

Significance
The study's significance is in its novelty along several lines. First, the ion channel studied, a Fluc F− channel, is recently discovered and has been only sparsely studied yet; this work adds a piece of the puzzle to the channel's mechanistic landscape. Second, this is the first channel, to our knowledge, for which two-sided blocking has been observed; this type of blocking is a consequence of the unusual dual-topology assembly of Fluc channels. Third, the mathematical analysis of this kind of block is novel (necessarily so). Fourth, the main result—that the two ends of the pore can be simultaneously occupied by blockers but with negative cooperativity in binding—provokes future structural and mechanistic experiments.
Keywords: ion channel, monobody, block, cooperativity
Abstract
The Fluc family is a set of small membrane proteins forming F−-specific electrodiffusive ion channels that rescue microorganisms from F− toxicity during exposure to weakly acidic environments. The functional channel is built as a dual-topology homodimer with twofold symmetry parallel to the membrane plane. Fluc channels are blocked by nanomolar-affinity fibronectin-domain monobodies originally selected from phage-display libraries. The unusual symmetrical antiparallel dimeric architecture of Flucs demands that the two chemically equivalent monobody-binding epitopes reside on opposite ends of the channel, a double-sided blocking situation that has never before presented itself in ion channel biophysics. However, it is not known if both sites can be simultaneously occupied, and if so, whether monobodies bind independently or cooperatively to their transmembrane epitopes. Here, we use direct monobody-binding assays and single-channel recordings of a Fluc channel homolog to reveal a novel trimolecular blocking behavior that reveals a doubly occupied blocked state. Kinetic analysis of single-channel recordings made with monobody on both sides of the membrane shows substantial negative cooperativity between the two blocking sites.
Several years ago, Baker et al. (1) discovered that many microorganisms harbor in their membranes anion-exporter proteins that keep cytoplasmic F− below the toxic concentrations encountered throughout the aqueous environment. The novelty of this previously unsuspected microbial physiology is mirrored in the unusual molecular architecture of one class of these exporters, the Fluc family. Flucs are highly F−-selective ion channels built as dual-topology homodimers, wherein the paired subunits of the functional channel assemble in antiparallel transmembrane topology (2, 3). This architecture demands that, if the twin subunits adopt identical conformations, the channel must present structurally identical ion entryways to the two sides of the membrane, in sharp contrast to the parallel assembly of conventional multisubunit membrane proteins. Antiparallel assembly of Flucs was established definitively (2) by the use of “monobodies,” small fibronectin-domain proteins of known structure engineered by random variation of amino acid sequences and selected in combinatorial libraries as nanomolar affinity-specific binders (4). In double-sided perfusion experiments, single Fluc channels were shown to be blocked with similar kinetics by monobodies added separately to the internal or external aqueous solution. The channel thus presents to each side of the membrane identical epitopes for the blocker, as required of symmetrical, antiparallel assembly (Fig. 1A), a circumstance that naturally raises the question can monobodies occupy both blocking sites simultaneously?
Fig. 1.

Symmetrical single-sided block of Fluc channels. (A) Cartoon scheme of a symmetric antiparallel Fluc homodimer illustrating the availability of both cis and trans epitopes to monobody block. The channel is rendered in gray with a red monobody-binding epitope, and the monobody is rendered in red. (B) Single Bpe channel recordings with 500 nM monobody L3 added to either cis or trans sides, with holding voltage indicated and opening shown upward in all traces. The top trace is a recording from a control channel in the absence of monobody. Dashed lines represent zero-current level. (C) Voltage dependence of monobody-blocking affinity. Dissociation constants were measured from single-channel open and block times in symmetrical solutions of 500 mM NaF, with 500 nM L3 added to the cis side of the bilayer. Each point represents a separate channel, and the solid curve has no theoretical meaning.
We address this question by direct binding and single-channel experiments with monobody on both sides of the channel. The results reveal a previously unobserved “trimolecular” channel-blocking behavior that reflects double occupancy. Quantitative features of this behavior show substantial negative cooperativity in blocker binding to these two equivalent sites, which we conjecture is electrostatic in origin.
