

Significance
Because of their unique ability to self-renew and generate all cell lineages of an organism (pluripotency), embryonic stem cells represent a versatile model for developmental biology, and a promising avenue for regenerative medicine. Understanding the molecular mechanisms regulating self-renewal and pluripotency provides a productive path to effectively use embryonic stem cells, and to improve current methods for induction/differentiation of pluripotent stem cells and direct somatic cell reprogramming. This study provides novel insights into transcriptional regulation of the stem cell state by characterizing interactions between key transcription factors SOX2 and OCT4, and a recently identified, multifunctional, stem cell coactivator—the xeroderma pigmentosum, complementation group C DNA repair complex—to control pluripotency gene-expression networks.
Keywords: transcription, pluripotency, protein–protein interactions, ChIP-seq, RNA-seq
Abstract
The embryonic stem cell (ESC) state is transcriptionally controlled by OCT4, SOX2, and NANOG with cofactors, chromatin regulators, noncoding RNAs, and other effectors of signaling pathways. Uncovering components of these regulatory circuits and their interplay provides the knowledge base to deploy ESCs and induced pluripotent stem cells. We recently identified the DNA-repair complex xeroderma pigmentosum C (XPC)-RAD23B-CETN2 as a stem cell coactivator (SCC) required for OCT4/SOX2 transcriptional activation. Here we investigate the role of SCC genome-wide in murine ESCs by mapping regions bound by RAD23B and analyzing transcriptional profiles of SCC-depleted ESCs. We establish OCT4 and SOX2 as the primary transcription factors recruiting SCC to regulatory regions of pluripotency genes and identify the XPC subunit as essential for interaction with the two proteins. The present study reveals new mechanistic and functional aspects of SCC transcriptional activity, and thus underscores the diversified functions of this regulatory complex.
Embryonic stem cells (ESCs) can be maintained in culture to retain their defining properties of self-renewal (propagation without loss of cellular identity) and pluripotency (ability to generate all embryonic lineages upon appropriate developmental stimuli). In the case of murine ESCs (mESCs), a minimal culture media supplemented with serum and LIF (leukemia inhibitory factor) can perpetuate the pluripotency state (1). Such culture conditions, among others, provide the external cues to counteract differentiation programs hardwired in ESCs [e.g., the autocrine FGF4 signaling (2)], by fueling an intricate network of transcription factors (TFs) at the core of which stands the autoregulated and self-sustained OCT4 (POU class 5 homeobox 1), SOX2 (sex determining region Y-box2), and NANOG (Nanog homeobox) circuit (3, 4). These “core” pluripotency factors orchestrate ESC transcriptional programs in conjunction with “ancillary” TFs (e.g., ESRRB, KLF2, KLF4, SALL4, TBX3, TFCP2L1), various cofactors (5), noncoding RNAs (6), histone modifiers, and chromatin remodelers (7), ultimately conveying regulatory inputs to specialized basal transcriptional machineries (8–10). Identifying components of these complex regulatory circuitries and their interplay has an obvious impact on both developmental biology and regenerative medicine by instructing the efficacious and safe use of ESCs, as well as the current methods for induction and differentiation of pluripotent stem cells and direct somatic cell reprogramming.
Unbiased approaches, such as transcriptional, epigenetic, and proteomic profiling, siRNA screenings, large-scale proteomics, and genome-wide TF occupancy studies (ChIP-seq), have been used by many investigators to uncover new players in the maintenance of pluripotency. Although effective, such experimental strategies often fail to establish a direct role for identified elements in the transcriptional regulation of pluripotency. To circumvent this limitation, we recently established an unbiased in vitro transcription-biochemical complementation assay and used it to search for the minimal components required for OCT4- and SOX2-mediated transcriptional activation in murine and human ESCs. We uncovered three distinct activities, one of which has been identified as the xerodoma pigmentosum, complementation group C (XPC)-RAD23B-CETN2 trimeric complex, referred to as the stem cell coactivator (SCC) in our studies (11, 12).
SCC complex was previously well established as a DNA damage sensor in the global genome nucleotide excision repair pathway (NER) (13, 14). Within SCC, the XPC subunit scans the genome and recognizes helix-distorting lesions (e.g., UV-induced pyrimidine dimers) through its DNA-binding domain. CETN2 (centrin, EF-hand protein, 2) and RAD23B (RAD23 homolog B) stabilize the XPC protein and potentiate its interaction with damaged DNA (15–17). XPC also initiates the NER repair cascade through direct recruitment of XPA and general transcription factor IIH (TFIIH), which mediate damage verification and DNA unwinding, respectively (18, 19). Subsequent DNA damage excision, DNA synthesis through the gap, and ligation complete the repair process (14).
Concomitantly to our study that identified SCC as an OCT4/SOX2 ESC coactivator (11), SCC was shown to assemble onto promoters of activated genes in HeLa cells, along with the entire NER machinery, to facilitate DNA demethylation and gene looping (20, 21). In ESCs, however, the SCC coactivator activity we uncovered is uncoupled from DNA repair, and no other NER factors emerged from our biochemical assays, suggesting a certain degree of specificity for OCT4/SOX2 transcription. In fact, a preliminary analysis of RAD23B genomic occupancy highlighted a striking overlap with OCT4/SOX2 binding sites, and depletion of SCC by RNA interference showed that it contributes in vivo to maintaining the mESC state and to generating induced pluripotent stem cells from somatic cells (11). Nevertheless, several key issues remained unaddressed in our initial study: is SCC working as a coactivator in ESCs for TFs other than OCT4 and SOX2? What is the transcriptional network orchestrated by SCC in ESCs? Which transcriptional changes occur globally upon SCC depletion? Are these resulting from SCC transcriptional activity or deriving from a DNA damage response? How is SCC recruited to chromatin? What are the biochemical bases for SCC interaction with OCT4, SOX2, and possibly other TFs? To address some of these issues, we delved into a more thorough analysis of SCC chromatin occupancy in ESCs, combined with whole transcriptome profiling of SCC-depleted ESCs. These genome-wide scale studies revealed that SCC is indeed recruited to gene regulatory regions mostly through OCT4 and SOX2. Transcriptional perturbations following SCC depletion resemble those seen upon OCT4 and SOX2 knockdown, rather than reflect a p53-mediated DNA damage response. We also demonstrate that, within SCC, XPC is the major subunit directly interacting with OCT4 and SOX2, and truncation experiments indicate that nonoverlapping XPC domains are responsible for interaction with the core TFs.
At a more fundamental level, our studies also underscore the importance of multifunctional complexes serving as key regulatory molecular machines that cells have evolved to coordinate different biological processes. By investigating mechanistic and functional aspects of the interaction between OCT4, SOX2, and the XPC-RAD23B-CETN2 DNA repair complex, our findings provide new insights into how this multisubunit factor executes and integrates functions beyond DNA repair.
Results
RAD23B Targets Gene Promoters and Distal Regulatory Regions in mESCs.
To more thoroughly investigate how SCC works as a transcriptional coactivator responsive to OCT4/SOX2 and possibly other sequence-specific TFs in mESCs, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) using murine D3 ESCs. Because none of the commercially available antibodies gave reliable ChIP signals, we raised and affinity-purified polyclonal antibodies against the RAD23B and XPC subunits of SCC. Although antibodies against both proteins were specific and efficient at immunoprecipitating SCC complex (Fig. S1 A–C), only the anti-RAD23B antibody showed robust and reproducible enrichment in ChIP experiments (Fig. S1 D and E; see also Fig. 2C, Upper). We therefore used RAD23B-specific antibodies to immunoprecipitate the holo-SCC complex from D3 cells. Two RAD23B ChIP-seq biological replicates identified 59,768 and 42,364 RAD23B binding sites in the mESC genome, respectively. Of these, 40,671 of the RAD23B binding sites were common between the two independent experiments (R = 0.86), indicative of high experimental reproducibility (Fig. S1 D and E). The top 50%, high-confidence RAD23B binding sites with the strongest enrichment (P < 10−5, 29,884 peaks) were selected for further analysis (see Dataset S1 for a full list). To determine where RAD23B binds with respect to genes, for each ChIP-seq peak midpoint we calculated the distance to the transcription start site (TSS) of the closest RefSeq gene. About 18% of RAD23B binding occurs within 200 bp of a TSS, a significant enrichment with respect to the control dataset (preimmune IgGs) (Fig. 1A). This genome-wide distribution is consistent with our previous biochemical data suggesting that SCC facilitates transcriptional initiation by OCT4 and SOX2 (11). However, promoters (±500 bp from TSS) only account for one-fourth (24%) of all RAD23B binding sites, whereas proximal (500 bp < TSS < 5 kb) and distal (5 kb < TSS < 50 kb) gene regions contain the majority of RAD23B target sites (Fig. 1B). To probe whether these other RAD23B binding regions are, in fact, distal regulatory elements, we compared them to prototypic enhancer features mapped by others in mESCs such as H3K4me1 and H3K27ac chromatin marks, as well as p300 coactivator binding sites (22). We observed a significant (twofold and higher) overlap between RAD23B proximal and distal binding sites at all of the tested enhancer features, irrespective of whether they mark active (H3K27ac) or poised (H3K4me1, p300) enhancers (Fig. S2 A and D). In line with this observation, we found both active and inactive genes targeted by RAD23B (i.e., genes with at least one RAD23B ChIP-seq peak within 5 kb of their TSS) (Fig. 1C).
