

Significance
More than 300 mutations in the genes encoding the cardiac IKs channel have been identified in patients with cardiac arrhythmia. These mutations cause either loss of function or gain of function of the IKs channel. This study describes how polyunsaturated fatty acids and their analogues activate or inhibit the IKs channel. These modulators can restore rhythmic firing in arrhythmic firing cardiac myocytes and restore prolonged QT interval in guinea pig hearts. The study also describes a mechanism by which an auxiliary β-subunit alters the pharmacological sensitivity of the IKs channel. Our findings may form the basis for future design of antiarrhythmic compounds that target IKs channels for treating different cardiac arrhythmias caused by mutations in the IKs channel.
Keywords: Kv7.1, KCNQ1, KCNE1, IKs, antiarrhythmic
Abstract
Polyunsaturated fatty acids (PUFAs) affect cardiac excitability. Kv7.1 and the β-subunit KCNE1 form the cardiac IKs channel that is central for cardiac repolarization. In this study, we explore the prospects of PUFAs as IKs channel modulators. We report that PUFAs open Kv7.1 via an electrostatic mechanism. Both the polyunsaturated acyl tail and the negatively charged carboxyl head group are required for PUFAs to open Kv7.1. We further show that KCNE1 coexpression abolishes the PUFA effect on Kv7.1 by promoting PUFA protonation. PUFA analogs with a decreased pKa value, to preserve their negative charge at neutral pH, restore the sensitivity to open IKs channels. PUFA analogs with a positively charged head group inhibit IKs channels. These different PUFA analogs could be developed into drugs to treat cardiac arrhythmias. In support of this possibility, we show that PUFA analogs act antiarrhythmically in embryonic rat cardiomyocytes and in isolated perfused hearts from guinea pig.
The cardiac action potential is initiated and maintained by inward sodium and calcium currents and terminated by outward potassium currents (1). The IKs channel, formed by four α subunits (voltage-gated potassium channel subunit Kv7.1, originally called KCNQ1 or KvLQT1) and two to four auxiliary β subunits (Kv channel beta subunit KCNE1, originally called minK) (1, 2), contributes a major component of the repolarizing potassium current. More than 300 mutations in the genes encoding Kv7.1 and KCNE1 have been identified in patients with cardiac arrhythmia (1). Loss-of-function mutations of the IKs channel prolong the QT interval as observed in long QT syndrome, leading to ventricular arrhythmias, ventricular fibrillation, and sudden death (1). Gain-of-function mutations of the IKs channel shorten the QT interval, possibly leading to arrhythmia such as short QT syndrome or atrial fibrillation (1). Pharmacological augmentation (in the case of long QT syndrome) or inhibition (in the case of short QT syndrome) of IKs channel activity is a logical pharmacological strategy to treat these forms of cardiac arrhythmias.
Kv7.1 is a tetrameric voltage-gated K (Kv) channel with six transmembrane segments (called S1–S6) per subunit (3). S5 and S6 from all four subunits together form the pore domain with the central ion-conducting pore. In Kv channels, S6 has been shown to function as the activation gate, shutting off the intracellular access to the pore for K+ ions in the closed state of the channel (3–5). Most reported activators or inhibitors of Kv7.1 channels target the ion-conducting pore domain of the channel, opening or blocking the ionic pathway (6–10). S1–S4 of each subunit form a voltage-sensor domain (VSD). In Kv channels, each S4 segment has several positively charged residues and has been shown to move in response to changes in the transmembrane voltage (3, 11). In response to membrane depolarization, the S4 segments move outward with respect to the membrane, which causes channel opening. Although four Kv7.1 subunits per se form a functional channel, Kv7.1 needs to coassemble with the auxiliary β-subunit KCNE1 to recapitulate the biophysical properties of the native cardiac IKs channel (12, 13). KCNE1, a single transmembrane helix protein, has been proposed to associate with Kv7.1 in the lipid cleft between adjacent VSDs, making contact with VSD transmembrane segments S1 and S4 and pore transmembrane segment S6 (14–16).
In this study, we explore the prospects of polyunsaturated fatty acids (PUFAs) and PUFA analogs as small molecules enhancing or inhibiting the activity of the cardiac IKs channel by changing IKs channel voltage dependence. We previously suggested that PUFAs facilitate opening of the related Shaker Kv channel via electrostatic attraction of S4 (17–20). The pharmacological sensitivity of IKs to small-molecule activators has been shown to depend on the Kv7.1:KCNE1 stoichiometry (21–23). We therefore also determine the impact of Kv7.1:KCNE1 stoichiometry on PUFA sensitivity.
Below we show that PUFAs affect the Kv7.1 channel by an electrostatic effect on the voltage sensor movement. We also show that KCNE1 abolishes the PUFA sensitivity of the Kv7.1 channel at physiological pH, suggesting that physiologically occurring PUFAs do not act on IKs channels in vivo. Furthermore, we identify PUFA analogs that have effects on the IKs channel at physiological pH, increase IKs in cardiomyocytes, restore rhythmic firing in arrhythmic cardiomyocytes, and shorten the QT interval in isolated perfused guinea pig hearts. These results may form the basis for development of pharmacological drugs that target the IKs channel to prevent cardiac arrhythmias.
Results
n-3 PUFAs Open Kv7.1 Channels via an Electrostatic Mechanism.
Extracellular application of the n-3 (or ɷ-3) PUFA docosahexaenoic acid (DHA) (in the concentration range of total circulating plasma PUFAs) (24–26) activates Kv7.1 channels by shifting the conductance vs. voltage [G(V)] curve of human Kv7.1 in the negative direction along the voltage axis (Fig. 1 A and B) and changing Kv7.1 channel kinetics (Fig. S1 A and B). DHA (70 µM) shifts the G(V) by −9.3 ± 0.9 mV (n = 3). DHA (7 μM) induces significant G(V) shifts (−3.0 ± 0.5 mV; n = 3; P < 0.05). The estimated maximum shift is −15.8 ± 2.3 mV (from Eq. 2), and 50% of the maximum shift is caused by 50 ± 20 μM DHA (Fig. 1C).