Results
The single-channel recordings in Fig. 1B illustrate block of the bacterial Fluc homolog Bpe by the L3 monobody, a previously described channel–monobody pair (2, 3). In the absence of blocker, Bpe adopts a conducting state with open probability greater than 99%; infrequent closings of millisecond durations are filtered out in the recordings presented here, and in all figures, channel traces are displayed opening upward, regardless of voltage polarity. Addition of monobody to either side of the membrane produces long-lived nonconducting events, which we refer to as blocks, each representing the binding of a single-monobody molecule to the single channel (2). As with several other monobodies previously examined, the dissociation constant of block is on the order of 100 nM, and block times are typically in the range of tens of seconds. The recordings indicate the symmetrical character of the two blocking sites: similar blocking behavior is observed with L3 added on either side of the membrane (cis or trans). The recordings in Fig. 1B also show that block is voltage-dependent. Monobody on the cis side blocks the channel more strongly at −100 mV than at +100 mV, and the reverse effect is seen with monobody on the trans side. This mirrored voltage dependence—quantitative similarity of cis block at +100 mV to trans block at −100 mV and vice versa—is a dramatic manifestation of the chemical symmetry of the monobody receptors on the two sides of the channel. The source of this voltage dependence is unknown, but Fig. 1C shows that it is significant only at high voltages; between ±50 mV, block is voltage-independent. This property will become crucial for experiments reported below.
The low-throughput nature of electrical recording motivated us to supplement this blocking assay with more conventional biochemical methods using Bpe channels in detergent solution. With monobody and channel molecular masses of ∼10 and ∼30 kDa, respectively, fluorescence anisotropy is well-suited as a readout for Fluc–monobody association in the range of 100–1,000 nM Kd. Accordingly, the L3 monobody mutated to place a unique cysteine far away from its binding surface (A12C) was labeled with 5-((2-aminoethyl)amino)naphthalene-1-sulfonic acid (EDANS) fluorophore, a maneuver that did not affect block characteristics. Addition of Bpe channels to labeled L3 increases the initially low anisotropy along a simple binding curve (Fig. 2A), with an affinity similar to that deduced from single-channel block experiments (Kd ∼ 230 nM). Affinity is insensitive to the presence or absence of F− and decreases less than fivefold (Kd ∼ 55–260 nM) as ionic strength increases from 100 to 500 mM, an effect substantially weaker, for example, than that observed for scorpion toxin binding to K+ channels (5). This somewhat lackluster ionic strength dependence implies that electrostatic attraction between channel and monobody does not dominate the interaction.
Fig. 2.

Fluorescence anisotropy assay of monobody–Bpe association. (A) Anisotropy change on adding Bpe channel dimer at indicated concentrations to a fixed concentration (5 µM) of EDANS-labeled L3. Red points and curves mark data from 100 mM ionic strength (NaCl and NaF are indicated by open and filled circles, respectively); black points and curves similarly represent 500 mM ionic strength data. Solid curves are single-site isotherms according to Eq. 1, with Kd values of 59, 66, 120, and 272 nM for 100 mM Cl− and F− and 500 mM Cl− and F−, respectively. Each data point represents the mean ± SEM of three determinations. (B) Fluorescence anisotropy stoichiometry-binding assay. Dotted lines represent the expected results for a 2:1 monobody-channel stoichiometry. Solid lines and filled circles show 1:1 Fluc channel stoichiometry.
