

Significance
IgG molecules are capable of inducing pro- and antiinflammatory responses dependent on their fragment crystallizable domain (Fc) glycan composition. Antiinflammatory responses are specifically triggered upon Fc sialylation, which decreases the binding affinity for type I Fc receptors but enhances binding to type II Fc receptors such as SIGN-R1, CD23, or human DC-SIGN. Structural analyses revealed that sialylation induces conformational changes in the Fc portion, which is a prerequisite for the selective binding to type II Fc receptors. Here we generated an Fc variant, F241A, that mimics the conformational state of sialylated Fc. F241A, even when nonsialylated, mediated protection from autoantibody- and T cell-mediated inflammation in a type II Fc receptor-dependent manner.
Keywords: IgG Fc sialylation, conformational change, antiinflammatory, Treg cells
Abstract
The antiinflammatory activity of intravenous immunoglobulin (IVIG) is dependent on the presence of sialic acid in the core IgG fragment crystallizable domain (Fc) glycan, resulting in increased conformational flexibility of the CH2 domain with corresponding modulation of Fc receptor (FcR) binding specificity from type I to type II receptors. Sialylated IgG Fc (sFc) increases the activation threshold of innate effector cells to immune complexes by stimulating the up-regulation of the inhibitory receptor FcγRIIB. We have found that the structural alterations induced by sialylation can be mimicked by specific amino acid modifications to the CH2 domain. An IgG Fc variant with a point mutation at position 241 (F→A) exhibits antiinflammatory activity even in the absence of sialylation. F241A and sFc protect mice from arthritis in the K/BxN-induced model and, in the T cell-mediated experimental autoimmune encephalomyelitis (EAE) mouse model, suppress disease by specifically activating regulatory T cells (Treg cells). Protection by these antiinflammatory Fcs in both antibody- and T cell-mediated autoimmune diseases required type II FcRs and the induction of IL-33. These results further clarify the mechanism of action of IVIG in both antibody- and T cell-mediated inflammatory diseases and demonstrate that Fc variants that mimic the structural alterations induced by sialylation, such as F241A, can be promising therapeutic candidates for the treatment of various autoimmune disorders.
Intravenous immunoglobulin (IVIG), although initially developed as an Ig replacement therapy in patients with hypogammaglobulinemia (1), has gained widespread use for its immunomodulatory activities. It is an approved therapeutic for the treatment of autoimmune disorders such as immunothrombocytopenia (ITP), chronic inflammatory demyelinating polyneuropathy, Kawasaki’s disease, and Guillain-Barre syndrome (2, 3), and is used in a growing number of autoimmune and inflammatory disorders. Its antiinflammatory activity has been shown to result from the presence of a specific glycan, the α2,6-sialylated, complex biantennary structure present on the CH2 domain of the fragment crystallizable domain (Fc) and found in a small proportion of heterogeneous antibody preparations in IVIG (4). Sialylation of the Fc glycan on the CH2 domain results in IgGs that can engage type II Fc receptors (FcRs) such as specific ICAM-3 grabbing non-integrin-related 1 (SIGN-R1), dendritic cell-specific ICAM-3 grabbing non-integrin (DC-SIGN), and CD23 (5–8), while reducing their binding affinity to type I FcRs (9–11). Studies in mouse models of serum-induced arthritis, antibody-dependent ITP, nephrotoxic nephritis, and autoimmune blistering diseases confirmed the antiinflammatory activity of the sialylated Fc, whether from IVIG or generated from recombinantly expressed IgG1 (5, 9, 12, 13). Moreover, increasing the percentage of sialylated Fc fragments either in IVIG or recombinant expressed IgG1 resulted in an enhanced therapeutic potency of these preparations (6, 9, 12, 14). Elucidation of the mechanism by which sialylated IgG Fc (sFc) induces an antiinflammatory response was first reported in murine models of arthritis, demonstrating that selective binding of sialylated Fc to type II FcRs resulted in the production of interleukin (IL) 33 by regulatory macrophages, which in turn stimulated IL-4 secretion from basophils. IL-4 induced the up-regulation of the inhibitory receptor FcγRIIB on effector macrophages, thereby increasing the activation threshold of these cells and suppressing inflammation (15, 16). Subsequent studies have confirmed that IVIG treatment of human populations resulted in both increased serum IL-33 levels and FcγRIIB expression on lymphoid and myeloid cells, consistent with murine data (17–19).
Crystallographic and biophysical studies on sialylated and asialylated IgG Fc fragments have provided insights into the structural basis for the antiinflammatory activity of sialylated Fc. Sialylation of the complex, biantennary glycan of the IgG Fc results in increased conformational flexibility of the CH2 domain (20), thereby sampling the closed conformations of the CH2 domain required for type II FcR binding (11). In contrast, asialylated Fc structures uniformly result in open Fc conformations, consistent with their binding specificity for type I FcRs (21). Glycan interactions with amino acid residues of the CH2 domain are disrupted upon sialylation, providing a basis for the observed conformational changes seen in the protein structure and consistent with a model proposed for the binding specificity of sialylated Fc for type II FcRs (11). Based on these observations, we generated a series of Fc variants targeting the amino acids of the CH2 domain that interact with the glycan, with the goal of determining their impact on type II FcR binding and the resulting antiinflammatory activity. Both gain- and loss-of-function mutants were examined in this study. The identification of a gain-of-function variant, which could mimic the conformational state induced by sialylation, without requiring this specific carbohydrate modification, may potentially simplify the development of antiinflammatory IgG Fc for therapeutic use (20). We succeeded in identifying a mutation (F241A) predicted to increase mobility of the α1,3-arm, and which replicates the antiinflammatory activity of sialylated Fc even in the absence of sialylation. We have characterized this variant, in comparison with sialylated Fc, in both antibody and T-cell models of autoimmune inflammation.
Although the basis for IVIG protection in antibody-mediated models of inflammation has been extensively studied, as summarized above (9, 12, 13, 16), recent studies have demonstrated that IVIG can also protect in classical T cell-mediated autoimmune disorders, such as experimental autoimmune encephalomyelitis (EAE) (22), as well as in a model of airway hyperresponsiveness (23, 24). This therapeutic effect of IVIG is proposed to result from the activation and expansion of regulatory T cells (Treg cells), thus suppressing T-cell responses by IFN-γ–secreting TH1 and IL-17–secreting TH17 cells (25, 26). We therefore sought to investigate whether the Treg-cell activation and expansion were also the result of sialylated Fcs or the F241A variant. Using F241A and sialylated and asialylated IVIG, we investigated the mechanisms of action of their immunomodulatory effects on Treg-cell activation and suppression of T cell-dependent autoimmunity. We demonstrate that the sialylation of IVIG is critically required for Treg-cell activation and expansion and that the F241A variant is sufficient to suppress T cell-dependent inflammation in the EAE mouse model. Furthermore, both sFc as well as F241A stimulate the production of IL-33 that in turn activates Treg cells through the ST2 receptor, contributing to the suppression of T cell-mediated autoimmune responses.