Fig. 1.
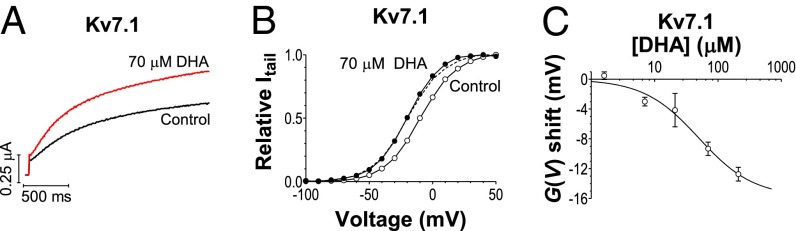
DHA shifts the voltage dependence of Kv7.1. (A) DHA (70 μM) increases current amplitude of Kv7.1 in response to a −20-mV voltage step. (B) DHA (70 μM) shifts the G(V) of Kv7.1. DHA (●), control (○). Dashed curve in B is control curve shifted −10 mV. (C) Concentration dependence of DHA effect. Mean ± SEM. ΔVmax = −15.8 mV, c0.5 = 50 μM. n = 3–5.
To determine the molecular mechanism of how DHA facilitates Kv7.1 channel opening, we explore the structural requirements of the DHA molecule to facilitate channel opening. We test the importance of the DHA head charge (i–iii) and the DHA acyl tail (iv) in four sets of experiments:
-
i)
We test the effect of the uncharged PUFA analog DHA methyl ester (70 μM) and find that it does not change the voltage dependence of Kv7.1 (Fig. 2A and Table S1). In contrast, the positively charged arachidonyl amine (AA+, 70 μM) and the positively charged docosahexaenyl amine (DHA+, 70 μM) shift the G(V) curve by +9.7 ± 2.1 (n = 8) and +7.8 ± 3.0 mV (n = 10), respectively (Fig. 2A). These data suggest that a charged PUFA head group is required for shifting the G(V) curve of the Kv7.1 channel. The valence of that charge determines the direction of the G(V) shift, as if the PUFA– Kv7.1 channel interaction is electrostatic.
-
ii)
We test the effect of pH on the G(V) shifts induced by PUFAs. Unsaturated fatty acids in a lipid environment are protonated/deprotonated with an apparent pKa ∼ 7 (18, 27) (illustrated in Fig. S1C). The apparent pKa value for DHA on Kv7.1 is ∼7.7 as reported by the shift of the G(V) relation (Fig. 2B). Increasing the external pH from 7.4 to 9 or 10 increases the shift in negative direction of the G(V) curve protocol by DHA, whereas reducing the external pH to 6.5 abolishes the DHA effect (Fig. 2C).
-
iii)
We coapply 70 µM DHA and 70 µM DHA methyl ester and find that DHA methyl ester reduces the shift of the G(V) relation (−5.5 ± 0.6 mV, n = 4) compared with application of 70 µM DHA alone (−9.3 ± 0.9 mV; Fig. S2 A and B). The induced G(V) shift from coapplication of 70 µM DHA and 70 µM DHA methyl ester (−5.5 mV) is close to that produced by 35 µM DHA alone (−6.5 mV; Fig. S2C), as if the uncharged DHA methyl ester competes with DHA for binding but has by itself no effect on Kv7.1 channel activation.
-
iv)
We test the effects of another PUFA with a slightly shorter tail [eicosapentaenoic acid (EPA)] and of the monounsaturated oleic acid (Fig. 2A). The length of the acyl chains of EPA and oleic acid differ by only two carbons. The main difference between EPA and oleic acid is the number of double bonds in the acyl chain where EPA contains five double bonds and oleic acid only one double bond. The polyunsaturated EPA is similar in its effect on Kv7.1 as DHA, whereas the monounsaturated oleic acid has no effect (Fig. 2A and Table S1). These data suggest that a polyunsaturated acyl tail is required for shifting the Kv7.1 channel voltage dependence.
Fig. 2.

A charged head group and a polyunsaturated acyl tail are necessary. (A) Induced G(V) shifts for 70 μM of indicated substance. Means ± SEM n = 3–8. (B) pH dependence of the G(V) shift caused by the application of 70 µM DHA on Kv7.1. Means ± SEM. ΔVmax = −28.0 mV, c0.5 = 1.8 × 10−8 = pH 7.7. n = 3–5. (C) Concentration-response curves for DHA on Kv7.1 at different pHs. pH 9.0: ΔVmax = −27.7 mV, c0.5 = 10 μM; pH 7.4, see Fig. 1C. n = 3–8. (D) Schematic illustration of PUFA modulation. PUFAs incorporate in the lipid cleft close to the channel’s VSD and electrostatically affect the S4 movement. +++, S4 gating charges.
In summary, both a negatively charged head group and a polyunsaturated acyl tail of the PUFA are required for shifting the G(V) curve of the Kv7.1 channel in the negative direction along the voltage axis. These structural requirements of the PUFA molecule to modulate Kv7.1 channel activity are similar to the structural requirements of PUFAs for modulating the Shaker Kv channel (17–19). In analogy with the mechanism for PUFA suggested for the Shaker Kv channel (18, 20), we hypothesize that the negatively charged head group of PUFA interacts electrostatically with the VSD of Kv7.1 in facilitating channel opening and that the polyunsaturated acyl tail of PUFA is important to locate the PUFA molecule to the otherwise phospholipid filled space between two neighboring VSDs (Fig. 2D).
The Auxiliary KCNE1 Subunit Reduces the DHA Effect on Kv7.1.
KCNE1 has been proposed to locate in the space between two neighboring VSDs (14–16) of tetrameric Kv7.1 channels, close to where PUFAs have been proposed to interact with Kv channels (17). One might thus expect that the DHA effect on Kv7.1 is altered in the presence of KCNE1. To explore this, we coexpress Kv7.1 with high concentrations of KCNE1 and test the DHA sensitivity of the heteromeric Kv7.1/KCNE1 channel. Coexpression with KCNE1 prevents 70 μM DHA from shifting the G(V) of Kv7.1 (Fig. 3 A and B and Table S1).