Fig. 2.

RAD23B and OCT4/SOX2 extensively colocalize in mESCs. (A) Percentage of RAD23B ChIP-seq peaks that do (O/S) or do not (NA) overlap with OCT4/SOX2 (24) binding sites in D3 mESCs. RAD23B peaks are split in two groups depending on their enrichment over IgG control (Partek P value < 10−9 and 10−9 < P < 10−5). The black pattern identifies the location of peaks with respect to gene TSS. (B) Sequence analysis of OCT4/SOX2-overlapping RAD23B binding sites (peak midpoint ±125 bp) by MEME and TOMTOM motif discovery and comparison tools. (C) Co-occupancy of RAD23B, OCT4, and SOX2 proteins at Nanog (Left) and Oct4/Pou5f1 (Right) loci evaluated by ChIP-qPCR (Upper) (11), and ChIP-seq (Lower). IGV-computed ChIP-seq tracks (71, 72) are plotted as (number of reads) × [1,000,000/(total read count)]; data range for each track is specified on the y axis. Genomic coordinates are in kilobases. Normal IgG (gray) controls for specificity. See Fig. S3 for additional loci. E, distal enhancer; I, intron; TSS, proximal promoter.
Fig. 1.

Genome-wide distribution of RAD23B binding. (A) Distribution of RAD23B and control (IgG) ChIP-seq peaks around RefSeq gene TSS in D3 mESCs (annotated list of RAD23B binding sites in Dataset S1). Distance to the closest TSS is binned in 200-bp intervals. The two samples show a significantly different distribution up to 800 bp away from the TSS (***P < 10−14, two-sample test for equality of proportions with continuity correction). (B) Percent distribution of RAD23B binding between promoters (0–500 bp from TSS), proximal (500–5,000 bp from TSS) and distal (5–50 kb from TSS) gene regions. Fifteen percent of binding events are more than 50 kb away from any annotated TSS (gray bar). (C) Percent of active (ON, red shades) and inactive (OFF, gray shades) genes in D3 mESCs (8) with a RAD23B binding site within 5 kb from their TSS. (D) GO of active (ON, Upper) and inactive (OFF, Lower) genes with a RAD23B binding site within 5 kb from their TSS compared with all ON/OFF genes in D3 mESCs. In parentheses is the number of target genes in each GO term. Bonferroni P value < 10−5 for all categories (complete table in Dataset S1; Polycomb binding in Fig. S2B). (E) Overlap between RAD23B binding sites and ChIP-seq peaks of major transcription factors in mESCs (24, 70). Plotted is the percentage of RAD23B peaks overlapping with a given TF relative to the overlap in control (IgG) ChIP-seq peaks (dashed line: ratio to IgG = 1, background levels). In blue is the overlap when considering all TF binding sites, in gray is the overlap when considering only those TF binding sites that do not overlap with OCT4/SOX2 peaks. Additional overlap analyses in Fig. S2. Data in A–E are from one of two highly overlapping RAD23B ChIP-seq experiments in D3 mESCs (Fig. S1).
A gene ontology (GO) analysis of these putative targets revealed an overrepresentation of genes involved in transcriptional regulation among both active and inactive genes (Fig. 1D; full list in Dataset S1). There was also a specific enrichment of developmentally regulated, Polycomb-bound genes among the inactive target genes (Fig. 1D and Fig. S2B). Interestingly, RAD23B binding sites are enriched for regions co-occupied by cohesin and Mediator, but not CTCF (Fig. S2 C and D), which have been reported to favor enhancer-promoter looping and transcriptional activation in mESCs (23). Taken together, our genome-wide analyses reveal that RAD23B binds to promoter and distal gene regulatory regions of both actively transcribed and silenced genes in mESCs.
RAD23B Extensively Colocalizes with OCT4/SOX2 Binding Sites in mESCs.
As part of the DNA-damage recognition and repair machinery, the XPC subunit of SCC has been reported to bind DNA directly (13), although the interaction is neither sequence-specific nor required for SCC transcriptional activity (11). We therefore hypothesized that the observed RAD23B recruitment to regulatory regions was more likely to depend on protein–protein-mediated transactions involving other sequence-specific TFs. Compared with a series of mESC enriched TFs, RAD23B binding sites showed the strongest colocalization with OCT4/SOX2 (>sevenfold enrichment over control), followed by NANOG (>fivefold), and STAT3, ESRRB, TFCP2L1, and KLF4 (two- to threefold) (Fig. 1E, blue bars). Importantly, RAD23B overlap with TFs other than OCT4 and SOX2 dropped to background levels when we subtracted from the analysis peaks located nearby OCT4/SOX2 sites (Fig. 1E, gray bars), suggesting that the observed colocalization between these TFs and RAD23B mostly depended on concurrent OCT4/SOX2 binding.
Given the strong correlation observed between OCT4 and SOX2 (O/S) binding and RAD23B enrichment at mESC regulatory regions, we further characterized their interaction with RAD23B. About 60% of the strongest (P < 10−9) RAD23B binding sites do, in fact, overlap with O/S, and for the most part the colocalization occurs away from core promoters (>500 bp from TSS) (Fig. 2A), in line with preferential binding of O/S at distal regulatory regions (24). This finding still holds true for weaker (10−9< P < 10−5) RAD23B binding sites, although the O/S overlap drops to ∼25%, indicating a direct correlation between RAD23B enrichment and O/S colocalization. De novo motif discovery within DNA sequences surrounding RAD23B peaks (±125 bp from peak midpoint) identified two prominent motifs: the top-ranking one was virtually identical to the O/S composite recognition element (P < 10−10) (Fig. 2B); a second motif showed moderate resemblance to both KLF4 (P < 10−5) and SP1 (P < 10−4) binding sites (Fig. S3A). The overlap between RAD23B and O/S binding is even more robust when superimposing their ChIP-seq tracks at specific loci such as Nanog, Pou5f1, Klf4, Sox2, Lefty2, and Stat3 (Fig. 2C, Lower, and Fig. S3), confirming the selective enrichment of RAD23B at pluripotency genes that we previously observed by ChIP-qPCR (Fig. 2C, Upper) (11).
RAD23B Recruitment Follows Activator Binding.