The above experiments reprise previous results with several different monobody–Fluc homolog pairs (2). The new question that we pose here is: Can the two identical sites—one on each side of the channel—be simultaneously occupied by blocker? To adjust the anisotropy method for this purpose, fluorescently labeled L3 monobody at 5 µM, far above Kd, is titrated with Bpe (Fig. 2B). At these high concentrations, nearly every channel molecule added will bind L3 as long as the monobody is in excess of the empty sites on Bpe. Accordingly, while unbound L3 sites are still available, the anisotropy signal should increase linearly with Bpe addition. After all of the L3 is bound, however, addition of Bpe can produce no additional anisotropy, and the anisotropy plot will abruptly flatten. The mole ratio of Bpe:L3 at this knee marks the stoichiometry at saturation. These expectations for the anisotropy titration are fulfilled (Fig. 2B), and the result is unexpected. An unambiguous knee is observed, but instead of the anticipated channel to monobody stoichiometry of 0.5 (two monobodies per channel), the data cleanly determine a 1:1 stoichiometry. Thus, at L3 concentrations above the knee, where monobody concentration is >20-fold higher than Kd, the second monobody site on the opposite end of the channel remains unoccupied.
This result was so surprising that we sought to test the apparent negative cooperativity by alternative means: single-channel inhibition by L3 monobody. We examine the concentration dependence of two-sided monobody block in experiments with monobody added symmetrically on both sides of the membrane, according to Scheme 1, which includes a single open state and three blocked states.
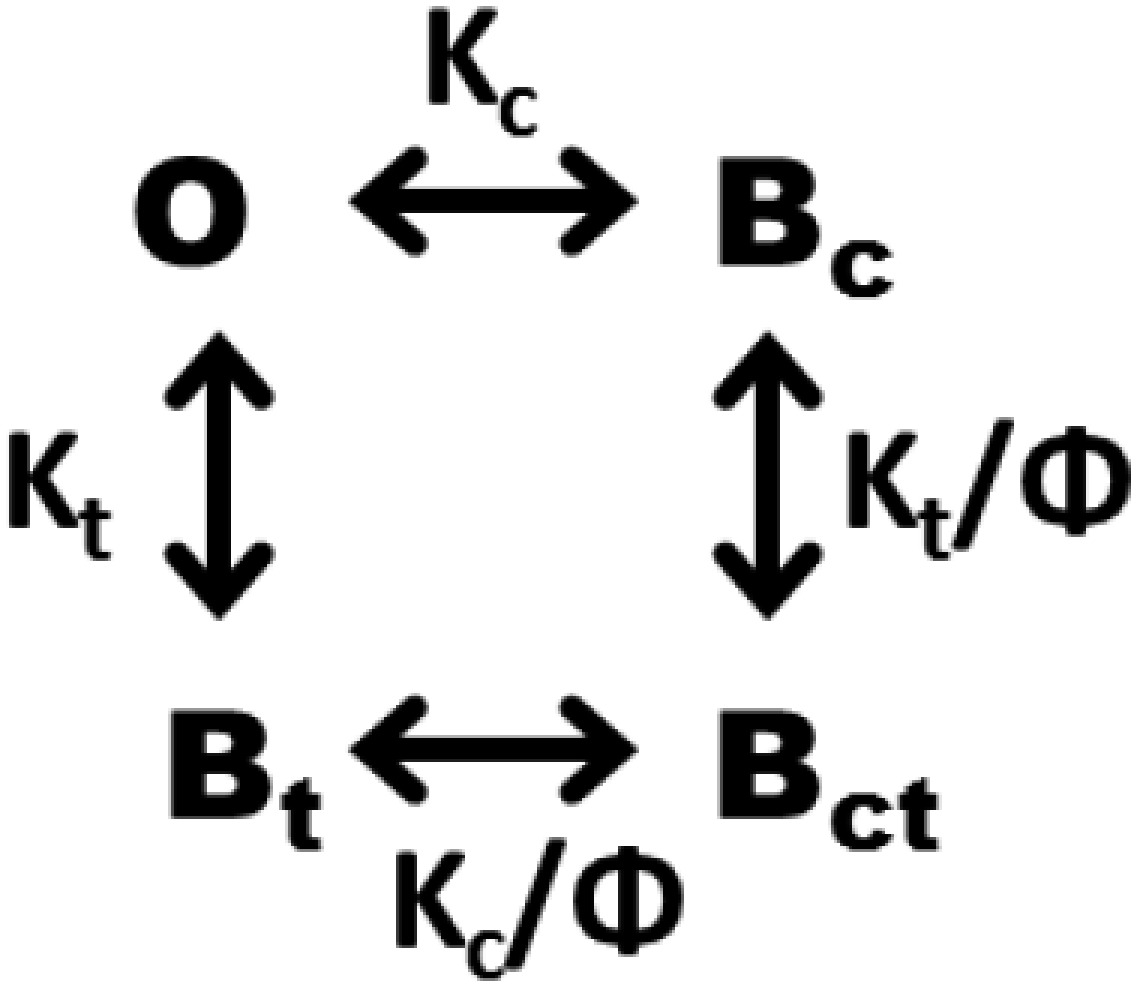
Here, K's represent dissociation constants from the cis or trans sides, and Φ is a cooperativity factor, with Φ = 1 reflecting independent binding and Φ < 1 reflecting negative cooperativity. Scheme 1 predicts that open probability should fall with monobody concentration according to
| [1] |