Results
F241A Mimics Sialylated IgG Fc and Protects from Autoantibody-Induced Inflammation.
The crystal structures of nonsialylated and sialylated IgG molecules show differences in the orientation of the heavy chains. Whereas nonsialylated IgG (G0F form) remains in an open conformation and provides a structure capable of interacting with type I Fc receptors, sialylated IgG (G2FS2 form) is more flexible, allowing alternate conformations (open and closed) (20) enabling it to bind to type II Fc receptors (11). As shown in Fig. 1A, the aromatic side chains of F241 in an asial Fc structure are stacked with respect to each other (Fig. 1A, Left). In this orientation, the phenylalanine side chain forms a hydrophobic interaction with sugar residues in the α1,3-arm of the Fc glycan. Surprisingly, the aromatic ring of F241 in the sial Fc structure exhibits a near-90° rotation relative to the aromatic ring of F241 in the asial Fc structure (Fig. 1A, Right). This suggests that upon sialylation, the interaction between F241 and the α1,3-arm of the Fc glycan may be disrupted, which potentially contributes to the observed changes in antibody structure and function. Hence, to mimic this disruption in protein–glycan interaction, we introduced an alanine point mutation at residue F241 (F241A) and determined how this mutation altered the activity of sial and asial Fc. We produced α2,6-sialylated F241A Fc by expressing the recombinant protein in 293 cells stably overexpressing the glycosyltransferases ST6GalI and β4GalTI. For comparison, a fraction of this sial F241A Fc preparation was subsequently treated with neuraminidase to remove sialic acid, yielding asial F241A Fc. We confirmed the sialylation status of both F241A Fc preparations by lectin blotting (Fig. S1A). We next verified that sial F241A Fc retained DC-SIGN binding activity in an ELISA format, as demonstrated by increased receptor binding affinity relative to asial WT Fc (Fig. S2). However, neuraminidase treatment of F241A Fc did not abolish DC-SIGN binding, establishing that the F241A mutation conferred DC-SIGN binding activity independent of sialic acid. To determine whether the F241A mutation resulted in functional DC-SIGN binding and signaling, we investigated the ability of F241A and sFc to induce IL-33 expression in DC-SIGN–expressing bone marrow-derived macrophages (BMMΦs). Only sFc induces IL-33 expression in these cells, whereas both sialylated and asialylated F241A Fc induced IL-33 expression in BMMΦs in a DC-SIGN–dependent manner (Fig. 1B). Furthermore, human (h)DC-SIGN+ BMMΦs stimulated with asial F241A Fc, as well as sFc, suppressed footpad swelling when transferred to mice that were then challenged with arthritogenic K/BxN serum. Protection was dependent on DC-SIGN expression (Fig. 1C). Furthermore, protection was only achieved with sialylated wild-type Fc, whereas the Fc mutant F241A was capable of protecting mice, even when nonsialylated (Fig. 1D). Thus, the F241A mutation recapitulates several Fc functions in assays developed to measure sial Fc activity.
Fig. 1.

F241A is capable of suppressing serum-induced arthritis. (A) Schematic depiction of the interaction between mannose residues of the nonsialylated core glycan (Asialo-Fc) with the phenylalanine residue at position 241 of the Cγ2 domain. Upon sialylation (Sial-Fc) this interaction no longer occurs, resulting in a higher conformational flexibility of the Fc portion to switch between the so-called open and closed conformations. (B) hDC-SIGN+ bone marrow-derived macrophages were pulsed with sialylated (sFc) or nonsialylated (Fc) wild-type Fc or F241A Fc. Whole-cell extracts were used to analyze IL-33 production by Western blotting. Actin served as loading control. (C) BMMΦs from either SIGN-R1−/− or hDC-SIGN+ mice were pulsed with nonsialylated wild-type Fc (asialo-Fc), sialylated wild-type Fc (sFc), or F241A. After extensive washes, the cells were adoptively transferred into C57BL/6 mice and then challenged with K/BxN serum. (D) hDC-SIGN+ BMMΦs were pulsed with either asialylated (asialo Fc) or sialylated (α2,6sFc) variants of wild-type Fc or F241A. The cells were adoptively transferred into C57BL/6 mice and then challenged with K/BxN serum. Means ± SEM are plotted; *P < 0.05; **P < 0.01 determined by Tukey’s post hoc test.
To further define the activity of F241A as an antiinflammatory molecule, we treated C57BL/6 mice either with PBS, IL-4ic, or asialylated F241A (0.033 g/kg) and then challenged them with K/BxN sera. Serum IL-6 levels were significantly reduced in mice that received IL-4ic or F241A (Fig. 2A). Consistent with this observation, IL-4ic– and F241A-treated mice showed reduced clinical signs of arthritis (Fig. 2B), showing that F241A is sufficient to suppress inflammation comparable to IVIG and sFc (9, 12). To confirm that this suppression by F241A is type II FcR-dependent, we used SIGN-R1−/− or SIGN-R1−/− hDC-SIGN+ recipients. Mice received either sialylated wild-type Fc or neuraminidase-treated nonsialylated F241A (Fig. S1A) (both 0.033 g/kg) and were challenged with K/BxN sera. Suppression of arthritic inflammation was achieved by both preparations; however, only mice that expressed human DC-SIGN (hDC-SIGN+) were protected (Fig. 2C), demonstrating that the presence of the type II Fc receptor SIGN-R1 or its human ortholog DC-SIGN, respectively, is required for the immunomodulatory effect induced by sialylated Fc and F241A.
Fig. 2.

F241A induces antiinflammatory responses through engagement of type II Fc receptors. (A) C57BL/6 wild-type mice were treated with PBS, IL-4ic, or F241A (0.033 g/kg) and challenged 1 h later with K/BxN sera. Blood was collected from these mice 6 d posttreatment, and IL-6 levels in the sera were analyzed by ELISA. (B) Clinical scores of mice were monitored reflecting the severity of serum-induced arthritis. (C) SIGN-R1−/− and hDC-SIGN+ mice were treated with sialylated wild-type Fc (sFc) or F241A (both 0.033 g/kg) and challenged 1 h later with K/BxN serum. Clinical examination of the mice was conducted daily until day 6 post K/BxN challenge, demonstrating the development and severity of arthritic inflammation. Means ± SEM are plotted; **P < 0.01; ***P < 0.001 determined by Tukey’s post hoc test.