If SCC works mostly as a canonical O/S transcription coactivator, one would expect it not to bind DNA directly but rather rely on OCT4 or SOX2 for recruitment onto chromatin. Indeed, our in vitro biochemical studies demonstrated that SCC DNA binding activity is dispensable for transcription (11). To confirm in vivo that SCC is not preloaded onto chromatin through its DNA-binding subunit XPC, but rather recruited to gene regulatory elements by activators, we performed ChIP-quantitative PCR (qPCR) of RAD23B in D3 mESCs at selected loci upon knockdown of activators by RNA interference. For OCT4, we selected two lentiviral shRNA constructs that gave us the strongest OCT4 mRNA and protein depletion with the lowest effect on SOX2 and RAD23B levels at 24, 48, and 72 h postinfection compared with uninfected cells (Fig. 3 A and B). Under these experimental conditions, OCT4 levels remained high enough to prevent overt differentiation of mESCs into trophectoderm, although we did observe a reduction in cell proliferation at the latest time point. As expected, OCT4 binding to DNA dropped almost to background levels 72 h postinfection with shRNA constructs, with different kinetics depending on the investigated genomic locus (Oct4/Pou5f1, Nanog, or Klf4) (Fig. 3C). To ensure that the drop was not a result of intrinsic differences in chromatin properties and processing across time points, we performed ChIP on the same samples with Pol II and TBP antibodies, and checked their enrichment at the promoter of the housekeeping β-actin gene (Actb) (Fig. 3C, Bottom Right). Both Pol II and TBP signals were actually somewhat higher in OCT4-depleted cells than in uninfected ones, at both 48 and 72 h postinfection, thus controlling for any experimental bias. We next proceeded to check for SOX2 and RAD23B binding at various pluripotency loci upon OCT4 knockdown. Consistent with cooperative OCT4/SOX2 binding to their target genes (25), SOX2 recruitment was significantly reduced 72 h after OCT4 knockdown, even though SOX2 protein levels remained unchanged (Fig. 3 A and C). Interestingly, at an earlier time point (48 h postinfection), SOX2 ChIP signal on Klf4 and Nanog enhancers was equal to uninfected cells, if not higher, indicating that at these loci OCT4 depletion is initially compensated by an increase in SOX2 binding. This result is not unreasonable, given that both OCT4 and SOX2 were shown to independently bind to the O/S composite motif (26) and that single-molecule imaging indicates that SOX2 engages the target DNA first, followed by OCT4 (27). Most importantly, when we checked SCC chromatin binding in OCT4-depleted cells using RAD23B antibody, we observed that it closely followed SOX2 kinetics at all tested loci, reaching background levels 72 h post OCT4 depletion (Fig. 3C). We also performed complementary experiments knocking down SOX2 and checking OCT4 and RAD23B levels on chromatin, but failed to obtain conditions where SOX2 levels could be reduced without also affecting RAD23B and OCT4 protein levels. Nonetheless, from the OCT4 knockdown experiments we can conclude that RAD23B requires at least a prebound SOX2 to be recruited to gene regulatory regions.
Fig. 3.

RAD23B recruitment follows OCT4/SOX2 binding. (A) Immunoblotting of D3 mESCs at different time points after lentiviral-mediated knockdown (KD) of OCT4. KD3 and KD4 constructs were chosen for further analyses. Uninfected cells (–) control for knockdown efficiency. (B) Oct4, Sox2, and Rad23b transcript levels in D3 mESCs at different time points after OCT4 KD compared with uninfected (–) cells. Values are average and SD of two independent experiments, normalized to Gapdh. (C) Binding of OCT4, SOX2, and RAD23B proteins at Oct4/Pou5f1, Nanog, and Klf4 enhancers in D3 mESCs at different time points after OCT4 KD, evaluated by ChIP-qPCR. Binding of Pol II and TBP on actin promoter (Actb) controls for cell viability and chromatin quality across time points. Plotted are average and SD from the two KD constructs in B.
Impaired Stemness of SCC-Depleted mESCs.
We previously reported that depletion of SCC by RNA interference compromises transcription of some pluripotency genes, resulting both in impaired pluripotency of ES cells and defective somatic cell reprogramming (11). To confirm these earlier results obtained by RNA interference and to gain a more comprehensive view of the transcriptional program coregulated by SCC in mESCs, we generated an independent Rad23b knockout mESC line (Rad23b−/− JM8.N4). Next, we depleted Xpc by RNA interference to obtain two Rad23b ablated/Xpc shRNA-depleted cell lines (SCC KD1 and SCC KD2 JM8.N4), and compared genome-wide transcription profiles of these lines to WT mESCs by poly(A)-RNA-seq (see Material and Methods and Fig. S4 for details on the cell line generation).
RNA-seq analysis revealed that ∼15% of protein-coding genes in mESCs are either up- or down-regulated (1.5-fold or more) in Rad23b−/− and SCC KD1/KD2 cells compared with WT cells (Fig. S5A; full gene list in Dataset S2). As expected, Rad23b and both Rad23b and Xpc are among the most dramatically down-regulated genes in Rad23b−/− and SCC KD1/KD2 mESCs, respectively (Fig. S5A). To assess the impact of SCC depletion on mESC properties, we selected the genes up- and down-regulated in both SCC KD1 and SCC KD2 mESCs, averaged their expression levels, and compared them to WT cells. We then manually curated a list of genes involved in ESC maintenance (“pluripotency signature”) or differentiation (“differentiation signature”) and compared their transcript levels in WT and SCC KD mESCs (see Materials and Methods for details). SCC KD cells showed a preferential down-regulation of “pluripotency signature” genes (e.g., Tfcp2l1, Klf4, Esrrb, Nanog, Lefty1, Lefty2) and a concomitant up-regulation of “differentiation signature” genes, including several trophectoderm markers (e.g., Hand1, Cdx2, Wnt7b, Gata3, Bmp4, Ascl2) (Fig. 4A; RT-qPCR validation in Fig. S5B). This phenotype is consistent with the underlying hypothesis of SCC working as an O/S transcriptional coactivator, because knockdown of either TF results in loss of pluripotency and induction of trophectoderm differentiation (26, 28–30). Down-regulation of pluripotency genes also translated to a reduction in protein levels (Fig. 4B and Fig. S5C) and impaired clonogenic ability of SCC KD cells compared with WT cells (Fig. 4C). Interestingly, three of the most down-regulated genes (Tfcp2l1, Klf4, Lefty2) were previously reported as downstream of the LIF/STAT3 pathway (31–33). A further analysis revealed that Stat3 itself was down-regulated in both Rad23b−/− and SCC KD1 samples, but did not pass the threshold in the SCC KD2 sample, and was thus initially designated as “not changed.” Indeed, RT-qPCR confirmed Stat3 down-regulation in Rad23b−/− and both SCC KD samples (Fig. S5B), and protein analysis revealed reduced levels of STAT3 and a consequent defect in STAT3 activation, as measured by decreased phosphorylation of tyrosine 705. These data suggest that SCC KD cells are defective in LIF/STAT3 signaling, possibly because of an altered transcriptional response.
Fig. 4.
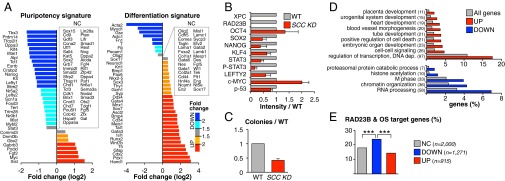
Transcription profile of SCC knockdown mESCs. (A) Comparative expression of pluripotency (Left) and differentiation (Right) markers between SCC KD and WT mESCs, evaluated by RNA-seq. Genes whose expression is not changed (NC) are listed in boxes. Plotted is the log2 ratio of SCC KD and WT RPKM counts (details on SCC KD generation in Fig. S4; global pairwise comparisons of RPKM values in Fig. S5A; qPCR validation of selected genes in Fig. S5B; full gene list in Dataset S2). (B) Protein levels of selected genes in SCC KD vs. WT mESCs, β-actin normalized. Plotted are average signal and SD of two independent Western blots quantified by ImageJ (representative blot in Fig. S5C). P-STAT3, antibody detecting active, Tyr705-phosphorylated STAT3. Analysis of p-53 targets in Fig. S5G. (C) Clonal assays in SCC KD compared with WT mESCs. Plotted is the number of AP+ colonies obtained from plating 300 cells in six-well plates (average and SD of three independent experiments). (D) GO analysis of genes up-regulated (UP) and down-regulated (DOWN) in SCC KD mESCs compared with all annotated genes. Bracketed is the number of deregulated genes belonging to each category. EASE (Expression Analysis Systematic Explorer) scores < 0.01 for all categories (complete table in Dataset S2). (E) Percentage of genes bound by RAD23B and OCT4/SOX2 within 5 kb from their TSS among genes either not changed (NC), down-regulated (DOWN), or up-regulated (UP) upon SCC KD. The number of genes in each category is specified (n). ***P < 10−3, two-sample test for equality of proportions with continuity correction. Data of SCC KD mESCs are average of two independent Xpc shRNA constructs in Rad23b−/− mESCs.