Given the rather low conductance of Bpe (∼5 pS), we typically record this channel at high voltages (100–200 mV) as in Fig. 1. However, we cannot do this for these experiments for one crucial reason. The slow blocking process makes the triple-exponential distribution of block times demanded by Scheme 1 impossible to analyze, because any one single-channel recording, typically lasting 1–2 h, will entail at most only a few hundred block events. We therefore require that a low voltage (<40 mV) be used for recording to ensure that the blocking parameters are identical from both sides of the channel. Under these conditions, Scheme 1 above reduces to a simpler model (Scheme 2).
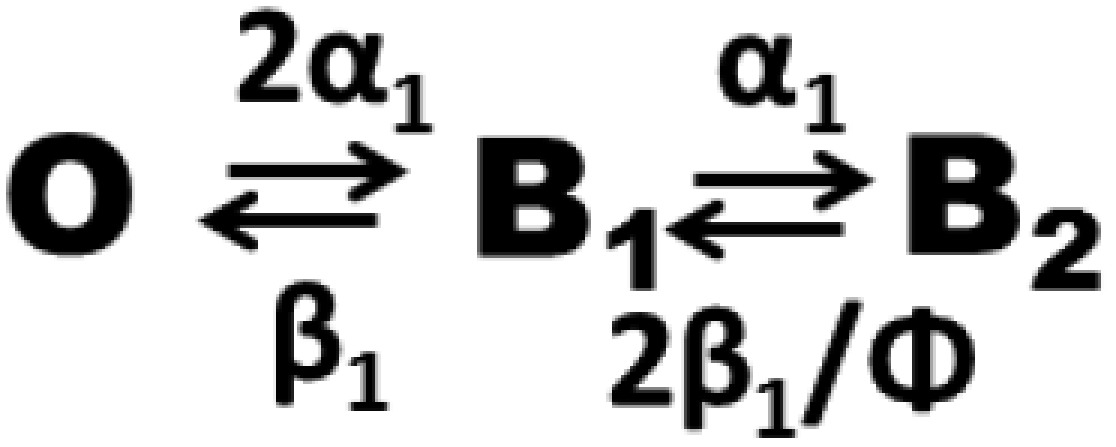
B1 represents the singly occupied blocked channel, without regard for which of the two sites is occupied, and B2 is the doubly occupied blocked state. Rate constants of monobody binding α1 and dissociation β1 refer to single-sided block. For simplicity, Scheme 2 assumes that cooperativity is expressed solely in the off rate, but very similar predictions follow, regardless of how that factor is parsed—in the on rate, off rate, or a combination of the two. The predicted monobody inhibition curve (i.e., the variation of open probability with blocker concentration) now becomes
| [2] |
Here, M represents monobody concentration symmetrically applied to both sides, and K1 is the single-sided dissociation constant at low voltage (identical from the two sides). Accordingly, we sacrificed signal to noise of the channel recordings for the confidence afforded by the simpler Scheme 2 and used a low holding voltage of −25 mV; to increase the channel amplitude at this voltage, we also used biionic conditions, with F− on only one side of the membrane and Cl−, which neither permeates nor blocks this channel, on the other side at a high concentration of 500 mM. Under these conditions, monobody L3 applied separately to either side follows strict bimolecular behavior up to concentrations 20-fold higher than the single-sided Kd (Fig. 3), with on rate increasing linearly and off rate independent of L3 concentration. Fig. 3 also confirms that, despite the chemical asymmetry imposed in solution by biionic conditions, the blocking behavior is quantitatively identical from the two sides, an absolute requirement for using Scheme 2.