Fig. 3.

KCNE1 abolishes PUFA effect on Kv7.1/KCNE1 at physiological pH. (A–D) DHA (70 μM) applied on Kv7.1/KCNE1 at pH 7.4 (A and B) and pH 9 (C and D). DHA (●), control (○). Bold traces = 0 mV (A) or +10 mV (C). Dashed curve in D is control curve shifted −35 mV. (E) pH dependence for 70 µM DHA. Kv7.1/KCNE1: ΔVmax = −29.0 mV, c0.5 = 2.5 × 10−9 = pH 8.6. n = 3–6. For Kv7.1, see Fig. 2B. (F) Model: KCNE1 changes probability of protonation of negatively charged acidic (Upper) and positively charged amine (Lower) PUFA analogs, thereby changing PUFA-Kv7.1 voltage sensor (+++) interactions. (G) G(V) shift induced by 70 µM DHA or AA+. n = 3–8.
We test the following three possible explanations of why KCNE1 reduces the DHA efficacy on Kv7.1 channels: (i) DHA binds to the Kv7.1/KCNE1 channel, but its effects are masked by the strong KCNE1 effects on Kv7.1 activation gating; (ii) KCNE1 excludes DHA from binding to a shared location on Kv7.1; and (iii) KCNE1 modifies the local environment of the head group of the PUFAs, thereby reducing DHA potency (for instance, by altering the local proton concentration). We use different Kv7.1 and KCNE1 constructs to distinguish among these possible explanations.
KCNE1-induced reduction of DHA sensitivity is not due to altered activation gating.
We coexpress Kv7.1 with a KCNE1 construct in which the C terminus is truncated (KCNE1ΔC). This construct was previously shown to associate with Kv7.1 (28) but to alter channel kinetics and voltage dependence only to a small degree: Kv7.1/KCNE1ΔC kinetics and activation voltage dependence are similar to these of Kv7.1 alone (Fig. S3A). The Kv7.1/KCNE1ΔC channel is, however, insensitive to 70 µM DHA (Fig. S3A and Table S1). A strong alteration of activation gating by KCNE1 is thus not required for suppression of DHA action.
KCNE1-induced reduction of DHA sensitivity is not dependent on Kv7.1:KCNE1 stoichiometry.
KCNE1 has been shown to eliminate the effect of Kv7.1 activators, such as R-L3 and ML277. These compounds potentiate Kv7.1 homotetrameric channels expressed in Xenopus oocytes, but not Kv7.1 coexpressed with high levels of KCNE1 (21, 22). A gradual decrease in the potency of these compounds is observed depending on the number of KCNE1 subunits in the Kv7.1/KCNE1 channel complex. We test whether the potency of DHA on Kv7.1/KCNE1 channels increases if the number of KCNE1 subunits in the Kv7.1/KCNE1 complex is reduced. To this end, we construct two concatemers, KCNE1-Kv7.1-Kv7.1 and KCNE1-Kv7.1, forcing either a 4:2 or a 4:4 stoichiometry of Kv7.1 and KCNE1 subunits in assembled channels. DHA (70 µM) applied to either one of the concatemeric channels only shifts the G(V) relation by a small positive voltage (Fig. S3 B and C). This positive shift (observed at external pH 7.4) is unique for the concatemeric channels, because it is not seen in WT Kv7.1/KCNE1 channels (Table S1). This positive shift does not require the negative charge of the DHA head group because the uncharged DHA methyl ester induces a similar positive shift in both concatemeric channels (Table S1). Experiments on these concatemeric channels show that a Kv7.1 channel with only two KCNE1 subunits is also not sensitive to DHA.
KCNE1-induced reduction of DHA sensitivity is due to protonation of DHA.
To test whether KCNE1 reduces the DHA potency by altering the local proton concentration close to the head group of the PUFAs, we test the DHA effect on Kv7.1/KCNE1 at pH 9. In contrast to the experiments at pH 7.4 where the G(V) of WT Kv7.1/KCNE1 (or concatemers Kv7.1-KCNE1) is not shifted (or slightly in positive direction for the concatemers), 70 µM DHA at pH 9.0 shifts the G(V) curves by −26 mV (−28 to −30 mV for the concatemers; Fig. 3 C–E and Table S1). These shifts are comparable to the DHA-induced G(V) shift of Kv7.1 alone at pH 9 (Table S1). These data suggest that DHA binds to Kv7.1 channels coexpressed with KCNE1 and that KCNE1 lower the local pH close to the head group of the PUFAs. KCNE1 alters the apparent pKa value for DHA from 7.7 to 8.6 (Fig. 3E). The KCNE1-induced protonation of DHA close to Kv7.1 eliminates the electrostatic effect of DHA on the Kv7.1 channel.
Increasing external pH shifts the G(V) curve of Kv7.1/KCNE1 toward more positive voltages (29, 30). However, this pH-induced shift in G(V) does not per se increase the DHA sensitivity of Kv7.1/KCNE1 because the Kv7.1/S225L/KCNE1 mutant (shifted about +30 mV compared with WT Kv7.1/KCNE1) is not sensitive to 70 µM DHA at pH 7.4 (mean shift: +0.5 ± 0.3 mV, n = 4).
PUFA Analogs Effective on Kv7.1/KCNE1.
If KCNE1 decreases the G(V) shifting potency of DHA by lowering the local pH close to the head group of the PUFAs and thereby neutralizing acidic PUFAs by protonation (Fig. 3F, Upper), then we expect KCNE1-induced protonation to increase the effect of amino-analogs of PUFAs (Fig. 3F, Lower). This expectation is met by experimental data; 70 µM arachidonyl amine at pH 7.4 shifts the G(V) curve of the Kv7.1/KCNE1 channel by +22.6 ± 1.9 mV (n = 10; Fig. 3G, Table S1, and Fig. S4A), compared with only +9.7 ± 2.1 mV (n = 8) for Kv7.1 alone (Fig. 3G and Table S1). In addition, DHA and arachidonyl amine have opposite pH dependence of their effect on Kv7.1/KCNE1: the G(V) shifting potency of DHA increases with increasing pH, whereas the potency of arachidonoyl amine decreases with increasing pH (Fig. S5A).