To further validate these results, we also performed an unbiased GO analysis on the list of deregulated transcripts in SCC KD mESCs (Fig. 4D; full table in Dataset S2). In accordance with the accentuated “differentiation signature” of SCC KD cells, among the up-regulated genes we observed a significant overrepresentation of categories related to tissue development and morphogenesis (placenta, urogenital system, heart, blood vessels, and so forth). The same GO analysis performed on down-regulated genes was less informative, with overrepresentation of gene categories like RNA processing, chromatin organization, and M-phase regulation.
Interestingly, the GO analysis also highlighted an overrepresentation of genes involved in the positive regulation of cell death among the up-regulated transcripts (Fig. 4D). This finding agrees with our observation that SCC KD cells exhibit reduced cell growth (Fig. S5F). Knowing that SCC KD mESCs are defective for DNA repair of UV-induced damage (34), we became concerned that some of the observed phenotypes (reduced pluripotency, increased differentiation, and cell death) could result from a DNA damage response mediated by the tumor protein p53 rather than a direct transcriptional defect. Indeed, several reports suggest that p53 can suppress pluripotency and self-renewal in ESCs and activate differentiation programs (reviewed in ref. 35). To control for potential complicating p53 effects in our analysis, we checked p53 RNA and protein levels, as well as p53 activation and induction of p53-response genes (p21, Mdm2, Gadd45α) in SCC KD mESCs (Fig. 4B and Fig. S5G). We did not detect any elevated p53 activation in SCC KD cells relative to WT cells, and under normal culture conditions, no p53-mediated DNA damage response was elicited, suggesting that the phenotypes we observed are likely p53-independent.
To identify genes that might be direct SCC transcriptional targets, we correlated transcriptional deregulation in SCC KD mESCs with RAD23B binding by juxtaposing ChIP-seq and RNA-seq data. Globally, there is no preferential RAD23B binding within 5 kb of TSSs of genes, either unchanged, down-regulated, or up-regulated upon SCC knockdown (Fig. S5D). Up-regulated genes are actually somewhat underrepresented among RAD23B ChIP targets compared with unchanged or down-regulated genes, suggesting that the up-regulation of differentiation genes is likely an indirect effect of depleting SCC. However, when we repeated the same analysis to include only those genes with both RAD23B and O/S binding within 5 kb of their TSS, there was clear down-regulation upon SCC knockdown (Fig. 4E), suggesting a direct, positive role for SCC and O/S in the transcriptional regulation of these target genes. This finding prompted us to investigate whether genes affected by SCC KD are also direct targets of O/S. For this purpose, we used a previously described list of OCT4 direct targets in ESCs (36) and checked how many of them are deregulated in our SCC KD cell lines. Again, we observed that genes positively regulated by OCT4, and hence down-regulated upon OCT4 KD, are more likely to be down-regulated than up-regulated upon SCC KD. The reverse does not hold true: genes repressed by OCT4 (i.e., up-regulated in OCT4 KD ESCs) are not preferentially up-regulated in SCC KD cells (Fig. S5E), consistent with these genes being perhaps indirect, downstream targets of OCT4 and thus not necessarily SCC-dependent.
Collectively, these RNA-seq data confirm that SCC indeed functions as an O/S coactivator in vivo on a genome-wide scale, directly fueling the expression of a subset of pluripotency genes. Loss of SCC partially impairs mESC self-renewal and pluripotency, leading to a derepression of differentiation genes, mostly related to the trophectoderm lineage, via a mechanism that seems to be independent of SCC DNA repair activity.
SCC Interaction with OCT4/SOX2 Requires XPC.
Having established OCT4 and SOX2 as the key TFs driving SCC recruitment to specific genes in mESCs, we set out to further characterize the molecular mechanism underpinning the relationship between SCC and its partner TFs. As previously shown (11), mouse SCC coimmunoprecipitates (co-IP) with OCT4 and SOX2 when all four proteins (RAD23B, XPC, OCT4, and SOX2) are ectopically expressed in 293T cells by transient transfection (Fig. 5 A–D, set 1). To assess which protein subunits dictate this interaction, we repeated the co-IP assays excluding one protein at a time in all possible combinations (Fig. 5 A–D, sets 2–5). We did not evaluate the requirement of CETN2 for the interaction, because previous data suggested that this small subunit is dispensable for SCC transcriptional activity (11). Without the large XPC subunit of SCC, RAD23B failed to pull-down either SOX2 or OCT4 (Fig. 5B, compare sets 1–3 to 4). Similarly, in the absence of XPC, neither SOX2 (Fig. 5C, compare sets 1 to 3–4) nor OCT4 (Fig. 5D, compare sets 1–2 to 4) were able to interact with RAD23B. As expected, loss of XPC had no effect on the ability of OCT4 to bind SOX2 (Fig. 5 C and D, sets 1 and 4). On the other hand, excluding RAD23B from the transfection mixture did not significantly alter the interaction of XPC with either SOX2 or OCT4 (Fig. 5A, set 5). We conclude that within holo-SCC, the large XPC subunit is necessary and likely sufficient for interaction with OCT4 and SOX2 (Fig. 5E). This finding agrees well with our previous observation that XPC alone—but not RAD23B—can partly serve to coactivate OCT4 and SOX2 transcription (11).
Fig. 5.

SCC interacts with OCT4/SOX2 mainly via XPC. (A–D) Pull-down assays with XPC (A), RAD23B (B), SOX2 (C), and OCT4 (D) antibodies in 293T overexpressing different combinations of SOX2, OCT4, XPC, and RAD23B FLAG-tagged proteins, followed by immunoblotting with anti-FLAG antibody. IN, 0.5% input; Ig, IgG control pull-downs; IP, specific pull-downs. IgG controls were intentionally omitted from the gel when pulling-down through the protein excluded from the transfection reaction (set 4 in A, set 5 in B, set 2 in C, and set 3 in D). (E) Schematic of SCC interaction with OCT4/SOX2 as inferred from A–D. (F) Boxplot showing the enrichment of OCT4/SOX2 (O/S) at RAD23B binding sites in WT and Xpc knockout (Xpc−/−) mESCs. O/S binding strength is calculated as log10 of scaled fold ratios between O/S and control IgG ChIP-seq signals. The analysis only includes RAD23B ChIP-seq peaks overlapping with O/S sites (24). ***P < 10−10, ANOVA. Complete list of RAD23B peaks in Xpc−/− mESCs in Dataset S1; see Fig. S6B for specific examples. (G) Sequence analysis of RAD23B binding sites in Xpc−/− mESCs as in Fig. 2B.
To confirm in vivo that SCC interaction with OCT4 and SOX2 occurs via XPC, we again mapped RAD23B binding sites by ChIP-seq in XPC-depleted mESCs (Xpc−/−) and compared them to the sites of RAD23B interaction found in WT cells (see Materials and Methods for details on the cell line generation). ChIP efficiency of RAD23B in Xpc−/− mESCs was overall much lower than in WT cells, revealing a total of only 828 binding sites, about 65% of which (534) corresponding to the RAD23B ChIP-seq peaks mapped in WT cells. These 534 peaks were used for further analysis (full list in Dataset S1). About 30% of the RAD23B binding sites retained in Xpc−/− mESCs still colocalize with O/S, but OCT4 and SOX2 signal at these overlapping peaks is much weaker than what was normally found in WT cells (Fig. 5F). In fact, XPC depletion abolishes RAD23B binding at strong, prototypic O/S targets like pluripotency genes (Oct4/Pou5f1, Sox2), whereas it only partially affects RAD23B binding at those genomic regions with little O/S enrichment (Fig. 5F and Fig. S6B). Concordantly, de novo motif discovery within DNA sequences surrounding the RAD23B peaks retained in Xpc−/− mESCs identifies only a motif that matches either SP1 (P < 10−7) or KLF4 (P < 0.01) binding sites but no O/S composite recognition elements (Fig. 5G). These observations confirm on a genome-wide scale the biochemical evidence that XPC is the main and direct determinant of SCC interactions with OCT4 and SOX2.
OCT4 and SOX2 Independently Interact with XPC.