Fig. 3.

Symmetrical monobody block. Single Bpe channels were recorded under biionic conditions—500 mM NaF cis/NaCl trans at −25 mV holding voltage. (A) Representative traces from either side at indicated L3 concentrations. (B) Bimolecular kinetics of single-sided block. Effective rate constants 1/τ of binding or (Inset) dissociation are plotted against L3 concentration to show linear dependence of the on rate and constancy of the off rate. Filled and open circles indicate cis or trans monobody additions, respectively. Each point represents the mean and SEM of three to seven separate single-channel runs, except for points without error bars, which represent single runs.
We are now in position to compare single- with double-sided monobody inhibition (Fig. 4). Single channels were recorded as a function of L3 concentration under each condition, and as expected, single-sided inhibition falls along a simple rectangular hyperbola, with K1 ∼ 270 nM. The double-sided data fall below this curve, as expected for two-sided binding, but not as far below as predicted (Eq. 2) for independent binding on both sides (Φ = 1). Instead, these data cannot be convincingly distinguished from the infinite negative-cooperativity curve (Φ = 0). However, we are unwilling to use this experiment to quantify the effect, because its dynamic range—the separation of the predicted curves—is rather small. It is clear, however, that these equilibrium inhibition results qualitatively rule out double-sided independent binding.
Fig. 4.

Single- vs. double-sided inhibition by monobody. (Left) Representative single-channel traces under the conditions in Fig. 3 at indicated L3 concentration and sidedness. (Right) Inhibition curves predicted by various models: single site (dashed curve; Kd = 270 nM), double-sided with independent binding (Φ = 1; solid curve), or infinite negative cooperativity (Φ = 0; dotted curve). Closed circles represent cis and trans single-side data, and open circles represent double-side data. Each point represents the mean and SEM of three to five runs.
Blocking kinetics provides a more sensitive analysis of double-sided block. Scheme 2 demands a double-exponential distribution of block times, which, in contrast with single-sided bimolecular behavior, should vary with monobody concentration. The cumulative block-time distribution P(t) for Scheme 2 is (6)
| [3a] |
where subscripts refer to fast (f) and slow (s) fractions:
| [3b] |
| [3c] |
| [3d] |
For our purposes, this distribution has a most felicitous property. For independent binding or negative cooperativity, Φ ≤ 1, the fast-fraction amplitude can never exceed 9.8% of the distribution, and it diminishes rapidly with increasing blocker concentration, such that it is <1% for all conditions used in our experiments. Thus, block-time distributions here are completely dominated by the slow-fraction time constant. The limited number of blocking events in a given run can therefore be validly approximated by a single-exponential distribution.