To further test whether KCNE1 reduces the effect of PUFAs on the Kv7.1 channel via a local pH reduction, we use the PUFA analog docosahexaenoyl glycine (DHA-Gly), whose lower pKa value should keep the DHA-Gly molecule deprotonated at physiological pH; 70 µM DHA-Gly shifts by a similar amount the G(V) curves of channels formed by Kv7.1 alone and by Kv7.1 together with KCNE1 (−25 mV; Fig. 4A, Fig. S4B, and Table S1). DHA-Gly has an apparent pKa value of 7.1 for Kv7.1/KCNE1, which is about 1.5 pH units lower than that of DHA (Fig. 4B). Moreover, a permanently charged PUFA analog, which is not protonated or deprotonated by changes in the local pH in our experimental setting, should be equally effective on Kv7.1 alone as on Kv7.1/KCNE1; 70 µM of the permanently negatively charged N-arachidonoyl taurine shifts by a similar amount the G(V) curves of Kv7.1 channels with and without KCNE1 (−26 to −27 mV; Table S1, Fig. 4C, and Fig. S4C). N-arachidonoyl taurine also increases the maximum conductance of Kv7.1/KCNE1 (Fig. S4C). The mechanism for this is unclear.
Fig. 4.

PUFA analogs are effective on Kv7.1/KCNE1. (A) DHA-Gly (70 μM) induces a negative G(V) shift of Kv7.1/KCNE1. DHA-Gly (●), control (○). Dashed line is control curve shifted −20 mV. (B) pH dependence for 70 µM DHA-Gly or 70 µM DHA applied on Kv7.1/KCNE1. DHA-Gly: ΔVmax = −38.8 mV, c0.5 = 7.6 × 10−8 = pH 7.1. n = 3–6. For DHA, see Fig. 3E. (C) N-arachidonoyl taurine (N-AT; 70 µM) induces a negative G(V) shift of Kv7.1/KCNE1. N-AT (●), control (○). Dashed line is control curve shifted −24 mV. (D) DHA-Gly (70 µM) does not shift Kv7.1/R228Q and has a smaller effect on Kv7.1/K218C and Kv7.1/G219C. Means ± SEM; n = 4–14. (E) Normalized fluorescence during an activating test pulse to −60 mV from a holding voltage of −120 mV. (F) N-AT (70 μM) shifts the F(V) of Kv7.1/G219C*. N-AT (●), control (○).The continuous lines are Boltzmann curves (Eq. 1) fitted to experimental data. For control: V50 = −41.1 mV; s = 11.7. For N-AT: V50 = −61.1 mV; s = 16.3. Means ± SEM; n = 9.
These results show that even in the presence of KCNE1, Kv7.1 is activated by a PUFA analog with a lower pKa value and a PUFA analog with a permanent negative charge, supporting the hypothesis that KCNE1 reduces the local pH and thereby modifies the sensitivity to PUFAs. A simple model can recapitulate the pH dependence of DHA and DHA-Gly on Kv7.1 and Kv7.1/KCNE1 (Fig. S5 B and C). Furthermore, we also note that the G(V) shifts of (i) Kv7.1 induced by 70 μM DHA at pH 9 and 10 (almost fully deprotonated), (ii) Kv7.1/KCNE1 by 70 μM DHA at pH 9 (almost fully deprotonated), (iii) Kv7.1 and Kv7.1/KCNE1 by 70 μM DHA-Gly at pH 7.4 (almost fully deprotonated), and (iv) Kv7.1 and Kv7.1/KCNE1 by 70 μM N-arachidonoyl taurine at pH 7.4 (negatively charged) are all about −30 mV, suggesting that, as long as the PUFA is charged, the effect of the PUFA is independent of KCNE1.
PUFAs Target the VSD.
Because PUFAs require a charged head group to affect Kv7.1 channel activity, we hypothesize that the PUFAs electrostatically facilitate Kv7.1 channel opening via interaction with the extracellular-facing part of the positively charged voltage sensor S4, as previously suggested for PUFA-induced opening of the Shaker channel (17, 20). To test this hypothesis, we neutralize the outermost positive gating charge R228 in S4 of Kv7.1 by generating a R228Q mutant channel; 70 µM DHA-Gly does not shift the G(V) curve of R228Q (Fig. 4D, Table S1, and Fig. S6A). This lack of effect of DHA-Gly on R228Q cannot be explained by an alteration in local pH caused by the R228Q mutation because 70 µM N-arachidonoyl taurine (which is not subject to protonation) is also ineffective on R228Q (mean shift: +2.4 ± 3.5 mV, n = 5). The voltage dependence of the R228Q mutant is shifted toward more negative voltages compared with WT Kv7.1. This shift in G(V) toward more negative voltages caused by the R228Q mutation does not per se reduce the DHA sensitivity of R228Q, because the opening of the R228Q/F351A mutant (shifted about +25 mV toward more positive voltages compared with Kv7.1; Fig. S6B) is not facilitated by 70 µM DHA-Gly (Fig. S6B; mean shift: +4.0 ± 1.0 mV, n = 5). In addition to S4 gating charge residues, other residues in the top of S3 and S4 were previously shown to be important for the PUFA effect on the Shaker channel (17). We therefore also test the effect of K218C and G219C, located in the S3–S4 loop of Kv7.1, on PUFA potency. Both these mutations reduce the effect of 70 µM DHA-Gly on Kv7.1 (Fig. 4D) and reduce the affinity of DHA-Gly by a factor of ∼3–4 (Fig. S6C).
