), 1664–1666 10.1126/science.287.5458.1664. [DOI] [PubMed] [Google Scholar]
- Gire S. K.; Goba A.; Andersen K. G.; Sealfon R. S. G.; Park D. J.; Kanneh L.; Jalloh S.; Momoh M.; Fullah M.; Dudas G. (2014) Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345(6202), 1369–1372 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murin C. D.; Fusco M. L.; Bornholdt Z. A.; Qiu X.; Olinger G. G.; Zeitlin L.; Kobinger G. P.; Ward A. B.; Saphire E. O. (2014) Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc. Natl. Acad. Sci. U.S.A. 10.1073/pnas.1414164111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye J. M.; Herbert A. S.; Kuehne A. I.; Barth J. F.; Muhammad M. A.; Zak S. E.; Ortiz R. A.; Prugar L. I.; Pratt W. D. (2012) Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc. Natl. Acad. Sci. U.S.A. 109(13), 5034–5039 10.1073/pnas.1200409109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Koellhoffer J. F.; Zak S. E.; Frei J. C.; Liu N.; Long H.; Ye W.; Nagar K.; Pan G.; Chandran K. (2014) Synthetic antibodies with a human framework that protect mice from lethal Sudan Ebolavirus challenge. ACS Chem. Biol. 9(10), 2263–2273 10.1021/cb5006454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald W. B.; Geisbert T. W.; Davis K. J.; Geisbert J. B.; Sullivan N. J.; Jahrling P. B.; Parren P. W. H. I.; Burton D. R. (2007) Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathogens 3(1), e9 10.1371/journal.ppat.0030009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias J. o. M.; Kuehne A. I.; Abelson D. M.; Bale S.; Wong A. C.; Halfmann P.; Muhammad M. A.; Fusco M. L.; Zak S. E.; Kang E. (2011) A shared structural solution for neutralizing ebolaviruses. Nat. Struct. Mol. Biol. 18(12), 1424–1427 10.1038/nsmb.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koellhoffer J. F.; Chen G.; Sandesara R. G.; Bale S.; Ollmann Saphire E.; Chandran K.; Sidhu S. S.; Lai J. R. (2012) Two synthetic antibodies that recognize and neutralize distinct proteolytic forms of the Ebola virus envelope glycoprotein. ChemBioChem 13(17), 2549–2557 10.1002/cbic.201200493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T.; Rodriguez L. L.; Jahrling P. B.; Sanchez A.; Khan A. S.; Nichol S. T.; Peters C. J.; Parren P. W. H. I.; Burton D. R. (1999) Ebola virus can be effectively neutralized by antibody produced in natural human infection. J. Virol. 73(7), 6024–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S.; Liu T.; Li S.; Wang Y.; Abelson D.; Fusco M.; Woods V. L. Jr; Saphire E. O. (2011) Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Neglected Trop. Dis. 5(11), e1395 10.1371/journal.pntd.0001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecher M.; Schornberg K. L.; Delos S. E.; Fusco M. L.; Saphire E. O.; White J. M. (2012) Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J. Virol. 86(1), 364–372 10.1128/JVI.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt J.; Zeitlin L.; Kim D. H.; Working C.; Johnson J. C.; Bohorov O.; Bratcher B.; Hiatt E.; Hume S. D.; Johnson A. K. (2013) Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci. Transl. Med. 5(199), 199ra113 10.1126/scitranslmed.3006608. [DOI] [PubMed] [Google Scholar]
- Marzi A.; Wegele A.; Pöhlmann S. (2006) Modulation of virion incorporation of Ebolavirus glycoprotein: effects on attachment, cellular entry and neutralization. Virology 352(2), 345–356 10.1016/j.virol.2006.04.038. [DOI] [PubMed] [Google Scholar]
- Matsuno K.; Nakayama E.; Noyori O.; Marzi A.; Ebihara H.; Irimura T.; Feldmann H.; Takada A. (2010) C-type lectins do not act as functional receptors for filovirus entry into cells. Biochem. Biophys. Res. Commun. 403(1), 144–148 10.1016/j.bbrc.2010.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. (2013) Synthesized peptide inhibitors of HIV-1 gp41-dependent membrane fusion. Curr. Pharm. Design 19(10), 1800–1809 10.2174/1381612811319100004. [DOI] [PubMed] [Google Scholar]