Sialic acid can be linked to the penultimate galactose of the complex, biantennary Fc glycan in either α2,3-, α2,6-, or α2,8-conformations. We have previously reported that only the α2,6-linked glycoform of sialic acid is biologically active (12). Our previous modeling data on the structural analysis of different Fc sialoforms (11) predicted that the Glu318/Lys340 pocket at the Cγ2–Cγ3 interface was required for the biological activity of α2,6-sial Fc and could uniquely accommodate this glycoform, whereas the α2,3-linked sialic acid would be sterically inhibited from fitting into this pocket. To test this prediction, we introduced an E318N point mutation into IgG1 Fc and compared its properties, when α2,6-sialylated, to wild-type α2,6-sial Fc. Although comparable degrees of sialylation were achieved with the mutant compared with the wild type, only the wild-type sialylated Fc was capable of stimulating IL-33 expression in hDC-SIGN+ BMMΦs (Fig. S3A). Mice receiving BMMΦs stimulated with α2,6-sial wild-type Fc, but not α2,6-sial E318N Fc, exhibited reduced clinical signs of disease in the K/BxN serum transfer arthritis model (Fig. S3B), as well as lower levels of IL-6 (Fig. S3C). Together, these results define both F241 and E318 as residues that contribute to the antiinflammatory activity of α2,6-sial Fc.
Sialylated Fc/F241A Activates Treg Cells.
Recent studies in patients and in animal models suggest that administration of IVIG can result in an expansion of Treg cells (22, 27), effectively dampening T cell-dependent autoimmune reactions by increasing the number and the suppressive capacity of Treg cells (28). To determine which component of an IVIG preparation may be responsible for this effect, we used IVIG (1 g/kg), F(ab′)2 (0.66 g/kg), or Fc (0.33 g/kg) preparations of IVIG and administered them i.v. at equimolar concentrations into C57BL/6 wild-type mice. Four days postinjection, we analyzed the percentage of splenic CD4+CD25+Foxp3+ Treg cells by flow cytometry. In comparison with PBS-treated mice, administration of intact IVIG or its Fc fragments led to a twofold expansion of Treg cells and was abrogated by using IVIG F(ab′)2 (Fig. 3A). We next determined the role of Fc sialylation in Treg-cell expansion in an ongoing inflammatory response. C57BL/6 mice were treated either with PBS, IVIG (1 g/kg), neuraminidase-treated nonsialylated IVIG (NA-IVIG) (1 g/kg), or F241A (0.033 g/kg) (Fig. S1B), challenged with K/BxN serum, and evaluated for disease progression and Treg-cell expansion. As observed previously (13, 29), clinical scores of arthritis showed that IVIG and F241A, but not nonsialylated IVIG, protected mice from arthritis (Fig. 3B). Treg-cell expansion was observed in IVIG- and F241A-treated mice that were subsequently challenged with K/BxN serum, but not in PBS- or asial IVIG-treated mice (Fig. 3C), suggesting that the Treg-cell subset becomes selectively expanded by sialylated Fc and F241A, respectively, in this model of arthritis.
Fig. 3.

sFc/F241A activates and expands Treg cells. (A) C57BL/6 wild-type mice received i.v. injections of IVIG (1 g/kg), IVIG F(ab′)2 (0.66 g/kg), IVIG Fc (0.33 g/kg), or PBS as control. On day 4 postinjection, Treg-cell numbers in spleens were analyzed by flow cytometry. (B) C57BL/6 wild-type mice were given IVIG (1 g/kg), NA-IVIG (1 g/kg), nonsialylated F241A (0.033 g/kg), or PBS as control and then challenged with K/BxN serum. Clinical signs of arthritis were monitored. (C) On day 5 the mice in B were euthanized and Treg cells in spleens were analyzed by flow cytometry. Means ± SEM are plotted; *P < 0.05; **P < 0.01; ***P < 0.001 determined by Tukey’s post hoc test.
IVIG- or F241A-Activated Treg Cells Suppress Pathogenic CD4+ T-Cell Responses in Vivo.
To evaluate whether Treg-cell expansion in response to IVIG, sFc, or F241A is able to suppress pathogenic CD4+ T-cell responses, we induced EAE in C57BL/6 wild-type mice by immunization with MOG35–55 peptide emulsified in complete Freund's adjuvant (CFA). Starting 5 d postinduction, the mice were treated either with PBS, IVIG, or NA-IVIG every 5 d (at 1 g/kg). Clinical scores of EAE showed that mice that received IVIG had significantly reduced clinical scores compared with PBS-treated mice (Fig. 4A). However, when asial IVIG (NA-IVIG) (Fig. S1B) was administered, the protective effect was abolished. To determine the potential mechanistic basis for this effect, we characterized cells from draining lymph nodes at day 25 postinduction and analyzed the percentages of various T-cell subsets. All subpopulations of CD4+ effector T cells, TH1 (IFN-γ+), TH17 (IL-17A+), and IFN-γ+IL-17A+CD4+ T cells, were present at similar percentages (Fig. 4B); however, the percentages of CD4+CD25+Foxp3+ Treg cells were significantly elevated in mice that were given IVIG compared with PBS- or NA-IVIG–treated groups (Fig. 4C).
Fig. 4.

IVIG/F241A-activated Treg cells efficiently suppress T cell-mediated autoimmunity in EAE mice. (A) C57BL/6 wild-type mice were immunized with MOG35–55 peptide to induce EAE. Starting on day 5 post EAE induction, mice received i.v. injections of PBS, IVIG (1 g/kg), or NA-IVIG (1 g/kg) every 5 d. Clinical scores of EAE are shown. (B and C) Mice were euthanized and CD4+ effector T cells (B) and Treg cells (C) from draining lymph nodes were analyzed by flow cytometry. (D) EAE was induced in C57BL/6 wild-type mice by immunization with MOG35–55 peptide. Starting on day 5 and every 5 d thereafter, mice received nonsialylated F241A (0.033 g/kg) or PBS intravenously. For Treg-cell depletion, mice were given the Treg-cell depletion antibody PC61 (400 µg) 3 d before EAE induction as well as every fifth day postinduction. Clinical scores of disease are depicted. (E and F) Cells from draining lymph nodes of EAE mice were isolated and analyzed for the percentages of CD4+ effector T cells (E) and Treg cells (F) by flow cytometry. Means ± SEM are plotted; *P < 0.05; **P < 0.01; ***P < 0.001 determined by Tukey’s post hoc test.
To assess whether the protection from EAE we observed in IVIG-treated animals was specifically mediated through activation and expansion of Treg cells, we tested the protective potential of F241A as a surrogate for IVIG in untreated and Treg cell-depleted mice. Treg-cell depletion was achieved by administration of an anti-CD25 antibody (PC61) 3 d before EAE induction and every 5 d thereafter. Mice treated with F241A (0.033 g/kg) displayed reduced disease severity compared with PBS-treated mice (Fig. 4D), demonstrating that F241A was effective at protecting animals from EAE at a 35-fold reduced dose compared with IVIG. However, in Treg cell-depleted mice, this protective effect of F241A treatment was lost. FACS analysis of draining lymph nodes harvested at the conclusion of the study indicated that neither F241A nor PC61 treatment significantly affected CD4+ effector T cells (Fig. 4E), whereas Treg-cell levels were reduced by PC61 treatment (Fig. 4F). Depletion of Treg cells thus correlated with the loss of protection from EAE observed in F241A-treated animals. To distinguish between natural (nTreg) and inducible (iTreg) Treg cells, we analyzed the Treg cells in IVIG- and PBS-treated EAE mice for their expression of the nTreg-specific transcription factor Helios (30). The expansion of Treg cells did not correlate with an increase in Helios expression (Fig. S4C), which indicates that IVIG (1 g/kg), which protected mice from EAE (Fig. S4 A and B), specifically induces CD4+CD25+Foxp3+Helios− iTreg cells.