In addition to identifying XPC as the major driver of SCC interaction with O/S, the co-IP experiments above also revealed that SCC can independently bind to either SOX2 or OCT4, because the absence of one has no effect on the pull-down efficiency of the other through either XPC or RAD23B (Fig. 5 A and B, sets 1–3). Searching for domains within XPC that might mediate interactions with these two distinct TFs, we overexpressed either full-length XPC or progressive C-terminal truncations in 293T cells and individually tested their binding to cotransfected SOX2 or OCT4 by co-IP (Fig. 6A). Removal of the C-terminal CETN2/TFIIH-interaction domain of XPC (17, 19) did not impair its interaction with either SOX2 or OCT4 (Fig. 6 B and C, 1–808: “–Benz” panels). This finding was consistent with previous observations that this region is dispensable for SCC transcriptional activity in vitro (11). In contrast, deletion of a region encompassing the DNA binding domain and part of the RAD23B interacting residues (18, 19) drastically reduced XPC interaction with SOX2 but not with OCT4 (Fig. 6 B and C; 1–599, “–Benz” panels). OCT4 binding to XPC was only abolished upon removal of the whole RAD23B interaction domain (Fig. 6 B and C, 1–511, “–Benz” panels). XPC thus independently binds OCT4 and SOX2, whereas its N terminus is dispensable for interaction with both. We note that the first 200 amino acids of XPC, rather than its C terminus, were recently reported to recruit TFIIH onto damaged chromatin (37). To further verify that SCC coactivation of O/S transcription occurs independently of TFIIH recruitment, we performed in vitro transcription assays with recombinant human SCC containing either WT or N-terminal truncated XPC (Δ1–195, hXPCΔN) (Fig. 6 A and D) (11). ΔN-SCC enhanced OCT4/SOX2-dependent activation of the Nanog promoter to levels comparable with using WT-SCC, confirming that SCC transcriptional activity is unlikely related to the ability of XPC to bind TFIIH.
Fig. 6.
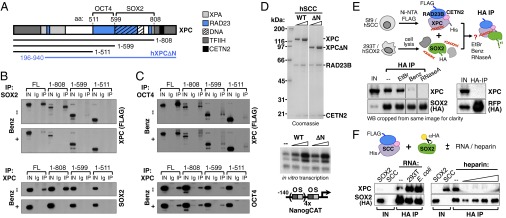
OCT4 and SOX2 independently interact with XPC. (A) Schematic representation of XPC protein and its functionally relevant domains (17–19, 37, 73), and of the truncations used below. OCT4- and SOX2-interaction domains as inferred by truncation experiments are annotated. (B) Pull-down assays in 293T overexpressing SOX2 and either full-length (FL) or truncated XPC FLAG-tagged proteins by SOX2 (Upper) and XPC (Lower) antibodies, followed by immunoblotting with anti-FLAG and anti-SOX2 antibodies, respectively. Lysates were (+) or were not (−) treated with benzonase (Benz) to digest nucleic acids. IN, 0.5% input; Ig, IgG control pull-downs; IP, specific pull-downs. (C) Same as B but in 293T overexpressing OCT4 and either full-length (FL) or truncated XPC FLAG-tagged proteins. Additional pull-down assays with XPC DNA-binding mutant and XPC internal fragments in Fig. S7. (D, Upper) Coomassie-stained SDS/PAGE gel of purified recombinant human SCC containing WT or N terminus deleted (ΔN: amino acids 196–940) XPC; twofold concentration range tested. (Lower) Reconstituted in vitro transcription reactions supplemented with OCT4 and SOX2 only (−) or with WT or ΔN-SCC. All reactions contain a Nanog template with four extracopies of the O/S composite element (NanogCAT) and a partially purified Q0.3 fraction from NT2 nuclear extracts. Fourfold concentration range tested. See ref. 11 for details. (E) Pull-down assays of human SCC complex and SOX2 protein with or without nucleic acids. SCC purified from Sf9 cells was mixed with lysates of 293T cells overexpressing HA-tagged SOX2; complexes were immunoprecipitated with HA resin and blotted with anti-XPC and anti-HA antibodies. Reactions were either left untreated (–) or treated with ethidium bromide (EtBr), benzonase (Benz), or RNaseA. HA pull-down of purified SCC incubated with lysates from 293T cells overexpressing HA-tagged red fluorescent protein (RFP, Right) controls for specificity. Nucleic acids mediating SOX2:XPC interaction might be already associated with SCC from Sf9 cells or derive from HeLa cells. IN, 0.5% input. (F) Pull-down assays of human SCC complex and SOX2 protein with or without RNA and with or without heparin. SCC purified from Sf9 cells was incubated with HA resin-captured tagged SOX2 protein purified from 293T cells in the presence or absence (–) of total RNA extracted from 293T and E. coli cells, or of a 10-fold concentration range of heparin (0.5–50 μg). After extensive washes, complexes were eluted from the resin and blotted with anti-XPC and anti-HA antibodies. SCC IN, input of purified recombinant SCC from Sf9 cells; SOX2 IN, whole 293T lysate input before affinity purification with HA-resin.
For SOX2, we recapitulated the above truncation results using internal XPC fragments and verified that the XPC DNA binding domain is critical for its interaction with SOX2 (Fig. S7 A and B). We next tested whether nucleic acids play any role in XPC binding to SOX2. Benzonase treatment of cell lysates before co-IP, as well as co-IP assays with the XPC DNA-binding mutant W683S (38), confirmed that XPC binding to nucleic acids is indeed important for its interaction with SOX2 but not OCT4 (Fig. 6 B and C, “+ Benz” panels, and Fig. S7 C and D). Interestingly, the W683S mutation enhanced XPC binding to OCT4 (Fig. S7D); we surmise that this alteration may compensate for the loss of SOX2 interaction and could explain why the XPC W683S DNA-binding mutant retains near normal transcription coactivator activity in vitro (11).
To further confirm that nucleic acids mediate SCC/SOX2 interaction, and to investigate which class might be involved, we carried out reconstitution experiments using an orthologous system in which the human SSC complex was recombinantly expressed in Sf9 insect cells, purified, and mixed with lysates of 293T cells overexpressing human SOX2 (Fig. 6E). Reactions were either left untreated or treated with ethidium bromide, benzonase, or RNase A to assess dependence of SCC/SOX2 interaction on double-stranded DNA, nucleic acids in general, or RNA, respectively. Although benzonase treatment was confirmed to abrogate SOX2 interaction with SCC, ethidium bromide had minimal, if any, effect on this interaction. More interestingly, RNase A treatment completely abolished SOX2 binding to SCC, suggesting that an RNA component might be involved in bridging the two proteins or influencing either the stability or the conformation of one for binding to the other. Finally, we verified that RNA can boost SCC interaction with SOX2 in the absence of any other protein component, performing co-IPs with purified proteins in the presence or absence of total RNA extracted from 293T cells (Fig. 6F). The addition of extra RNA increased SCC pull-down efficiency through SOX2 by a factor of 2- to >10-fold, depending on the experiment. Importantly, addition of equal amounts of total RNA extracted from Escherichia coli also enhanced SCC/SOX2 interaction, suggesting that the required RNA species are not mammalian, or even eukaryotic-specific. To exclude that the boost of SCC/SOX2 interaction is simply a result of RNA acting as an ion exchanger, we performed a similar experiment in the presence of increasing amounts of heparin, and observed that this ionic polymer actually hampered, rather than facilitated, SCC binding to SOX2, probably competing away the required RNA. Taken together, these experiments point to a model in which different domains of XPC independently interact with OCT4 and SOX2, and highlight the potential involvement of an RNA scaffold in mediating SCC/SOX2 interactions.
Discussion
In this study we analyzed transcriptional function and mechanism of action of the XPC-RAD23B-CETN2 DNA repair complex, which we recently established as an O/S selective coactivator in embryonic stem cells (SCC) (11).