The experiment in Fig. 5A illustrates qualitatively that double-occupancy block can occur. A channel was recorded with 3 µM monobody on the trans side only, and the block-time distribution yields a time constant of ∼45 s. Then, with the same channel in the bilayer, an equal concentration of monobody was added to the opposite side, and the distribution was again determined. The double-sided distribution is, as expected, well-fit by a single exponential, with a clearly lengthened time constant of 94 s. This lengthening directly reflects double occupancy, an effect strictly forbidden in bimolecular kinetics. However, the observed doubling of the time constant is much less than predicted by Eq. 3b at this concentration for independent binding to the two sites (approximately eightfold), again implying negative cooperativity in the second binding step. A crucial control experiment (Fig. 5B), necessary in light of the heavy filtering required for these recordings, shows that this time constant lengthening does not arise from missed short opening events, a circumstance that would, on average, artifactually increase the measured block times. The experiment was therefore repeated, except that the second addition of monobody was made to the same side as the first addition, a maneuver that doubles the single-sided L3 concentration and correspondingly shortens the open times twofold, as in the double-sided experiment. In this case, no change in block-time distribution is observed. At monobody concentrations greater than 12 µM, however, missed opening events become significant, and therefore, we limit these double-sided experiments to 5 µM or lower.
Fig. 5.

Double-sided kinetic block analysis. (A) Double-sided L3 block. Left shows representative traces for (Upper) single-sided addition of monobody and in same channel, (Lower) double-sided addition. Right displays cumulative block-time distributions for single- (black) and double-sided (red) block times for the same single channel with single exponential fits (τ = 45 and 94 s, respectively). (B) Control experiment ruling out missed events artifact. Experimental maneuvers and results are displayed as in A, except that the second addition of monobody was made to the same side as the first additional. Solid curve indicates fit with τ = 51 s. (C) Double-sided block time as a function of symmetrical L3 concentration. Curves are expectations of slow-fraction block time (Eq. 3b), with different cooperativity parameters: Φ = 1 (dotted line), Φ = 0 (dashed line), and Φ = 0.11 (solid line).
To quantify the extent of negative cooperativity, block times were measured as a function of two-sided symmetrical variation of monobody in many single-channel recordings (Fig. 5C). According to Eq. 3b, if the two sites were to bind monobody independently (Φ = 1), at a double-sided addition of 0.5 µM (about two times the single-site Kd) block times would approximately double, and at 5 µM, they would increase ∼12-fold to over 5 min. The observed increase in block time at all concentrations is plainly much smaller than this, a direct manifestation of negative cooperativity. It was therefore necessary to use high concentrations of monobody (>5 Kd) to quantify the lengthening of blocked-state time constant. The results (Fig. 5C) produce a cooperativity factor Φ = 0.11, which was derived from the fit of Eq. 3b to the data. Thus, for the second monobody to bind to the channel, it must overcome an extra free energy penalty of 1.3 kcal/mol. However, this value is very sensitive to small errors in the best-fit curve; error analysis shows that cooperativity factors ranging from 0.06 to 0.14, equivalent to free energy penalties of 1.2–1.7 kcal/mol, account for our block-time data within 95% confidence limits.
Discussion
The main inference of this study is that the two monobody-binding sites located on opposite sides of the Bpe channel can both be occupied at the same time as long as a sufficiently high concentration of monobody is used. The most powerful evidence leading to this conclusion is qualitative: that block times arising from monobody addition to one side of the membrane lengthen when the blocker is also added to the other side. This effect reflects the second-order monobody-binding reaction that occurs within the composite blocked intervals (B1 ˗ B2); the time required for both monobodies to dissociate, thus terminating the blocked interval through two sequential stochastic events, is longer than that needed for a single blocker to dissociate. This effect of trimolecular kinetics intensifies as monobody concentration is raised and the doubly occupied state becomes increasingly populated. Quantitative analysis of this effect shows negative cooperativity, in which the second monobody binds approximately ninefold more weakly than the first monobody. This cooperativity reveals itself in three types of experiment. First, as measured by fluorescence anisotropy, binding stoichiometry is very close to one monobody per channel homodimer (Fig. 2B) rather than the value of two that we initially anticipated from single-sided block identical from both sides. This inference is not in conflict with our double-sided block results, which clearly reflect two monobodies occupying the channel simultaneously, because the range of cooperativity factors consistent with the block-time data is rather broad. The absence of any indication of double occupancy in the fluorescence anisotropy experiments would favor a cooperativity factor closer to ∼0.03 (equivalent to ∼2 kcal/mol). In such a case, impractically high concentrations of channel and monobody would be required to observe double occupancy in the fluorescence assay. Second, the equilibrium monobody-inhibition curve (Fig. 4) falls between the single-site and independent double-site predictions, although not with sufficient precision to quantify the effect. Third, the block times lengthen with double-sided monobody concentration much less steeply than predicted for independent binding to yield a cooperative free energy of 1.2–1.7 kcal/mol.