Together, these results indicate that sialylated IVIG as well as F241A lead to the activation and expansion of Treg cells, resulting in the suppression of pathogenic CD4+ effector T-cell responses and clinical disease in EAE.
Type II Fc Receptors Are Required for sFc-Mediated Protection from EAE.
The requirement for the type II Fc receptor SIGN-R1 or hDC-SIGN for sFc-induced suppression of inflammation has been extensively studied in the context of autoantibody-mediated diseases and for the stimulation of IL-33 production (29). We examined the requirement for the type II Fc receptor SIGN-R1 for sFc-mediated protection in EAE. EAE was induced in C57BL/6 wild-type or SIGN-R1−/− mice and then treated with IVIG (1 g/kg) or PBS as control. Whereas wild-type mice were protected from EAE by IVIG, this protective effect was significantly reduced in SIGN-R1 knockout mice (Fig. 5A). This is consistent with our observations that in the SIGN-R1−/− background, IVIG (1 g/kg) neither protects from K/BxN-induced arthritis (Fig. S5A) nor induces Treg-cell activation (Fig. S5B). By contrast, administration of exogenous IL-33 partially restored protection in SIGN-R1−/− mice from EAE (Fig. 5B). Similarly, transgene expression of hDC-SIGN partially complements the loss of SIGN-R1 (29) and results in reduced EAE clinical scores in both IVIG- (1 g/kg) and F241A- (0.033 g/kg) treated mice (Fig. 5 C and D).
Fig. 5.

Requirement for type II Fc receptors for IVIG/F241A-mediated protection in EAE. EAE was induced by immunization with MOG35–55 peptide. Mice received i.v. injections on days 5, 10, 15, and 20 postinduction. Clinical scores of disease are displayed. (A) C57BL/6 wild-type and SIGN-R1−/− mice were treated with IVIG (1 g/kg) or PBS. (B) SIGN-R1−/− mice were given IVIG (1 g/kg) as mentioned above or IL-33 (0.5 µg) i.p. every 2 d starting on day 5 post EAE induction. (C) SIGN-R1−/− and SIGN-R1−/− hDC-SIGN+ mice were treated with PBS or IVIG (1 g/kg). (D) SIGN-R1−/− hDC-SIGN+ mice received nonsialylated F241A (asialo-F241A) (0.033 g/kg) or PBS control. Means ± SEM are plotted; *P < 0.05; **P < 0.01; ***P < 0.001 determined by Tukey’s post hoc test.
IL-33 Is a Critical Mediator of sFc-Triggered Treg-Cell Activation.
We have previously reported that the up-regulation of FcγRIIB on effector macrophages by sialylated Fc critically depends on production and secretion of IL-33 (29). Recent findings have indicated that IL-33 has a positive effect on Treg-cell stimulation and activation (31, 32) and thereby contributes to the suppression of inflammation in a mouse model of experimental colitis (33). To test the possibility that sFc-induced production of IL-33 may also contribute to Treg-cell stimulation, we confirmed that naïve CD4+ T cells isolated from C57BL/6 wild-type mice, cultured for 3 d in the presence of anti-CD3 and anti-CD28 antibodies and TGF-β to specifically drive Treg-cell differentiation and then treated with IL-33, had a synergistic effect on Treg-cell differentiation as well as on Foxp3 expression (Fig. S6), as was previously reported by Schiering and coworkers (33). Moreover, IL-33 induced up-regulation of the IL-33 receptor ST2 on Treg cells. Addition of IL-23 to the Treg-cell culture counteracted the effect of IL-33 (Fig. S6), consistent with IL-23 being a negative regulator of ST2 (33, 34).
We next determined whether administration of IL-33 also affects Treg cells in vivo. IL-33 (0.5 µg) was given to C57BL/6 wild-type mice daily for 4 consecutive days. On day 5, spleens were analyzed for Treg-cell numbers. IL-33 administration resulted in a significant increase of Treg cells compared with PBS-treated control mice (Fig. 6 A and B). hDC-SIGN+ BMMΦs were pulsed either with PBS, IVIG, or nonsialylated F241A, and IL-33 expression was measured by quantitative real-time PCR, showing that IVIG and F241A clearly induced IL-33 expression in a DC-SIGN–dependent manner (Fig. 6C). After treatment, BMMΦs were subsequently transferred into C57BL/6 wild-type mice. Five days post cell transfer, only mice that received IVIG- or F241A-pulsed hDC-SIGN+ BMMΦs had significantly higher levels of Treg cells (Fig. 6 D and E). This phenotype correlated with enhanced expression of the IL-33 receptor ST2 on these cells (Fig. 6D). In addition, IL-33 treatment of EAE resulted in a significant amelioration of EAE symptoms, which correlated with an increase of Treg cells in draining lymph nodes (Fig. 6 F and G).
Fig. 6.
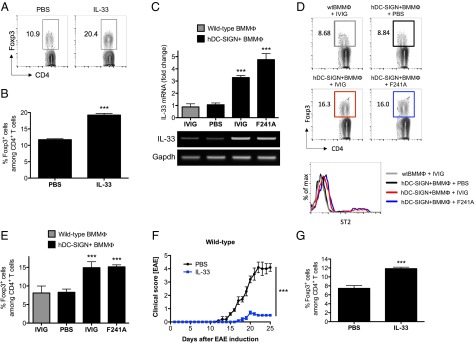
IVIG- or F241A-induced IL-33 production activates Treg cells. (A and B) C57BL/6 wild-type mice were given IL-33 (0.5 µg) i.p. on 4 consecutive days. On day 5, mice were euthanized and the percentages of splenic Treg cells were analyzed by flow cytometry. (C) Bone marrow-derived macrophages from C57BL/6 wild-type or hDC-SIGN+ mice were pulsed with PBS, IVIG, or nonsialylated F241A. Total RNA was isolated and used for quantitative real-time PCR to measure IL-33 mRNA levels. The housekeeping gene Gapdh was used for normalization. (D and E) After treatment, BMMΦs were extensively washed and adoptively transferred into C57BL/6 wild-type recipient mice. On day 5 posttransfer, mice were killed and Treg cells and ST2 expression were analyzed by flow cytometry. (F) EAE was induced in C57BL/6 wild-type mice by immunization with MOG35–55 peptide. Every 2 d postinduction, mice received IL-33 (0.5 µg i.p.). Clinical scores of disease are shown. (G) The percentages of Treg cells from draining lymph nodes of EAE mice were analyzed by flow cytometry. Means ± SEM are plotted; ***P < 0.001 determined by Tukey’s post hoc test.