Our high-throughput genome-wide mapping of RAD23B binding sites in mESCs reveals enrichment at TSSs and enhancers of both active and developmentally poised genes. Such a pattern is more consistent with SCC acting as a TF rather than as a repair complex randomly scanning the genome. One alternative explanation we considered was the possibility that RAD23B recruitment at regulatory regions might represent a byproduct of XPC interacting with TFIIH (39), the latter being an intrinsic component of transcription initiation platforms assembled at promoters and tissue-specific enhancers (40). Two lines of evidence suggest that TFIIH is not the driver: (i) truncated versions of XPC lacking putative TFIIH interaction domains (19, 37) remain competent to interact with O/S (Fig. 6 and Fig. S7) and stimulate their transcriptional activity (Fig. 6D) (11); (ii) after probing a variety of ESC TFs, RAD23B was found to predominantly colocalize with O/S (Fig. 1E), whereas O/S bipartite motifs are faithfully retrieved from RAD23B peaks (Fig. 2B) and RAD23B binding relies upon O/S (Fig. 3C). These results strongly suggest that SCC is primarily recruited by sequence-specific TFs rather than by components of the basal transcription machinery, such as TFIIH.
Using an inducible knockout strategy, Niwa and coworkers recently reported that XPC may be dispensable for mESC pluripotency and O/S transcription, and that its loss has a modest impact on global gene expression, as measured by microarray analyses (41). This finding is in agreement with previous studies showing that Xpc knockout mice are viable with no overt developmental defects, although they are UV sensitive (42, 43). Similarly, our transcriptome analysis of SCC-depleted mESCs cells shows a relatively mild gene deregulation (1.5- to 4-fold). We caution, however, that SCC is only one of three coactivators required for fully activating O/S transcription in vitro, and its knockdown alone might not be sufficient to severely hamper transcription in vivo (11, 12). Nonetheless, we observed a clear trend upon SCC depletion in mESCs, with down-regulation of pluripotency genes and concomitant up-regulation of differentiation markers, accompanied by reduced clonogenic ability (Fig. 4 and Fig. S5). It is also worth pointing out that in Ito et al. (41), floxing the Xpc gene reduces its mRNA level by half even before Cre excision; in these Xpcfl/fl ESCs, Nanog transcripts are reduced compared with WT cells as much as we observe upon SCC depletion. These results taken in aggregate suggest that, although SCC might be dispensable for mESC self-renewal and pluripotency because of partial functional redundancy, SCC remains an important player in stabilizing stem cell transcriptional programs.
To confirm the results from our biochemical assays showing that SCC interacts with specific chromatin sites mainly via direct interactions of O/S and XPC, here we performed ChIP-seq of RAD23B in Xpc−/− mESCs. Indeed, in the absence of XPC we observed a reduced colocalization of RAD23B with O/S, and we retrieved no obvious O/S motif from sequences surrounding the remaining RAD23B binding sites. However, to our surprise we found regions that retained RAD23B binding even in the absence of XPC, pointing at an XPC-independent loading of RAD23B onto chromatin. We speculate that this might reflect RAD23B functions beyond NER (i.e., its role as a proteasome shuttling factor) (44). Indeed, several studies have documented a role for the 26S proteasome in transcription, through proteolytic as well as nonproteolytic activities (45). In mESCs, the proteasome safeguards pluripotency by degrading preinitiation complexes preassembled at tissue-specific enhancers to allow transcription at later stages of development (46). Proteasome recruitment was verified at a few permissive loci, and our SCC ChIP-seq reveals that the very same sites are also bound by RAD23B. A transcriptional role for RAD23B has also been suggested in yeast, again through its interaction with the proteasome regulatory subunit (47). Further studies are needed to test the contribution of RAD23B alone to transcriptional regulation, which might partially explain the developmental abnormalities observed in Rad23b−/− mice (48, 49).
Among the various mechanistic aspects of SCC and O/S interactions revealed by our studies, the most intriguing one is probably that RNA species might help mediate direct SOX2–XPC interactions. Interestingly, it was reported by others that binding to a noncoding RNA allows SOX2 recruitment to promoter regions of neurogenic TFs (50). It is tempting to speculate that a similar role for RNA exists at pluripotency genes regulated by SOX2 in ESCs. Our preliminary data indicate that the RNA facilitating SOX2–XPC binding is neither ESC- nor eukaryotic-specific, because we observed comparable results when using total RNA extracted from either 293T or bacterial cells. However, heparin failed to boost SCC association with SOX2, indicating that some property of RNA other than its strong charge (e.g., secondary structure) is responsible for the effect. Although our data are very preliminary and more experiments will be needed to fully address this issue, we regard it with particular interest, in light of recent emerging evidence for a more pervasive role of both specific and promiscuous RNA binding in the regulation of Pol II-dependent transcription (51–54).
In conclusion, consistent with observations that ESCs are exceptionally sensitive to reduced levels of transcriptional cofactors and chromatin regulators (23, 55–57), herein we provide evidence that the DNA repair trimeric complex XPC-RAD23B-CETN2 participates in the maintenance of mESC identity by working as a transcriptional coactivator targeted by the core TFs O/S. The mechanistic and functional insights presented herein add another level of insight into how multiprotein complexes can accommodate and provide seemingly unrelated and orthogonal functions that stand at the point of convergence between diverse biological processes (i.e., transcription and DNA repair) to generate the coordinated responses required to maintain cellular identity.
Materials and Methods
Antibodies.
Antibodies to immunoprecipitate RAD23B and XPC were raised in guinea pigs (Covance) against GST-fused peptides (pGEX-4T-3; GE Healthcare) encompassing residues 108–177 and 155–217 of murine RAD23B (NP_033037) and XPC (NP_033557) proteins, respectively. Antisera were affinity-purified using the same peptides tagged with MBP (pMAL-c5X, New England Biolabs) or SUMO (pET SUMO, Life Technologies) and immobilized to Affygel resins (Bio-Rad). Monoclonal anti-Pol II antibody (clone 8WG16) (58) was purified from hybridoma cell culture media using Protein G Sepharose (GE Healthcare). Commercial antibodies were as follows: ChromPure goat, rabbit and mouse normal IgG from Jackson ImmunoResearch; anti-OCT4 (sc-8628), and anti-KLF4 (sc-20691) from Santa Cruz Biotechnology; anti-SOX2 (AB5603) from Millipore; anti-ACTB (A2228), and anti-FLAG M2 (F3165) from Sigma-Aldrich; anti-TBP (ab51841), anti-c-MYC (ab32072), anti-HA C5 (ab18181), and anti-LEFTY2 (ab124952) from Abcam; anti-XPC (A301-122A), anti-RAD23B (A302-305A), and anti-NANOG (A300-397A) from Bethyl Laboratories; anti-STAT3 (4904), antiphospho-STAT3 (9131), and anti-P53 1C12 (2524) from Cell Signaling; anti-CETN2 (15977-1-AP) from ProteinTech Group. Anti-HA affinity gel (E6779) was from Sigma-Aldrich.
Cells.
WT D3 (ATCC) and JM8.N4 (trans-NIH Knockout Mouse Project Repository, https://www.komp.org) mESCs were cultured on gelatin-coated plates in ESC media as previously described (11). The Rad23btm1a(KOMP)Wtsi JM8.N4 clone from KOMP (see below) was cocultured with irradiated mouse embryonic fibroblasts before and after electroporation; hygromycin-resistant clones were selected and expanded on mouse embryonic fibroblast feeders for few passages before adaptation to gelatin-coated plates. The 293T cells were grown in DMEM high-glucose with GlutaMAX (Invitrogen) supplemented with 10% (vol/vol) FBS (HyClone).
Primers.
Primers used in this study are listed in Table S1.
ChIP and ChIP-seq Analysis.
ChIP and ChIP-seq experiments were performed as previously described (11). ChIP-seq reads from our RAD23B data as well as other publicly available mESC TFs were analyzed with Partek Flow (v1.1.1219) and Genomics Suite (v6.12.0103) softwares (Partek), using mm9 as a reference genome. All RefSeq genes with a RAD23B/SCC binding site within 5 kb from their TSS were considered putative SCC targets. Poly-A RNA-seq performed in D3 mESCs by Liu et al. (8) was used to assess target gene expression levels; RefSeq genes were classified as active when having a reads per kilobase and million mapped reads (RPKM) ≥ 1 and inactive when having an RPKM < 1. For GO analysis we used DAVID 6.7 Functional Annotation Tool (59, 60) (accessed March 2014). Overlaps between RAD23B/SCC and other mESC TFs were calculated with Galaxy (61–63), requiring a minimum 1-bp overlap between the ChIP-seq peaks.