What is the source of this negative cooperativity? Two mechanisms suggest themselves as possibilities: allosteric or electrostatic. Binding of the first blocker to the initially symmetrical channel might cause a distant structural perturbation on the other site, weakening its affinity. Alternatively, because the monobody carries a net charge of −4 (2), electrostatic repulsion emanating from the first monobody might account for the weakened affinity suffered by the second. Without a high-resolution structure of the Bpe–L3 complex, we cannot even begin to approach this issue in a serious way, but a crude and certainly unrealistic calculation can indicate whether the electrostatic mechanism is remotely plausible. We represent the monobodies in the doubly blocked configuration as point charges of −4 each separated by an effective distance of 70 Å with a protein-like homogeneous dielectric constant of 30 between them. Then, the cooperative free energy arising from Coulombic repulsion would be given by
| [4] |
equivalent to strong negative cooperativity (Φ = 0.02) that our measurements would not be able to distinguish from Φ = 0. We offer this estimate only to suggest that an electrostatic mechanism is not implausible, as it agrees unreasonably well with the experimental results. If a crystal structure of the doubly bound complex could be obtained in the future, we would also be able to ask if the two monobodies occupy structurally asymmetric sites as predicted by the allosteric mechanism.
Materials and Methods
Biochemical.
All chemicals were reagent-grade. Phospholipids (1-palmitoyl,2-oleoyl phosphaditylethanolamine or the analogous phosphatidyl glycerol) were obtained from Avanti Polar Lipids, fluorophore-maleimide was from Molecular Probes, and n-decylmaltoside was from Anatrace. Amino acid sequence, expression, purification, and reconstitution of the Fluc homolog used here, which we denote Bpe, and monobody L3 were as described (2, 3). The Bpe construct used here carries two functionally nonperturbing mutations (R29K/E94S) to enhance biochemical tractability. For fluorescence anisotropy measurements, the L3 monobody substituted with a unique cysteine (A12C) located far from the Fluc-binding surface was used. Immediately after purification, the L3 cysteine mutant was treated with 1 mM EDANS-C2-maleimide at pH 7.0 for 90 min. Excess reactant was quenched with 10 mM l-cysteine and separated from L3 through a PD-10 G20 Resin-Desalting Column (GE Healthcare). Finally, the labeled L3 was isolated by size-exclusion chromatography and stored in aliquots at −80° at a concentration (30–50 μM) determined by absorption at 280 nm, with a small correction for contribution by the fluorescent moiety.
Fluorescence Anisotropy Assay of Bpe–L3 Binding.
Labeled monobody L3 (100 nM) was mixed with varying concentrations of Bpe in 100 or 500 mM NaF or NaCl, 5 mM n-decylmaltoside, and 25 mM Hepes (pH 7) and equilibrated for 20 min in darkness at 21 °C. Fluorescence anisotropy measurements were carried out with a Horiba FluoroMax-4 spectrofluorometer equipped with calibrated emission and excitation polarizers. Binding isotherms were fit to Eq. 5, which was derived from one site-binding equilibrium with total ligand rather than free ligand as the experimental variable:
| [5] |
Here, A([Bpe]) is the anisotropy as a function of total Bpe concentration added to a fixed concentration of L3, and Af and Ai are final and initial anisotropy values, respectively. Fluorescence anisotropy reports the fraction of L3 complexed with Bpe. Although this titration yields a Kd value for the channel–monobody interaction, it does not provide the stoichiometry at saturation. However, conditions of the Bpe–L3 binding experiment may be adjusted to determine stoichiometry. If the total L3 monobody concentration is maintained far above Kd, an increase in anisotropy reflects the filling of available binding sites on Bpe by the excess L3 monobody. As Bpe is added, anisotropy increases linearly until all of the binding sites are saturated, at which point the increase abruptly ceases. The molar fraction of Bpe dimer to L3 monobody at this point yields the stoichiometry of the complex. These experiments require attenuation of the labeled L3 fluorescence signal, which would be far too large at these high concentrations; accordingly, the labeled L3 monobody (100 nM) was supplemented with unlabeled L3 monobody (4.9 μM) in all samples.