The fact that three point mutations in or close to the voltage sensor S4 alters DHA-Gly potency on Kv7.1 and that two of these mutations alter the affinity of DHA-Gly suggests that the VSD is important for the PUFA effect on Kv7.1 and may form part of a PUFA binding site. Neutralizing the outermost positive gating charge in S4 abolishes the effect of DHA-Gly on Kv7.1, supporting the hypothesis of an electrostatic interaction between the charged PUFAs and the S4 of Kv7.1. Apparently, R228 plays a special part in this electrostatic interaction, as the other gating charges cannot substitute for R228 when R228 is neutralized. To further explore whether PUFAs indeed affect S4 movement, we test the effect of N-arachidonyl taurine on the Kv7.1/G219C mutant in which G219C is labeled with the fluorophore Alexa488 maleimide (referred to as Kv7.1/G219C*). As previously reported (31–33), using voltage-clamp fluorometry, we can detect S4 movement in this construct as a change in fluorescence intensity; 70 µM N-arachidonoyl taurine speeds up the fluorescence kinetics during an activating test pulse (Fig. 4E) and shifts the F(V) curve toward more negative voltages (Fig. 4F). The mean F(V) shift induced by 70 µM N-arachidonoyl taurine is −20.0 ± 1.9 mV (n = 9). These data support the hypothesis that PUFAs facilitate Kv7.1 channel opening by promoting S4 movement.
PUFA Analog Restores Cardiac Rhythm.
We test the effect of N-arachidonoyl taurine on isolated embryonic rat cardiomyocytes gestation day 16, in which there is IKs expression but no IKr expression (34); 30 µM N-arachidonoyl taurine shortens the cardiomyocyte action potential duration (APD50) by 17 ± 3% (n = 5, P < 0.05) and increases the frequency of action potential firing by 26 ± 5% (n = 5, P < 0.05; Fig. 5 A and B and Table S2). N-arachidonoyl taurine (30 µM) has no effect on the resting membrane potential compared with control (Table S2) but decreases the action potential peak amplitude by −9 ± 1 mV (Table S2). Fig. S7A, Left, shows the current in an isolated embryonic rat cardiomyocyte during E4031 and nifedipine treatment (to block possible interfering IKr and ICa,L-type, respectively); 10 µM of the IKs channel blocker Chromanol 293B blocks this current by 91 ± 5% (n = 3) at +10 mV, suggesting that the main part of this current is IKs. Application of 30 µM N-arachidonoyl taurine increases this current by 103 ± 68% at +10 mV (n = 5, P = 0.005; Fig. 5C and Fig. S7A). To induce arrhythmia in the embryonic rat cardiomyocytes, we apply a subsaturating concentration of Chromanol 293B, which is known to prolong the cardiac action potential duration and induce arrhythmia (35); 5 µM Chromanol 293B increases the action potential duration (APD50) by 30 ± 11% (n = 6, P < 0.05; Fig. S7B). With time, two types of effects (sometimes within the same cell) are seen on 5 µM Chromanol 293B application
-
i)
In 4 of 10 cells, we see arrhythmic firing with pronounced variation in the frequency of action potential firing (Fig. 5 D and E and Fig. S7C). During this arrhythmic firing, there is a large variation in action potential duration (Fig. S7D) due to double peaks and irregular RR interval.
-
ii)
In 9 of 10 cells, we see a slowing of the frequency of spontaneous action potential firing (Fig. S7 E and F). The mean increase in RR interval is 387 ± 95% (n = 9, P < 0.05). The slowing in action potential firing induced by Chromanol 293B is in line with earlier studies of long QT mutations in Kv7.1 channels in patients for whom bradycardia has been reported (36, 37). The mechanism of how long QT mutations in Kv7.1 could cause bradycardia is not clear.
Fig. 5.

Antiarrhythmic effect of PUFA analogs. (A and B) Effect of 30 µM N-AT on action potential duration and frequency in isolated embryonic rat cardiomyocytes. (C) Example of the effect of 30 µM N-AT on current amplitude in isolated embryonic rat cardiomyocytes. Gray trace is current in control solution from a voltage 20 mV more positive than the N-AT trace. (D–F) Representative examples of arrhythmia induced by application of 5 µM Chromanol 293B or 5 µM Chromanol 293B + 30 µM N-arachidonoyl taurine (N-AT) in isolated embryonic rat cardiomyocytes. (G and H) Representative example of effect of 0.03 µM E4031 and 10 µM DHA-Gly on QT interval (G) and action potential duration (H) in isolated perfused guinea pig heart. Hearts are paced at 250 beats/min. (I) Summary of effect of 0.03 µM E4031 and 10 µM DHA-Gly on QT interval and action potential duration (APD90) in isolated perfused guinea pig heart. Means ± SEM; n =3. Statistical significance compared with control.
These two different effects of Chromanol 293B are most likely due to different subtypes of cardiomyocytes. Independent of the type of effect induced by Chromanol 293B application, 30 µM N-arachidonoyl taurine reverses the effect: abolishing the arrhythmic firing (Fig. 5F and Fig. S7C) or restoring the frequency of spontaneous action potential firing (Fig. S7G; the RR interval in coapplication of Chromanol 293B and N-arachidonoyl taurine is not significantly different to control, P > 0.05).
PUFA Analog Restores QT Interval.
To test the effect of a PUFA analog on an intact heart, we apply DHA-Gly on isolated perfused guinea pig hearts. To mimic a long QT setting, first, 0.03 µM of the IKr blocker E4031 is perfused into the guinea pig heart to prolong the QT interval and action potential duration (Fig. 5 G–I). Twenty-minute perfusion of 0.03 µM E4031 increases the QT interval by 23.6 ± 3.6 ms (n = 3, P < 0.05) and prolongs the action potential duration (APD90) by 14.3 ± 2.2 ms (n = 3, P < 0.05). Perfusion of the guinea pig heart with 0.03 µM E4031 together with 10 µM DHA-Gly reverses the E4031-induced QT prolongation and E4031-induced prolongation of action potential duration (Fig. 5 G–I). Perfusion with 0.03 µM E4031 or 10 µM DHA-Gly produces no significant effects on intrinsic heart rate (Fig. S7H).