DNA Constructs, Lentiviral Vector Production, and Infection of mESCs.
pLKO.1 shRNA lentiviral constructs (Sigma-Aldrich) were the following: TRCN0000009611 (KD1), TRCN0000009612 (KD2), TRCN0000009614 (KD3), and TRCN0000009615 (KD4) targeting Oct4; TRCN0000240683 (KD1) and TRCN0000240685 (KD2) targeting Xpc; TRCN0000127120 targeting Rad23b. As a control, we used a nontargeting pLKO.1 construct (hairpin sequence: 5′-CCGG-CAACAAGATGAAGAGCACCAA-CTCGAG-TTGGTGCTCTTCATCTTGTTGTTTTT-3′). Lentiviral particles for stable knockdown experiments in D3 mESCs were prepared by transient transfection of 293T cells, collected and concentrated as described (64), and titrated on 293T cells by real-time qPCR following the EPFL transgenic core facility protocol (tcf.epfl.ch/). Next, 4.5 × 106 D3 mESCs were seeded to 15-cm plates 24 h before infection and transduced overnight at a multiplicity of infection of 20 in the presence of 4 μg/mL polybrene. Twenty-four hours posttransduction infected cells were either collected (24-h time point) or selected with puromycin (1.5 μg/μL) for another 24 (48-h time point) or 48 (72-h time point) h before collection for Western blot, RNA, and ChIP analyses.
The PG00023_C_2_C03 Rad23b targeting vector was obtained from KOMP. The neomycin resistance was replaced with a hygromycin cassette by assembly PCR. We amplified from the targeting vector a fragment containing part of the LacZ reporter cassette (upstream of the BssHII restriction site) up to the self-cleaving T2A peptide. The fragment was then assembled with a PCR amplicon containing the hygromycin gene from pCoHygro (Life Technologies) preceded by the last 20 nucleotides of the T2A peptide and followed by two stop codons and a BssHI restriction site. The assembled PCR amplicon was then cloned into the original PG00023_C_2_C03 vector digested with BssHI. Before electroporation into mESCs, the hygromycin targeting vector was linearized with SalI and XhoI, and gel-purified from low-melting agarose through phenol-chloroform extraction.
A lentiviral vector coexpressing an improved Cre recombinase (iCre) and an EGFP was generated from the previously described pCCL.GFP/ΔLNGFR bidirectional construct (65), by replacing GFP and ΔLNGFR reporters with iCre and EGFP, respectively. For transient iCre expression in JM8.N4 mESCs, we used a third-generation, integrase-defective lentiviral packaging system (66), and titrated the viral supernatant on 293T cells by flow cytometry. Next, 3 × 106 JM8.N4 mESCs were seeded to 10-cm plates 24 h before infection and transduced overnight at multiplicity of infection of 10 in the presence of 4 μg/mL polybrene. Forty-eight hours posttransduction GFP-expressing cells were sorted by FACS and expanded for further analyses.
For 293T pull-down experiments, cDNAs for mouse Pou5f1/Oct4 (NM_013633.3), Sox2 (NM_011443.3), and Xpc (NM_009531.2) genes were cloned into the pFLAG-CMV-5a mammalian expression vector, and Rad23b (NM_009011.4) cDNA was cloned into the p3XFLAG-CMV-10 plasmid, both from Sigma-Aldrich. To overexpress mouse HA-SOX2 protein, the tag was introduced by PCR at the C terminus followed by two stop codons, and the PCR product again cloned into a pFLAG-CMV-5a construct. XPC truncations and internal fragments were generated by PCR from full-length Xpc cDNA and cloned into pFLAG-CMV-5a. The W683S XPC mutant was obtained by a c.2048G > C substitution from full-length, WT Xpc cDNA and cloned into pFLAG-CMV-5a. For reconstitution experiments, human HA-tagged SOX2 cDNA was cloned into pFLAG-CMV-5a and HA-tagged RFP into pLKO.1 expression plasmid (Life Technologies). Constructs for expression of recombinant SCC were previously described (11).
Generation of SCC Knockdown and Xpc−/− mESCs.
Xpc−/− mESCs, in which Xpc exon 11 and a portion of each of the flanking introns are replaced with a selectable marker (43), were a generous gift from J. H. Hoeijmakers (Department of Genetics, Erasmus University Medical Center, Rotterdam, The Netherlands). Detailed molecular analysis of the cell line showed that the targeted Xpc locus still produces two species of functional mRNAs, translated into truncated proteins (Fig. S6A). To avoid interference of these truncated products with our experiments, we further depleted Xpc mRNA with shRNA lentiviral constructs (see below). Infected cells were selected with puromycin (1.5 μg/μL) and used to perform ChIP-seq libraries with the RAD23B antibody.
To generate SCC-KD mESCs, we started from a KOMP-generated JM8.N4 mESC line (allele Rad23btm1a(KOMP)Wtsi, project CSD40373). In these cells, one of the two Rad23b alleles is targeted by a reporter-tagged insertion with conditional knockout potential, floxing exon 2 (Fig. S4A). To obtain a complete Rad23b knockout, we electroporated these cells with a modified targeting vector containing hygromycin instead of the original neomycin resistance to target the second Rad23b allele, and selected for hygromycin-resistant colonies. We obtained six clones, and analyzed their Rad23b locus by Southern blot. In five of six clones the modified, hygromycin-targeting cassette simply replaced the neomycin one, leaving an intact WT allele. In one clone (#6) we could target the WT Rad23b allele, resulting in a double-targeted, Rad23b conditional knockout mESC line (Fig. S4B). Notably, clone #6 had already reduced, but still detectable, Rad23b mRNA levels compared with WT cells (Fig. S4C), because the targeting cassette contains a splicing acceptor site and a polyadenylation signal that can splice with Rad23b exon 1 and terminate transcription early in the first intron. However, a fully functional Rad23b mRNA can still be formed whenever splicing jumps from exon 1–2. To obtain a full Rad23b knockout, we removed the floxed exon 2 by infecting the cells with an integrase-defective lentiviral vector that would transiently express the Cre recombinase and a GFP reporter gene. GFP-expressing cells were sorted by FACS and checked for complete exon 2 removal by genomic PCR (Fig. S4C). RT-qPCR and Western blot analyses confirm that very little Rad23b mRNA and no protein are present in these Rad23b−/− cells (Figs. S4C and S5C). XPC protein levels are already reduced in Rad23b−/− mESCs compared with WT cells (Fig. S5C), in accordance with previous data that RAD23B protects its partner XPC from proteasomal degradation (15, 67). Therefore, we could easily obtain an almost complete depletion of SCC by infecting Rad23b−/− cells with two independent shRNA lentiviral constructs targeting Xpc, and get two SCC depleted cell lines (SCC KD1 and 2) (Figs. S4 A and D and S5C). WT and Rad23b−/− JM8.N4 mESCs were also infected with a nontargeting shRNA construct and used as a control for RNA-seq and all further analyses.
RT-qPCR and RNA-Seq Analysis.
Total RNA was purified from cell pellets using RNeasy Plus Mini kit (Qiagen) and quantified by spectrophotometer. For RT-qPCR, 1 μg of total RNA was retrotranscribed to cDNA with oligo(dT) primers (Ambion, Life Technologies) and SuperScript III (Invitrogen). Two microliters of 1:20 cDNA dilutions were used for qPCR with SYBR Green PCR Master Mix (Applied Biosystems) on an ABI 7300 Real Time PCR system. For RNA-seq, 6 μg of total RNA were used to prepare poly-A libraries following the TruSeq RNA Sample Preparation Guide (Illumina). Libraries were indexed with the following TruSeq adapters:
WT JM8.N4 mESCs: AR005-ACAGTG(A);
Rad23b−/− JM8.N4 mESCs: AR006-GCCAAT(A);
SCC KD1 JM8.N4 mESCs: AR012-CTTGTA(A);
SCC KD2 JM8.N4 mESCs: AR019-GTGAAA(C).