- Badani H.; Garry R. F.; Wimley W. C. (2014) Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim. Biophys. Acta (BBA)–Biomembranes 1838(9), 2180–2197 10.1016/j.bbamem.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessi A.; Langella A.; Capitò E.; Ghezzi S.; Vicenzi E.; Poli G.; Ketas T.; Mathieu C.; Cortese R.; Horvat B. (2012) A general strategy to endow natural fusion-protein-derived peptides with potent antiviral activity. PLoS One 7(5), e36833 10.1371/journal.pone.0036833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netter R. C.; Amberg S. M.; Balliet J. W.; Biscone M. J.; Vermeulen A.; Earp L. J.; White J. M.; Bates P. (2004) Heptad repeat 2-based peptides inhibit avian sarcoma and leukosis virus subgroup a infection and identify a fusion intermediate. J. Virol. 78(24), 13430–13439 10.1128/JVI.78.24.13430-13439.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S.; Takada A.; Watanabe T.; Ito H.; Kida H.; Kawaoka Y. (2000) Functional importance of the coiled-coil of the Ebola virus glycoprotein. J. Virol. 74(21), 10194–10201 10.1128/JVI.74.21.10194-10201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. H.; Harrison J. S.; Radoshitzky S. R.; Higgins C. D.; Chi X.; Dong L.; Kuhn J. H.; Bavari S.; Lai J. R.; Chandran K. (2011) Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J. Biol. Chem. 286(18), 15854–15861 10.1074/jbc.M110.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins C. D.; Koellhoffer J. F.; Chandran K.; Lai J. R. (2013) C-peptide inhibitors of Ebola virus glycoprotein-mediated cell entry: effects of conjugation to cholesterol and side chain-side chain crosslinking. Bioorg. Med. Chem. Lett. 23(19), 5356–5360 10.1016/j.bmcl.2013.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton T. R.; Weinstock M. T.; Jacobsen M. T.; Szabo-Fresnais N.; Pandya M. J.; Whitby F. G.; Herbert A. S.; Prugar L. I.; McKinnon R.; Hill C. P.; Welch B. D.; Dye J. M.; Eckert D. M.; Kay M. S. (2014) Design and characterization of ebolavirus GP prehairpin intermediate mimics as drug targets. Protein Sci. 10.1002/pro.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoshitzky S. R.; Warfield K. L.; Chi X.; Dong L.; Kota K.; Bradfute S. B.; Gearhart J. D.; Retterer C.; Kranzusch P. J.; Misasi J. N.; Hogenbirk M. A.; Wahl-Jensen V.; Volchkov V. E.; Cunningham J. M.; Jahrling P. B.; Aman M. J.; Bavari S.; Farzan M.; Kuhn J. H. (2011) Ebolavirus Δ-peptide immunoadhesins inhibit Marburgvirus and Ebolavirus cell entry. J. Virol. 85(17), 8502–8513 10.1128/JVI.02600-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos L. G.; O’Keefe B. R.; Bray M.; Sanchez A.; Gronenborn A. M.; Boyd M. R. (2003) Cyanovirin-N binds to the viral surface glycoprotein, GP1,2 and inhibits infectivity of Ebola virus. Antiviral Res. 58(1), 47–56 10.1016/S0166-3542(02)00183-3. [DOI] [PubMed] [Google Scholar]
- Dey B.; Lerner D. L.; Lusso P.; Boyd M. R.; Elder J. H.; Berger E. A. (2000) Multiple antiviral activities of cyanovirin-N: blocking of human immunodeficiency virus type 1 gp120 Interaction with CD4 and coreceptor and inhibition of diverse enveloped viruses. J. Virol. 74(10), 4562–4569 10.1128/JVI.74.10.4562-4569.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helle F. o.; Wychowski C.; Vu-Dac N.; Gustafson K. R.; Voisset C. c.; Dubuisson J. (2006) Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J. Biol. Chem. 281(35), 25177–25183 10.1074/jbc.M602431200. [DOI] [PubMed] [Google Scholar]
- Warren T. K.; Wells J.; Panchal R. G.; Stuthman K. S.; Garza N. L.; Van Tongeren S. A.; Dong L.; Retterer C. J.; Eaton B. P.; Pegoraro G.; Honnold S.; Bantia S.; Kotian P.; Chen X.; Taubenheim B. R.; Welch L. S.; Minning D. M.; Babu Y. S.; Sheridan W. P.; Bavari S. (2014) Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508(7496), 402–405 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P. P.; Wang T.; Kaletsky R. L.; Myers M. C.; Purvis J. E.; Jing H.; Huryn D. M.; Greenbaum D. C.; Smith A. B.; Bates P.; Diamond S. L. (2010) A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and Ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 78(2), 319–324 10.1124/mol.110.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshabrawy H. A.; Fan J.; Haddad C. S.; Ratia K.; Broder C. C.; Caffrey M.; Prabhakar B. S. (2014) Identification of a broad-spectrum antiviral small molecule against severe acute respiratory syndrome coronavirus and Ebola, Hendra, and Nipah viruses by using a novel high-throughput screening assay. J. Virol. 88(8), 4353–4365 10.1128/JVI.03050-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misasi J.; Chandran K.; Yang J.-Y.; Considine B.; Filone C. M.; Côté M.; Sullivan N.; Fabozzi G.; Hensley L.; Cunningham J. (2012) Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J. Virol. 86(6), 3284–3292 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi A.; Reinheckel T.; Feldmann H. (2012) Cathepsin B & L are not required for Ebola virus replication. PLoS Neglected Trop. Dis. 6(12), e1923 10.1371/journal.pntd.0001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu K.; Inazuki K.; Hosoya J.; Satoh S. (1986) Beneficial effect of new thiol protease inhibitors, epoxide derivatives, on dystrophic mice. Exp. Neurol. 91(1), 23–29 10.1016/0014-4886(86)90022-1. [DOI] [PubMed] [Google Scholar]