Because FcγRIIB up-regulation on effector macrophages has been demonstrated to require the presence of basophils (29), we investigated whether basophils also play a role in the sFc-mediated Treg-cell activation pathway. Mice were treated with PBS or IVIG (1 g/kg) and challenged with K/BxN serum. Mice were also treated with an anti-FcεRI antibody for depletion of basophils or with an isotype control (35). Whereas basophil depletion (Fig. S7A), as we have previously shown (29), disrupted the protective effect of IVIG in the K/BxN arthritis model (Fig. S7B), the ability of IVIG to expand Treg cells was not affected (Fig. S7C). This indicates that whereas IL-33 is required for Treg-cell activation, basophils, however, are not required.
sFc/F241A Activates Inducible Treg Cells via the IL-33/ST2 Axis.
To further explore the mechanism of Treg-cell expansion and activation in response to sFc and type II FcR engagement, we focused on the role of the IL-33 receptor ST2. As reported, IVIG- (1 g/kg) treated mice challenged with K/BxN serum in combination with an ST2 blocking antibody reduced the protective effect of IVIG in this serum transfer arthritis model (29) (Fig. 7A). FACS analysis of the Treg-cell numbers in these mice revealed that inhibition of IL-33 engagement by ST2 reduced the IVIG-induced expansion of Treg cells (Fig. 7B). Similarly, the therapeutic effect of F241A (0.033 g/kg) was significantly reduced by blocking the IL-33 receptor (Fig. 7C), with a concomitant reduction in Treg-cell expansion (Fig. 7D).
Fig. 7.

IVIG/F241A induces Treg-cell activation by signaling through the IL-33/ST2 axis. (A and B) C57BL/6 wild-type mice were treated with PBS or IVIG (1 g/kg) and then challenged with K/BxN serum. To block IL-33 signaling, mice received an anti-ST2 blocking antibody (100 µg i.v.) or isotype control. Clinical scores of rheumatoid arthritis are shown (A). On day 5 post disease induction, mice were killed and splenic Treg cells were analyzed by flow cytometry (B). (C) EAE was induced in C57BL/6 wild-type mice by immunization with MOG35–55 peptide. Mice were treated i.v. with PBS or nonsialylated F241A (0.033 g/kg) four times, on days 5, 10, 15, and 20. Each injection was preceded by an i.v. injection of an anti-ST2 blocking antibody. Shown are clinical scores of EAE. (D) EAE mice were killed and cells were isolated from draining lymph nodes. Treg cells and their Foxp3 and ST2 expression was analyzed by flow cytometry. Means ± SEM are plotted; *P < 0.05; ***P < 0.001 determined by Tukey’s post hoc test.
Finally, to determine the generality of our observations on the effect of sFc on T cell-mediated diseases, we used the experimental colitis mouse model and treated these mice weekly, starting 4 wk post T-cell transfer, with IVIG (1 g/kg) or PBS control until the end of the experiment. Body weight loss was used as a measure of disease severity, which showed that IVIG treatment mediated protection (Fig. S8 A, C, and D), and was again accompanied by a significant enrichment of Treg cells (Fig. S8B), whereas CD4+ effector T-cell levels were comparable in PBS- and IVIG-treated groups. Because only CD4+CD25− T cells had been adoptively transferred to induce acute colitis, the IVIG-expanded Treg cells observed in these mice originated from peripheral CD4+ T cells and are thus consistent with iTreg cells.
Discussion
Antibodies mediate either proinflammatory or antiinflammatory effector functions through the differential engagement of type I or II Fc receptors, respectively. A complex, N-linked glycan attached to the CH2 domain of IgG Fc regulates the interaction with these classes of FcRs by modulating the structure of the Fc to alternate between type I and type II binding conformations (8, 11, 20). The presence of an α2,6-linked sialic acid in this Fc-associated glycan results in reduced affinity for type I FcRs and enhanced binding to type II FcRs, such as DC-SIGN and CD23 (8, 11, 15). This switch in receptor specificity coincides with a switch in effector function in vivo, as sialylated IgG suppresses inflammation in mouse models of autoimmunity. Recent structural data support an earlier computational model that sialylation increases the conformational flexibility of the CH2 domain, sampling conformations compatible with type II FcR binding (8, 20, 36).
Here we present functional data demonstrating that specific interactions between the Fc backbone and the Fc glycan influence the effector properties of sialylated IgG. We previously reported that the antiinflammatory activity of sial Fc depends strictly on the α2,6-linkage of sialic acid because only α2,6-sial Fc, but not α2,3-sial Fc, binds to DC-SIGN and suppresses autoantibody-induced arthritic inflammation (6). How can different sialic acid linkages on the Fc glycan influence Fc structure? We previously hypothesized that the proximity of the sialic acid sugar residue to the protein backbone may determine how sialic acid interacts with specific amino acid side chains on the Fc. Indeed, molecular modeling suggested that only α2,6-linked sialic acid could fit into a groove formed by Glu318 and Lys340 at the Cγ2–Cγ3 interface. To determine whether this groove plays a role in the antiinflammatory activity of α2,6-sial Fc, we characterized the immunosuppressive functions of sial Fc bearing an E318N point mutation. Interestingly, the E318N α2,6-sial Fc fails to initiate antiinflammatory pathways associated with WT α2,6-sial Fc, such as induction of IL-33 expression or protection from arthritic inflammation by adoptive transfer of stimulated DC-SIGN+ BMMΦs to K/BxN serum-challenged mice. Thus, as with the α2,3-linkage of sialic acid, we propose that the E318N mutation abolished the interaction between α2,6-linked sialic acid and the Fc backbone necessary for the Fc to adopt its closed, antiinflammatory state.