Discussion
In this study, we showed that small molecules (PUFAs and PUFA analogs) activate or inhibit the Kv7.1 channel. We describe a novel mechanism by which the auxiliary KCNE1 subunit alters the pharmacological sensitivity of the Kv7.1 channel. KCNE1 reduces the Kv7.1 channel sensitivity to regular PUFAs by promoting PUFA protonation. In contrast, KCNE1 increases the Kv7.1 channel sensitivity to the positively charged PUFA analog arachidonyl amine by promoting amine protonation. The effect on Kv7.1 of the PUFA analogs docosahexaenoyl glycine, which has a lower pKa value, and of N-arachidonoyl taurine, which is permanently charged, are not modified by KCNE1 coexpression. Thus, understanding the mechanism for how KCNE1 tunes the pharmacological sensitivity of Kv7.1 allows us to chemically bypass the auxiliary subunit interference of KCNE1.
The similarity in PUFA modulation of Kv7.1 (reported here) and the Shaker Kv channel (17, 18, 20) implies a similar PUFA binding site and similar PUFA mechanism of action on these two channels from different species. Circulating PUFAs may therefore speculatively play an important role in tuning the activity of several voltage-gated ion channels by directly affecting the voltage sensor movement. One of the key findings in the present work is, however, that the coexpression of Kv7.1 with KCNE1 modulates the PUFA effect on the Kv7.1 channel by altering PUFA protonation. This finding suggests that Kv channels in different tissues might be differentially sensitive to PUFAs depending on the auxiliary subunits with which they form a complex.
Cellular, animal, and clinical studies support a hypoexcitable and antiarrhythmic effect of fish oil and PUFAs on the heart (38–42). One main hypothesis put forward to explain the beneficial effect is PUFA-induced inhibition of cardiac sodium and calcium channels (41–46). Some studies also describe shortening of the cardiac action potential on PUFA administration (47, 48). PUFA-induced shortening of the cardiac action potential could theoretically be explained by PUFA-induced augmentation of the IKs channel. Our experiments in Xenopus oocytes show that whereas the voltage dependence of Kv7.1 expressed alone is affected by PUFAs, the voltage dependence of Kv7.1 in complex with KCNE1 is not affected by DHA. This finding suggests that PUFAs and fish oil do not act antiarrhythmically by altering the voltage dependence of the Kv7.1/KCNE1 channel. Recently, Moreno et al. found that acute DHA and EPA applications increased the Kv7.1/KCNE1 currents and that acute EPA application shifted the Kv7./KCNE1 activation moderately to more hyperpolarized voltages (49). However, chronic applications of DHA and EPA did not increase the currents, and EPA even shifted the activation moderately to more depolarized voltages. Chronic EPA and DHA application also redistributed the Kv7.1 subunits and decreased their expression levels (49). The complex results from Moreno et al. suggest that more studies are necessary to clarify the clinical relevance of the effects of PUFAs on IKs channels. However, our findings here show proof of principle that PUFA analogs can be designed as IKs channel activators or inhibitors targeting the VSD of IKs channels. Because PUFA analogs can be designed as either channel activators or inhibitors depending on the charge of the head group, these analogs have the potential to be useful to prevent cardiac arrhythmias caused by either loss-of-function or gain-of-function IKs mutations, as in long QT syndrome or short QT syndrome, respectively. The prospect of PUFA analogs as clinical IKs channel modulators is supported by our results showing antiarrhythmic effects of N-arachidonoyl taurine on isolated cardiomyocytes and DHA-Gly–induced shortening of a prolonged QT interval in guinea pig hearts.
Materials and Methods
For detailed description of material and methods, see SI Materials and Methods.
Experiments on Xenopus laevis Oocytes.
Xenopus laevis oocytes were isolated and maintained as previously described (9). Approximately 50 nL cRNA (50 ng Kv7.1, KCNE1-Kv7.1-Kv7.1, or KCNE1-Kv7.1 alternatively 25 ng Kv7.1 together with 8 ng KCNE1 or KCNE1ΔC) was injected into each oocyte. Coinjection is referred to as Kv7.1/KCNE1 and concatemers as KCNE1-Kv7.1 or KCNE1-Kv7.1-Kv7.1. Voltage-clamp fluorometry experiments were performed as previously described on oocytes labeled for 30 min with 100 µM Alexa488 maleimide (Molecular Probes) at 4 °C (31–33). Synthesis of arachidonyl amine and docosahexaenoyl amine was performed as described in Fig. S8.
Experiments on Rat Cardiomyocytes.
Embryonic rat cardiomyocytes were isolated from Sprague–Dawley rats on gestation day 16. Rat experiments were approved by the Linköping Animal Ethics Committee at Linköping University and guinea pig experiments were approved by The Animal Experiments Inspectorate under the Danish Ministry of Food, Agriculture and Fisheries (University of Copenhagen). For current measurements, 1 µM E-4031 and 10 µM nifedipine were added to the extracellular solution. All experiments were carried out at 35 °C. Spontaneous action potential characteristics were analyzed during a stable 60-s recording. Effects of Chromanol 293B on APD50 were analyzed from the first 30 s of the trace after Chromanol 293B application. To analyze current amplitude, the holding voltage was set to −70 mV, and the steady-state current at the end of test pulses ranging from −70 to +40 mV was measured.
Isolated Perfused Heart Preparations.
The excised heart from female Dunkin Hartley guinea pigs was mounted on a Langendorff perfusion apparatus and perfused at a constant pressure of 60 mmHg. Pacing periods consisted of 2-min pacing at 250 BPM. The heart was stabilized for a minimum of 30 min, followed by 2 min of pacing to record baseline electrocardiogram and APD90; 0.03 µM E4031 was added to the perfusate for 20 min, and data were acquired during 2-min atrial pacing. DHA-Gly (10 µM) was added to the perfusate, and the heart was paced for 2 min every 10th minute.