Single-Channel Recording of Block by Monobody.
Single Bpe channels were inserted into planar lipid bilayer membranes (70% 1-palmitoyl,2-oleoyl phosphaditylethanolamine/30% phosphatidyl glycerol) and recordings collected at 22° ± 1° as previously reported (3), with the trans chamber defined as the electrical ground. For recordings made at high voltage (100–200 mV), solutions were symmetrical (500 mM NaF, 50 μg/mL BSA, 15 mM Mops, pH 7.0); for low-voltage recordings, biionic conditions (500 mM NaF cis/NaCl trans) were used. Recordings were low-pass filtered at 50–200 Hz and digitally filtered further for analysis of blocking kinetics. For analysis of the low-amplitude (∼0.1 pA) channels recorded near zero voltage, 3-Hz filtering was confirmed to give accurate open and blocked times by hand analysis of the recording.
For most experiments using two-sided addition of monobody, a recording was first collected with L3 present only on one side, and then, the monobody was added to the other side at an equal concentration. L3 monobody dissociation constants were measured from the ratio of total blocked and open times at a given monobody concentration, and dwell-time kinetics was determined from the cumulative distributions calculated from 22 to 880 open or blocked events in a given run; all distributions were satisfactorily fit with single exponentials. Cooperativity factor Φ for double-sided monobody block was determined by varying monobody concentration symmetrically on both sides of the membrane and determining block-time distributions as above. At each concentration, block-time constants from three separate single channels were collected, and the full dataset was fit to Eq. 3b, with Φ as the only adjustable parameter. Confidence limits on Φ were determined by exhaustive fitting of Eq. 3b to 81 possible subsets of the data, and each subset was composed of one time constant at each blocker concentration.
Acknowledgments
We thank Dr. Ming-Feng Tsai for insightful comments on blocking analysis. This work was supported, in part, by NIH Grants R01-GM107023 and K99-GM11167. J.T.N. was supported by Howard Hughes Medical Institute Summer Undergraduate Fellowships.
Footnotes
The authors declare no conflict of interest.
References
- 1.Baker JL, et al. Widespread genetic switches and toxicity resistance proteins for fluoride. Science. 2012;335(6065):233–235. doi: 10.1126/science.1215063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stockbridge RB, Koide A, Miller C, Koide S. Proof of dual-topology architecture of Fluc F- channels with monobody blockers. Nat Commun. 2014;5:5120. doi: 10.1038/ncomms6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stockbridge RB, Robertson JL, Kolmakova-Partensky L, Miller C. A family of fluoride-specific ion channels with dual-topology architecture. eLife. 2013;2:e01084. doi: 10.7554/eLife.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koide S, Koide A, Lipovšek D. Target-binding proteins based on the 10th human fibronectin type III domain (¹⁰Fn3) Methods Enzymol. 2012;503:135–156. doi: 10.1016/B978-0-12-396962-0.00006-9. [DOI] [PubMed] [Google Scholar]
- 5.Anderson CS, MacKinnon R, Smith C, Miller C. Charybdotoxin block of single Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J Gen Physiol. 1988;91(3):317–333. doi: 10.1085/jgp.91.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piasta KN, Theobald DL, Miller C. Potassium-selective block of barium permeation through single KcsA channels. J Gen Physiol. 2011;138(4):421–436. doi: 10.1085/jgp.201110684. [DOI] [PMC free article] [PubMed] [Google Scholar]