Fc structures typically resolve the α1,3-arms of the Fc glycan within the internal cavity formed by the CH2 domains. By occupying this cavity, the α1,3-arms stabilize the CH2 domains at a distance apart in the open conformation to facilitate binding to type I FcRs. In order for our proposed model to be correct, the sialylated α1,3-arms have to move out of this cavity toward the CH2–CH3 interface so that the terminal sialic acid may contact the E318/K340 groove. Thus, by repositioning the sialylated α1,3-arms to the CH2–CH3 interface, the CH2 domains may draw closer together to fill the now unoccupied cavity and form the closed conformation. Unlike the α1,6-arm of the Fc glycan, which forms multiple noncovalent interactions with the Fc backbone, the α1,3-arm forms only one known contact in the absence of sialylation—the aromatic side chain of F241. However, in the crystal structure of sial Fc, the only amino acid side chain that contacts the Fc glycan to show a significant change in orientation is the ring structure of F241. We predict that the observed 90° rotation of F241 abrogates the hydrophobic stacking interaction it normally forms with the carbohydrate. We believe this to be structurally important, because the disruption of this stabilizing interaction should impart greater degrees of freedom, or mobility, to the sialylated α1,3-arms of the Fc glycan. We propose that with this greater mobility, the sialylated α1,3-arms will sample the space outside of the internal cavity with greater frequency to encounter the E318/K340 pocket at the CH2–CH3 interface. Consistent with the crucial role of F241 in the structure of α2,6-sial Fc, we found that an F241A mutation that specifically disrupts this protein–sugar contact point recapitulated the antiinflammatory activity of sial Fc independent of sialylation. Both α2,6-sial and -asial F241A Fc bound to DC-SIGN, induced IL-33 expression, and transferred antiinflammatory activity with stimulated DC-SIGN+ BMMΦs to K/BxN serum-challenged mice, recapitulating the antiinflammatory activity of IVIG and sialylated IgG (29). Recently, reports have been published that question the essential role of Fc sialylation for modulating immune responses (37–39). Our data, and that of several other groups, have confirmed the antiinflammatory role of sialylated Fc in multiple models of antibody-mediated inflammation (9, 12–14, 16, 40). These discrepancies are likely the result of nonlinear dosing of IVIG in selective models used in those studies and thus not reflective of the physiologically relevant conditions in which IVIG is used.
We have identified that sialylated Fc, as well as F241A, specifically stimulated Treg-cell expansion and was sufficient to suppress T cell-mediated immune responses in models of EAE and experimental colitis by selective engagement of the type II Fc receptor SIGN-R1 or its human ortholog DC-SIGN. Furthermore, we could identify IL-33 as a mediator of this pathway. IL-33, induced in response to type II FcR engagement by IVIG, sialylated Fc, or F241A, acts pleiotropically, as summarized in Fig. S9. It can mediate IL-4 secretion by basophils to polarize macrophages to an M2 phenotype and induce inhibitory FcγRIIB expression, a pathway that dominates in antibody-mediated autoimmune inflammation, or it can act directly on Treg cells to mediate their activation and expansion. We could demonstrate that Treg cells can become activated by treatment with sialylated Fc and subsequent signaling through the IL-33/ST2 axis. We cannot exclude the possibility that the IL-33–dependent Treg-cell activation is mainly mediated indirectly via dendritic cells, as previously described by Matta and coworkers (32). However, our data do not support a direct interaction of IVIG with any T-cell subset, as has been proposed (22), nor could we observe any evidence in support of IVIG providing “Tregitopes” (41–44).
Previous studies have established the connection between IL-33 and an amelioration of T cell-mediated inflammation in different mouse models that were always accompanied by an enrichment of Treg cells (31, 45–48). Our studies demonstrate that IVIG, through the presence of sialylated Fc interacting with type II FcRs, provides a source of IL-33 that can induce Treg-cell activation and expansion. Consistent with these observations, serum levels of IL-33 have been shown to be elevated upon IVIG administration in human autoimmune patients (19), thus making it a potent inducer of various antiinflammatory responses. When antibodies were used to block ST2 that prevented IL-33 signaling, we could observe that this treatment significantly compromised the protective effect of IVIG/F241A in both a serum transfer arthritis model as well as in an EAE model.
It is becoming increasingly clear that far from being a “constant” domain, the Fc region of antibodies exhibits heterogeneous structures and functions. This current study advances the view that the conformational diversity of the Fc fragment serves as a general strategy for antibodies to shift receptor specificity to effect different immunological outcomes. IgG Fc dynamics are fine-tuned by protein–glycan interactions, which are, in turn, regulated by the sugar composition of the Fc glycan. We find that a model of increased Fc glycan mobility accounts for the biophysical and functional properties associated with antiinflammatory activity of sialylated IgG. Based on these structural and mechanistic observations, we have developed a surrogate for sialylated IgG, F241A, which offers the benefit of greater potency and uniformity than IVIG and is a promising candidate for clinical development for both autoantibody- and T cell-mediated inflammatory diseases.
Materials and Methods
Mice.
Six- to 10-wk-old sex- and age-matched C57BL/6, SIGN-R1−/−, and SIGN-R1−/−hDC-SIGN+ mice were used for all experiments in compliance with federal laws and institutional guidelines approved by The Rockefeller University. SIGN-R1−/− mice were bred to CD11c-DC-SIGN+ transgenic mice to generate SIGN-R1−/− hDC-SIGN+ mice. KRN T-cell receptor transgenic mice were on a C57BL/6 background and bred to nonobese diabetic (NOD) mice to create K/BxN mice (49). K/BxN serum was prepared by collecting blood samples from K/BxN mice. The serum was separated from blood and pooled and frozen into aliquots for further use. To induce serum transfer arthritis, 200 µL of pooled K/BxN serum was injected intraperitoneally. Severity of arthritis was scored by clinical examination of the paws. The addition of all four paw indices reflected the severity of disease: 0, unaffected; 1, swelling of one joint; 2, swelling of more than one joint; 3, severe swelling of the entire paw. In all experiments, groups of four or five mice were used, and means and SEM are plotted in graphs.
Reagents and Treatments.
IVIG (Octagam; Octapharma) and F241A (Merck) were used at concentrations indicated in the text and figure legends. IVIG-Fc and IVIG-F(ab′)2 preparations were prepared by papain digestion of IVIG for 2 h at 37 °C. Desialylation of IVIG and F241A was performed by neuraminidase treatment. One hundred milligrams of antibody preparation was incubated with 700 U neuraminidase (New England BioLabs) for 20 h at 37 °C. Antibodies were purified by protein-G affinity purification and dialyzed against PBS before injection. Sialic acid contents of all antibody preparations were verified by Sambucus nigra (SNA) lectin blotting using SNA-biotin (Vector Laboratories). All IVIG and F241A preparations were administered intravenously.
Basophils were depleted by daily i.p. injection with 10 µg anti-FcεRI (MAR-1; eBioscience) or hamster IgG isotype control (Sigma) for 4 consecutive days.
Treg cells were depleted by i.p. administration of 400 µg anti-CD25 (50) (PC61; Bio X Cell) 3 d before EAE induction as well as 1 d after each IVIG/F241A injection until the end of the experiment.
Blocking of the IL-33 receptor ST2 was achieved by i.v. injections of 100 µg anti-T1/ST2 (DJ8; MD Biosciences) on day 0 as well as every fifth day post EAE induction.
For cytokine treatment, mice received a single i.v. injection of 2.5 µg IL-4ic (PeproTech) or 0.5 µg IL-33 i.p. on 4 consecutive days or every 2 d in EAE experiments.
IL-6 serum levels were measured by an in vivo cytokine capture assay as described (51). IL-33 in cell-culture supernatants was detected and measured by ELISA as suggested by the manufacturer (eBioscience).