Electrophysiological Analysis.
To quantify V1/2, tail currents were plotted against the prepulse (test) voltage, and the following Boltzmann relation was fitted to the data:
| [1] |
where V1/2 is the midpoint, and s the slope factor. To quantify the concentration dependence and pH dependence of the DHA-induced shift of voltage dependence, ΔV, the following equation was used:
| [2] |
where ΔVmax is the maximal shift, c0.5 is the concentration causing 50% of the maximal shift, and c is the concentration of DHA (or H+).
Statistical Analysis.
Data are means ± SEM. Mean values for G(V) shifts were analyzed using a two-tailed one-sample t test compared with a hypothetical value of 0 or where mean values compared with each other. PUFA effects on VSD mutants were analyzed using one-way ANOVA comparing the PUFA effect on each mutant with the PUFA effect on Kv7.1 WT (Dunnett’s multiple comparison test). Compound effects on cardiomyocytes were analyzed using a paired two-tailed t test. Compounds effects on guinea pig hearts were analyzed using one-way ANOVA with Turkey’s multiple comparison test. P < 0.05 is considered statistically significant.
Supplementary Material
Acknowledgments
We thank Xiongyu Wu (Linköping University) for synthesizing the DHA-amine. We thank Drs. W. Nonner, L. Bianci, and F. Qui (University of Miami) and Dr. A. Fridberger (Linköping University) for valuable comments on the manuscript. The original clones for concatemers were generously provided by Dr. R. S. Kass (Columbia University). This work was supported by the National Institutes of Health (R01GM109762), American Heart Association (14GRNT20380041), Swedish Research Council, the Swedish Heart-Lung Foundation, the County Council of Östergötland, Queen Silvia’s Anniversary Foundation, and the Academy of Finland. S.I.L. has a postdoctoral fellowship from the Swedish Research Council.
Footnotes
Conflict of interest statement: A patent application based on these results has been submitted by the University of Miami with S.I.L., H.P.L., and F.E. identified as inventors.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1503488112/-/DCSupplemental.
References
- 1.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85(4):1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 2.Nakajo K, Ulbrich MH, Kubo Y, Isacoff EY. Stoichiometry of the KCNQ1 - KCNE1 ion channel complex. Proc Natl Acad Sci USA. 2010;107(44):18862–18867. doi: 10.1073/pnas.1010354107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Börjesson SI, Elinder F. Structure, function, and modification of the voltage sensor in voltage-gated ion channels. Cell Biochem Biophys. 2008;52(3):149–174. doi: 10.1007/s12013-008-9032-5. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19(1):175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 5.Jiang Y, et al. The open pore conformation of potassium channels. Nature. 2002;417(6888):523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 6.Xiong Q, Sun H, Li M. Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat Chem Biol. 2007;3(5):287–296. doi: 10.1038/nchembio874. [DOI] [PubMed] [Google Scholar]
- 7.Blom SM, Schmitt N, Jensen HS. The acrylamide (S)-2 as a positive and negative modulator of Kv7 channels expressed in Xenopus laevis oocytes. PLoS ONE. 2009;4(12):e8251. doi: 10.1371/journal.pone.0008251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lange W, et al. Refinement of the binding site and mode of action of the anticonvulsant Retigabine on KCNQ K+ channels. Mol Pharmacol. 2009;75(2):272–280. doi: 10.1124/mol.108.052282. [DOI] [PubMed] [Google Scholar]
- 9.Bentzen BH, et al. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology. 2006;51(6):1068–1077. doi: 10.1016/j.neuropharm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414(6859):43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 11.Larsson HP, Baker OS, Dhillon DS, Isacoff EY. Transmembrane movement of the shaker K+ channel S4. Neuron. 1996;16(2):387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 12.Sanguinetti MC, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384(6604):80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 13.Barhanin J, et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 14.Shamgar L, et al. 2008. KCNE1 constrains the voltage sensor of Kv7.1 K+ channels. PLoS ONE 3:e1943.
- 15.Chung DY, et al. Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proc Natl Acad Sci USA. 2009;106(3):743–748. doi: 10.1073/pnas.0811897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu X, Jiang M, Hsu KL, Zhang M, Tseng GN. KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J Gen Physiol. 2008;131(6):589–603. doi: 10.1085/jgp.200809976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Börjesson SI, Elinder F. An electrostatic potassium channel opener targeting the final voltage sensor transition. J Gen Physiol. 2011;137(6):563–577. doi: 10.1085/jgp.201110599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Börjesson SI, Hammarström S, Elinder F. Lipoelectric modification of ion channel voltage gating by polyunsaturated fatty acids. Biophys J. 2008;95(5):2242–2253. doi: 10.1529/biophysj.108.130757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Börjesson SI, Parkkari T, Hammarström S, Elinder F. Electrostatic tuning of cellular excitability. Biophys J. 2010;98(3):396–403. doi: 10.1016/j.bpj.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ottosson NE, Liin SI, Elinder F. Drug-induced ion channel opening tuned by the voltage sensor charge profile. J Gen Physiol. 2014;143(2):173–182. doi: 10.1085/jgp.201311087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salata JJ, et al. A novel benzodiazepine that activates cardiac slow delayed rectifier K+ currents. Mol Pharmacol. 1998;54(1):220–230. doi: 10.1124/mol.54.1.220. [DOI] [PubMed] [Google Scholar]