- Morimoto M.; Tanabe F.; Kasai H.; Ito M. (2007) Effect of a thiol proteinase inhibitor, E-64-d, on susceptibility to infection with Staphylococcus aureus in Chediak-Higashi syndrome (beige) mice. Int. Immunopharmacol. 7(7), 973–980 10.1016/j.intimp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Trinchese F.; Liu S.; Zhang H.; Hidalgo A.; Schmidt S. D.; Yamaguchi H.; Yoshii N.; Mathews P. M.; Nixon R. A.; Arancio O. (2008) Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Invest. 118(8), 2796–2807 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M. C.; Freiberg A. N.; Zhang T.; Akyol-Ataman Z.; Grock A.; Hong P. W.; Li J.; Watson N. F.; Fang A. Q.; Aguilar H. C.; Porotto M.; Honko A. N.; Damoiseaux R.; Miller J. P.; Woodson S. E.; Chantasirivisal S.; Fontanes V.; Negrete O. A.; Krogstad P.; Dasgupta A.; Moscona A.; Hensley L. E.; Whelan S. P.; Faull K. F.; Holbrook M. R.; Jung M. E.; Lee B. (2010) A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. U.S.A. 107(7), 3157–3162 10.1073/pnas.0909587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. J. (2009) Inhibition of protein-protein interactions using designed molecules. Chem. Soc. Rev. 38(12), 3289–3300 10.1039/b807197g. [DOI] [PubMed] [Google Scholar]
- Lee K.; Ren T.; Côté M.; Gholamreza B.; Misasi J.; Bruchez A.; Cunningham J. (2013) Inhibition of Ebola virus infection: identification of Niemann-Pick C1 as the target by optimization of a chemical probe. ACS Med. Chem. Lett. 4(2), 239–243 10.1021/ml300370k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A.; Li B.; Mills D. M.; Panchal R. G.; Cardinale S. C.; Butler M. M.; Peet N. P.; Majgier-Baranowska H.; Williams J. D.; Patel I. (2011) Identification of a small-molecule entry inhibitor for filoviruses. J. Virol. 85(7), 3106–3119 10.1128/JVI.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen L. M.; Brannan J. M.; Delos S. E.; Shoemaker C. J.; Stossel A.; Lear C.; Hoffstrom B. G.; DeWald L. E.; Schornberg K. L.; Scully C. (2013) FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci. Transl. Med. 5(190), 190ra79 10.1126/scitranslmed.3005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker C. J.; Schornberg K. L.; Delos S. E.; Scully C.; Pajouhesh H.; Olinger G. G.; Johansen L. M.; White J. M. (2013) Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 8(2), e56265 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring G.; Rohrmann K.; Atenchong N.; Mittler E.; Becker S.; Dahlmann F.; Pöhlmann S.; Vondran F. W. R.; David S.; Manns M. P. (2014) The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J. Antimicrob. Chemother. dku091 10.1093/jac/dku091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yermolina M. V.; Wang J.; Caffrey M.; Rong L. L.; Wardrop D. J. (2011) Discovery, synthesis, and biological evaluation of a novel group of selective inhibitors of filoviral entry. J. Med. Chem. 54(3), 765–781 10.1021/jm1008715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mire C. E.; Geisbert J. B.; Marzi A.; Agans K. N.; Feldmann H.; Geisbert T. W. (2013) Vesicular stomatitis virus-based vaccines protect nonhuman primates against Bundibugyo ebolavirus. PLoS Neglected Trop. Dis. 7(12), e2600 10.1371/journal.pntd.0002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilyuk J.; Uehara H.; Otsubo N.; Hessell A.; Burton D. R.; Barbas C. F. (2010) Potent inhibition of HIV-1 entry with a chemically programmed antibody aided by an efficient organocatalytic synthesis. ChemBioChem 11(15), 2113–2118 10.1002/cbic.201000432. [DOI] [PubMed] [Google Scholar]
- Gavrilyuk J.; Ban H.; Uehara H.; Sirk S. J.; Saye-Francisco K.; Cuevas A.; Zablowsky E.; Oza A.; Seaman M. S.; Burton D. R.; Barbas C. F. (2013) Antibody conjugation approach enhances breadth and potency of neutralization of anti-HIV-1 antibodies and CD4-IgG. J. Virol. 87(9), 4985–4993 10.1128/JVI.03146-12. [DOI] [PMC free article] [PubMed] [Google Scholar]