Flow Cytometry.
To analyze lymphocytes and bone marrow cells, single-cell suspensions were prepared from spleens, lymph nodes, and bone marrow. After red blood cell lysis, cells were stained with the respective antibodies and analyzed using a FACSCalibur (BD Biosciences). The antibodies used for murine cell stainings were anti-CD4 (GK1.5), anti-Foxp3 (FJK-16s), and anti–IFN-γ (XMG1.2) from eBioscience, anti-CD25 (PC61), anti–IL-17A (TC11-18H10.1), anti-CD209 (9E9A8), anti-Helios (22F6), anti-CD11b (M1/70), and anti-F4/80 (BM8) from BioLegend, and anti-T1/ST2 (DJ8) from MD Biosciences.
Differentiation of Bone Marrow-Derived Macrophages and Transfers.
Bone marrow cells were isolated from femurs and tibias, and cultured in 10-cm plates in DMEM supplemented with 20% (vol/vol) FBS, 2% (vol/vol) penicillin/streptomycin (Invitrogen), 1% l-glutamine 200 mM (Invitrogen), 0.1% β-mercaptoethanol, and 40 ng/mL M-CSF (PeproTech) for 5–7 d at 37 °C. Flow cytometry was used to analyze bone marrow-derived macrophages in the cell cultures (>90% CD11b+F4/80+ cells). Cells were recovered from plates, washed, and seeded in fresh tissue-culture plates, and subsequently pulsed with PBS, IVIG (15 mg/mL), or F241A (80 µg/mL) for 3 h at 37 °C. Cells were then extensively washed and 1 × 106 macrophages were administered i.v. to recipient mice. One hour posttransfer, mice were challenged with K/BxN sera.
Quantitative Real-Time PCR.
Total RNA was isolated from bone marrow-derived macrophages using the RNeasy Mini Kit (Qiagen) and reverse-transcribed with SuperScript III Reverse Transcriptase (Invitrogen). Quantitative PCR was performed to measure IL-33 mRNA levels using a C1000 Touch Thermal Cycler (Bio-Rad) with primer sets for IL-33 [5′-TCACTGCAGGAAAGTACAGCA-3′ (forward) and 5′-AGTAGCACCTGGTCTTGCTC-3′ (reverse)] and Gapdh [5′-ACAGTCCATGCCATCACTGCC-3′ (forward) and 5′-GCCTGCTTCACCACCTTCTTG-3′ (reverse)]. Gene expression levels were calculated by normalization to Gapdh mRNA levels.
Experimental Autoimmune Encephalomyelitis.
Six- to 8-wk-old C57BL/6, SIGN-R1−/−, or SIGN-R1−/− hDC-SIGN+ mice were immunized s.c. with 200 µL of an emulsion consisting of 200 µg MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK; AnaSpec) emulsified in complete Freund’s adjuvant (Difco Laboratories). On days 0 and 2, mice received 200 µg of pertussis toxin (List Biological) intraperitoneally. Development of disease was monitored daily according to the following criteria: 0, no clinical signs; 0.5, partially limp tail; 1, paralyzed tail; 2, loss of coordinated movement, and hind limb paresis; 2.5, one hind limb paralyzed; 3, both hind limbs paralyzed; 3.5, hind limbs paralyzed and hunched back; 4, severely hunched back and weakness in forelimbs; 4.5, forelimbs paralyzed; 5, moribund (52).
In Vitro Treg-Cell Differentiation.
Single-cell suspensions were prepared from spleens and lymph nodes of naïve C57BL/6 wild-type mice. Naïve CD4+ T cells were isolated by magnetic cell separation (Miltenyi Biotec) and cultured for 3 d in RPMI supplemented with 10% (vol/vol) FBS, 2% (vol/vol) penicillin/streptomycin (Invitrogen), 1% l-glutamine 200 mM (Invitrogen), 0.1% β-mercaptoethanol, and anti-CD3 (17A2; eBioscience) and anti-CD28 (37.51; eBioscience) antibodies in 24-well cell-culture plates that were coated with anti-hamster IgG (MP Bio). For Treg-cell differentiation, TGF-β (2.5 ng/mL; PeproTech) was added. IL-33 (1 ng/mL; BioLegend) or IL-23 (20 ng/mL; PeproTech) was added to the cell cultures as indicated in the figure legends. On day 3, cells were recovered and Treg cells (CD4+Foxp3+) were analyzed by FACS.
T-Cell Transfer Colitis.
For T-cell transfer colitis, 5 × 105 naïve CD4+CD45RBhiCD25− T cells from C57BL/6 wild-type animals were sorted and injected i.p. into C57BL/6 Rag1−/− recipient mice. Body weight loss was measured twice a week and used as a measure of disease severity.
Supplementary Material
Acknowledgments
We gratefully acknowledge the technical contribution of Patrick Smith and U. Kaufmann. The research was funded in part by Merck Sharp & Dohme and the Deutsche Forschungsgemeinschaft (FI 2035/1-1 to B.M.F.). Support was also provided by generous donations from Eileen Greenland, the Ludwig Fellowship Program, and The Rockefeller University.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505292112/-/DCSupplemental.
References
- 1.Ballow M. Primary immunodeficiency disorders: Antibody deficiency. J Allergy Clin Immunol. 2002;109(4):581–591. doi: 10.1067/mai.2002.122466. [DOI] [PubMed] [Google Scholar]
- 2.Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: How does IgG modulate the immune system? Nat Rev Immunol. 2013;13(3):176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- 3.Anthony RM, Ravetch JV. A novel role for the IgG Fc glycan: The anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol. 2010;30(Suppl 1):S9–S14. doi: 10.1007/s10875-010-9405-6. [DOI] [PubMed] [Google Scholar]
- 4.Nimmerjahn F, Ravetch JV. Antibody-mediated modulation of immune responses. Immunol Rev. 2010;236(1):265–275. doi: 10.1111/j.1600-065X.2010.00910.x. [DOI] [PubMed] [Google Scholar]
- 5.Kaneko Y, Nimmerjahn F, Madaio MP, Ravetch JV. Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J Exp Med. 2006;203(3):789–797. doi: 10.1084/jem.20051900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci USA. 2008;105(50):19571–19578. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwab I, Biburger M, Krönke G, Schett G, Nimmerjahn F. IVIg-mediated amelioration of ITP in mice is dependent on sialic acid and SIGNR1. Eur J Immunol. 2012;42(4):826–830. doi: 10.1002/eji.201142260. [DOI] [PubMed] [Google Scholar]
- 8.Pincetic A, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. 2014;15(8):707–716. doi: 10.1038/ni.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313(5787):670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 10.Scallon BJ, Tam SH, McCarthy SG, Cai AN, Raju TS. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol Immunol. 2007;44(7):1524–1534. doi: 10.1016/j.molimm.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General mechanism for modulating immunoglobulin effector function. Proc Natl Acad Sci USA. 2013;110(24):9868–9872. doi: 10.1073/pnas.1307864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anthony RM, et al. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320(5874):373–376. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwab I, et al. Broad requirement for terminal sialic acid residues and FcγRIIB for the preventive and therapeutic activity of intravenous immunoglobulins in vivo. Eur J Immunol. 2014;44(5):1444–1453. doi: 10.1002/eji.201344230. [DOI] [PubMed] [Google Scholar]
- 14.Washburn N, et al. 2015. Controlled tetra-Fc sialylation of IVIg results in a drug candidate with consistent enhanced anti-inflammatory activity. Proc Natl Acad Sci USA 112(11):E1297–E1306.