- 22.Yu H, et al. Dynamic subunit stoichiometry confers a progressive continuum of pharmacological sensitivity by KCNQ potassium channels. Proc Natl Acad Sci USA. 2013;110(21):8732–8737. doi: 10.1073/pnas.1300684110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao Z, Xiong Q, Sun H, Li M. Desensitization of chemical activation by auxiliary subunits: Convergence of molecular determinants critical for augmenting KCNQ1 potassium channels. J Biol Chem. 2008;283(33):22649–22658. doi: 10.1074/jbc.M802426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraser DD, et al. Elevated polyunsaturated fatty acids in blood serum obtained from children on the ketogenic diet. Neurology. 2003;60(6):1026–1029. doi: 10.1212/01.wnl.0000049974.74242.c6. [DOI] [PubMed] [Google Scholar]
- 25.Conquer JA, Holub BJ. Effect of supplementation with different doses of DHA on the levels of circulating DHA as non-esterified fatty acid in subjects of Asian Indian background. J Lipid Res. 1998;39(2):286–292. [PubMed] [Google Scholar]
- 26.Brouwer IA, et al. SOFA Study Group Effect of fish oil on ventricular tachyarrhythmia and death in patients with implantable cardioverter defibrillators: The Study on Omega-3 Fatty Acids and Ventricular Arrhythmia (SOFA) randomized trial. JAMA. 2006;295(22):2613–2619. doi: 10.1001/jama.295.22.2613. [DOI] [PubMed] [Google Scholar]
- 27.Hamilton JA. Fatty acid transport: Difficult or easy? J Lipid Res. 1998;39(3):467–481. [PubMed] [Google Scholar]
- 28.Tapper AR, George AL., Jr MinK subdomains that mediate modulation of and association with KvLQT1. J Gen Physiol. 2000;116(3):379–390. doi: 10.1085/jgp.116.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peretz A, Schottelndreier H, Aharon-Shamgar LB, Attali B. Modulation of homomeric and heteromeric KCNQ1 channels by external acidification. J Physiol. 2002;545(Pt 3):751–766. doi: 10.1113/jphysiol.2002.028381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heitzmann D, et al. KCNE beta subunits determine pH sensitivity of KCNQ1 potassium channels. Cell Physiol Biochem. 2007;19(1-4):21–32. doi: 10.1159/000099189. [DOI] [PubMed] [Google Scholar]
- 31.Barro-Soria R, et al. KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat Commun. 2014;5:3750. doi: 10.1038/ncomms4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osteen JD, et al. Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc Natl Acad Sci USA. 2012;109(18):7103–7108. doi: 10.1073/pnas.1201582109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osteen JD, et al. KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc Natl Acad Sci USA. 2010;107(52):22710–22715. doi: 10.1073/pnas.1016300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danielsson C, et al. Exploration of human, rat, and rabbit embryonic cardiomyocytes suggests K-channel block as a common teratogenic mechanism. Cardiovasc Res. 2013;97(1):23–32. doi: 10.1093/cvr/cvs296. [DOI] [PubMed] [Google Scholar]
- 35.Aiba T, et al. Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. J Am Coll Cardiol. 2005;45(2):300–307. doi: 10.1016/j.jacc.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 36.Swan H, et al. Sinus node function and ventricular repolarization during exercise stress test in long QT syndrome patients with KvLQT1 and HERG potassium channel defects. J Am Coll Cardiol. 1999;34(3):823–829. doi: 10.1016/s0735-1097(99)00255-7. [DOI] [PubMed] [Google Scholar]
- 37.Lupoglazoff JM, et al. Long QT syndrome in neonates: Conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J Am Coll Cardiol. 2004;43(5):826–830. doi: 10.1016/j.jacc.2003.09.049. [DOI] [PubMed] [Google Scholar]
- 38.Burr ML, et al. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: diet and reinfarction trial (DART) Lancet. 1989;2(8666):757–761. doi: 10.1016/s0140-6736(89)90828-3. [DOI] [PubMed] [Google Scholar]
- 39.Investigators G-P Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet. 1999;354(9177):447–455. [PubMed] [Google Scholar]
- 40.Tavazzi L, et al. Gissi-HF Investigators Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): A randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9645):1223–1230. doi: 10.1016/S0140-6736(08)61239-8. [DOI] [PubMed] [Google Scholar]
- 41.Moreno C, et al. Effects of n-3 polyunsaturated fatty acids on cardiac ion channels. Front Physiol. 2012;3:245. doi: 10.3389/fphys.2012.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leaf A, Xiao YF, Kang JX, Billman GE. Membrane effects of the n-3 fish oil fatty acids, which prevent fatal ventricular arrhythmias. J Membr Biol. 2005;206(2):129–139. doi: 10.1007/s00232-005-0789-9. [DOI] [PubMed] [Google Scholar]
- 43.Danthi SJ, Enyeart JA, Enyeart JJ. Modulation of native T-type calcium channels by omega-3 fatty acids. Biochem Biophys Res Commun. 2005;327(2):485–493. doi: 10.1016/j.bbrc.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 44.Leaf A, Xiao YF, Kang JX, Billman GE. Prevention of sudden cardiac death by n-3 polyunsaturated fatty acids. Pharmacol Ther. 2003;98(3):355–377. doi: 10.1016/s0163-7258(03)00039-1. [DOI] [PubMed] [Google Scholar]
- 45.Boland LM, Drzewiecki MM. Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochem Biophys. 2008;52(2):59–84. doi: 10.1007/s12013-008-9027-2. [DOI] [PubMed] [Google Scholar]
- 46.Hong MP, et al. Effects of free fatty acids on sodium currents in rat dorsal root ganglion neurons. Brain Res. 2004;1008(1):81–91. doi: 10.1016/j.brainres.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 47.Verkerk AO, et al. Incorporated sarcolemmal fish oil fatty acids shorten pig ventricular action potentials. Cardiovasc Res. 2006;70(3):509–520. doi: 10.1016/j.cardiores.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 48.Billman GE, Kang JX, Leaf A. Prevention of ischemia-induced cardiac sudden death by n-3 polyunsaturated fatty acids in dogs. Lipids. 1997;32(11):1161–1168. doi: 10.1007/s11745-997-0149-2. [DOI] [PubMed] [Google Scholar]
- 49.Moreno C, et al. Marine n-3 PUFAs modulate IKs gating, channel expression, and location in membrane microdomains. Cardiovasc Res. 2015;105(2):223–232. doi: 10.1093/cvr/cvu250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.