- 15.Anthony RM, Wermeling F, Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci. 2012;1253:170–180. doi: 10.1111/j.1749-6632.2011.06305.x. [DOI] [PubMed] [Google Scholar]
- 16.Böhm S, Schwab I, Lux A, Nimmerjahn F. The role of sialic acid as a modulator of the anti-inflammatory activity of IgG. Semin Immunopathol. 2012;34(3):443–453. doi: 10.1007/s00281-012-0308-x. [DOI] [PubMed] [Google Scholar]
- 17.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291(5503):484–486. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 18.Tackenberg B, et al. Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci USA. 2009;106(12):4788–4792. doi: 10.1073/pnas.0807319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma M, et al. Intravenous immunoglobulin-induced IL-33 is insufficient to mediate basophil expansion in autoimmune patients. Sci Rep. 2014;4:5672. doi: 10.1038/srep05672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed AA, et al. Structural characterization of anti-inflammatory immunoglobulin G Fc proteins. J Mol Biol. 2014;426(18):3166–3179. doi: 10.1016/j.jmb.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-Å crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406(6793):267–273. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 22.Ephrem A, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: A critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111(2):715–722. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- 23.Massoud AH, et al. Intravenous immunoglobulin attenuates airway inflammation through induction of forkhead box protein 3–positive regulatory T cells. J Allergy Clin Immunol. 2012;129(6):1656–1665.e3. doi: 10.1016/j.jaci.2012.02.050. [DOI] [PubMed] [Google Scholar]
- 24.Massoud AH, et al. 2014. Dendritic cell immunoreceptor: A novel receptor for intravenous immunoglobulin mediates induction of regulatory T cells. J Allergy Clin Immunol 133(3):853–863.e5.
- 25.Korn T, Oukka M, Kuchroo V, Bettelli E. Th17 cells: Effector T cells with inflammatory properties. Semin Immunol. 2007;19(6):362–371. doi: 10.1016/j.smim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jäger A, Kuchroo VK. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand J Immunol. 2010;72(3):173–184. doi: 10.1111/j.1365-3083.2010.02432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bayry J, Mouthon L, Kaveri SV. Intravenous immunoglobulin expands regulatory T cells in autoimmune rheumatic disease. J Rheumatol. 2012;39(2):450–451. doi: 10.3899/jrheum.111123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kessel A, et al. Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J Immunol. 2007;179(8):5571–5575. doi: 10.4049/jimmunol.179.8.5571. [DOI] [PubMed] [Google Scholar]
- 29.Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475(7354):110–113. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thornton AM, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184(7):3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turnquist HR, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol. 2011;187(9):4598–4610. doi: 10.4049/jimmunol.1100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matta BM, et al. IL-33 is an unconventional alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. 2014;193(8):4010–4020. doi: 10.4049/jimmunol.1400481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiering C, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513(7519):564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Izcue A, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28(4):559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol. 2008;9(3):310–318. doi: 10.1038/ni1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu X, et al. Engineering hydrophobic protein-carbohydrate interactions to fine-tune monoclonal antibodies. J Am Chem Soc. 2013;135(26):9723–9732. doi: 10.1021/ja4014375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Käsermann F, et al. Analysis and functional consequences of increased Fab-sialylation of intravenous immunoglobulin (IVIG) after lectin fractionation. PLoS ONE. 2012;7(6):e37243. doi: 10.1371/journal.pone.0037243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leontyev D, et al. Sialylation-independent mechanism involved in the amelioration of murine immune thrombocytopenia using intravenous gammaglobulin. Transfusion. 2012;52(8):1799–1805. doi: 10.1111/j.1537-2995.2011.03517.x. [DOI] [PubMed] [Google Scholar]
- 39.Campbell IK, et al. Therapeutic effect of IVIG on inflammatory arthritis in mice is dependent on the Fc portion and independent of sialylation or basophils. J Immunol. 2014;192(11):5031–5038. doi: 10.4049/jimmunol.1301611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hess C, et al. T cell-independent B cell activation induces immunosuppressive sialylated IgG antibodies. J Clin Invest. 2013;123(9):3788–3796. doi: 10.1172/JCI65938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Groot AS, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes.”. Blood. 2008;112(8):3303–3311. doi: 10.1182/blood-2008-02-138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elyaman W, Khoury SJ, Scott DW, De Groot AS. Potential application of Tregitopes as immunomodulating agents in multiple sclerosis. Neurol Res Int. 2011;2011:256460. doi: 10.1155/2011/256460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cousens LP, et al. In vitro and in vivo studies of IgG-derived Treg epitopes (Tregitopes): A promising new tool for tolerance induction and treatment of autoimmunity. J Clin Immunol. 2013;33(Suppl 1):S43–S49. doi: 10.1007/s10875-012-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cousens LP, et al. Tregitope update: Mechanism of action parallels IVIg. Autoimmun Rev. 2013;12(3):436–443. doi: 10.1016/j.autrev.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 45.Brunner SM, et al. Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int. 2011;24(10):1027–1039. doi: 10.1111/j.1432-2277.2011.01306.x. [DOI] [PubMed] [Google Scholar]
- 46.Wasserman A, et al. Interleukin-33 augments Treg cell levels: A flaw mechanism in atherosclerosis. Isr Med Assoc J. 2012;14(10):620–623. [PubMed] [Google Scholar]
- 47.Jiang HR, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur J Immunol. 2012;42(7):1804–1814. doi: 10.1002/eji.201141947. [DOI] [PubMed] [Google Scholar]
- 48.Barbour M, et al. IL-33 attenuates the development of experimental autoimmune uveitis. Eur J Immunol. 2014;44(11):3320–3329. doi: 10.1002/eji.201444671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korganow AS, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10(4):451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 50.Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur J Immunol. 2010;40(3):780–786. doi: 10.1002/eji.200939613. [DOI] [PubMed] [Google Scholar]
- 51.Finkelman F, Morris S, Orekhova T, Sehy D. 2003. The in vivo cytokine capture assay for measurement of cytokine production in the mouse. Curr Protoc Immunol 54(6.28):6.28.1–6.28.10. [DOI] [PubMed]
- 52.Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1(4):1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.