Libraries were checked for quality and concentration by Bioanalyzer (2100 DNA, Agilent Technologies), Qubit (Life Technologies), and qPCR and sequenced in one lane of the HiSeq2000 platform (single-end reads, 50 bp; Illumina). Reads were mapped against Ensembl genes using mm9 as a reference genome by TopHat (v2.0.6), and differentially expressed genes between WT and Rad23b−/− or SCC KD cells were determined using Cufflinks (v2.0.2) (68). Genes with RPKM < 1 in both WT and KO/KD samples were not considered for further analysis. For all of the other genes, the cut-off for differential expression was set at 1.5-fold change. The list of genes involved in pluripotency and differentiation of Fig. 4A was manually curated starting from the TaqMan Array Mouse Stem Cell Pluripotency Panel (Applied Biosystems) and the Qiagen Mouse Embryonic Stem Cells PCR Array.
Clonal Assays.
WT and SCC KD JM8.N4 mESCs were trypsinized to single-cell suspension, counted, and diluted to 30,000 cells/mL. Ten microliters of this cell suspension (containing 300 cells) were then plated to six-well plates in standard mESC medium with LIF (1,000 U/mL) and grown for 6–10 d. Emerging colonies were stained for alkaline phosphatase activity (Millipore) and counted.
Pull-Down Assays.
Detailed experimental procedures are available in SI Materials and Methods.
In Vitro Transcription Assays.
Transcription reactions contained purified recombinant TFIIA, -B, -E, and -F, immunoaffinity-purified native RNA polymerase II, TFIID and -H, and FLAG-tagged OCT4 and SOX2 purified from transiently transfected HeLa cells. Human recombinant His6-WTXPC:FLAG-RAD23B:CETN2 and His6-ΔNXPC:FLAG-RAD23B:CETN2 complexes were purified from Sf9 cells. The transcription template was the Nanog human promoter (−312 to +24) containing four additional OCT4/SOX2 binding sites (NanogCAT). Transcription reactions were supplemented with a partially purified Q0.3 fraction from nuclear extracts of N-TERA2 human embryonal carcinoma cells. See ref. 11 for detailed experimental procedures.
Datasets and Accession Numbers.
The ChIP-seq and RNA-seq data discussed in this publication have been deposited in National Center for Biotechnology Information’s Gene Expression Omnibus (69) and are accessible through GEO Series accession number GSE64040. OCT4 and SOX2 ChIP-seq data were from (24) (GSE11724); other mESC TFs from ref. 70 (GSE11431); p300, H3K4me1 and H3K27ac from ref. 22 (GSE24165), and CTCF, cohesin and Mediator from ref. 23 (GSE22557).
Supplementary Material
Acknowledgments
We thank D. Cado and C. Kang at the University of California, Berkeley Cancer Research Laboratory Gene Targeting Facility for generating the Rad23b−/− mESCs; H. Nolla and K. Heydari of the University of California, Berkeley flow cytometry facility; S. Zheng, S. Zhou, and G. Dailey for technical assistance; D. Risso and D. Pietrobon for suggestions on the bioinformatics analyses; and J. Ho and all laboratory members for fruitful discussions and critical reading of the manuscript. This work was supported by California Institute for Regenerative Medicine Research Grant RB4-06016 (to R.T.). C.C. and Y.W.F. have been California Institute for Regenerative Medicine postdoctoral fellows (training programs TG2-01164 and T1-00007). The Vincent J. Coates Genomics Sequencing Laboratory at University of California, Berkeley is supported by National Institutes of Health S10 Instrumentation Grants S10RR029668 and S10RR027303. E.T.Z. was a National Science Foundation Graduate Research Fellow and a University of California, Berkeley Distinguished Fellow. K.C. is a Nakajima Foundation Fellow.
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE64040).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505569112/-/DCSupplemental.
References
- 1.Hirai H, Karian P, Kikyo N. Regulation of embryonic stem cell self-renewal and pluripotency by leukaemia inhibitory factor. Biochem J. 2011;438(1):11–23. doi: 10.1042/BJ20102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunath T, et al. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development. 2007;134(16):2895–2902. doi: 10.1242/dev.02880. [DOI] [PubMed] [Google Scholar]
- 3.Hackett JA, Surani MA. Regulatory principles of pluripotency: From the ground state up. Cell Stem Cell. 2014;15(4):416–430. doi: 10.1016/j.stem.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Martello G, Smith A. The nature of embryonic stem cells. Annu Rev Cell Dev Biol. 2014;30:647–675. doi: 10.1146/annurev-cellbio-100913-013116. [DOI] [PubMed] [Google Scholar]
- 5.Fong YW, Cattoglio C, Yamaguchi T, Tjian R. Transcriptional regulation by coactivators in embryonic stem cells. Trends Cell Biol. 2012;22(6):292–298. doi: 10.1016/j.tcb.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosal S, Das S, Chakrabarti J. Long noncoding RNAs: New players in the molecular mechanism for maintenance and differentiation of pluripotent stem cells. Stem Cells Dev. 2013;22(16):2240–2253. doi: 10.1089/scd.2013.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145(6):835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Scannell DR, Eisen MB, Tjian R. Control of embryonic stem cell lineage commitment by core promoter factor, TAF3. Cell. 2011;146(5):720–731. doi: 10.1016/j.cell.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maston GA, et al. Non-canonical TAF complexes regulate active promoters in human embryonic stem cells. eLife. 2012;1:e00068. doi: 10.7554/eLife.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pijnappel WW, et al. A central role for TFIID in the pluripotent transcription circuitry. Nature. 2013;495(7442):516–519. doi: 10.1038/nature11970. [DOI] [PubMed] [Google Scholar]
- 11.Fong YW, et al. A DNA repair complex functions as an Oct4/Sox2 coactivator in embryonic stem cells. Cell. 2011;147(1):120–131. doi: 10.1016/j.cell.2011.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong YW, Ho JJ, Inouye C, Tjian R. The dyskerin ribonucleoprotein complex as an OCT4/SOX2 coactivator in embryonic stem cells. eLife. 2014;3:3. doi: 10.7554/eLife.03573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugasawa K, et al. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2(2):223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 14.Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res. 2008;18(1):73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- 15.Ng JM, et al. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 2003;17(13):1630–1645. doi: 10.1101/gad.260003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergink S, et al. Recognition of DNA damage by XPC coincides with disruption of the XPC-RAD23 complex. J Cell Biol. 2012;196(6):681–688. doi: 10.1083/jcb.201107050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishi R, et al. Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Mol Cell Biol. 2005;25(13):5664–5674. doi: 10.1128/MCB.25.13.5664-5674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bunick CG, Miller MR, Fuller BE, Fanning E, Chazin WJ. Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry. 2006;45(50):14965–14979. doi: 10.1021/bi061370o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchida A, et al. The carboxy-terminal domain of the XPC protein plays a crucial role in nucleotide excision repair through interactions with transcription factor IIH. DNA Repair (Amst) 2002;1(6):449–461. doi: 10.1016/s1568-7864(02)00031-9. [DOI] [PubMed] [Google Scholar]
- 20.Le May N, Fradin D, Iltis I, Bougnères P, Egly JM. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol Cell. 2012;47(4):622–632. doi: 10.1016/j.molcel.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 21.Le May N, et al. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell. 2010;38(1):54–66. doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marson A, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134(3):521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loh YH, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38(4):431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 26.Chew JL, et al. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol Cell Biol. 2005;25(14):6031–6046. doi: 10.1128/MCB.25.14.6031-6046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell. 2014;156(6):1274–1285. doi: 10.1016/j.cell.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24(4):372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 29.Fong H, Hohenstein KA, Donovan PJ. Regulation of self-renewal and pluripotency by Sox2 in human embryonic stem cells. Stem Cells. 2008;26(8):1931–1938. doi: 10.1634/stemcells.2007-1002. [DOI] [PubMed] [Google Scholar]
- 30.Adachi K, Suemori H, Yasuda SY, Nakatsuji N, Kawase E. Role of SOX2 in maintaining pluripotency of human embryonic stem cells. Genes Cells. 2010;15(5):455–470. doi: 10.1111/j.1365-2443.2010.01400.x. [DOI] [PubMed] [Google Scholar]
- 31.Cinelli P, et al. Expression profiling in transgenic FVB/N embryonic stem cells overexpressing STAT3. BMC Dev Biol. 2008;8:57. doi: 10.1186/1471-213X-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye S, Li P, Tong C, Ying QL. Embryonic stem cell self-renewal pathways converge on the transcription factor Tfcp2l1. EMBO J. 2013;32(19):2548–2560. doi: 10.1038/emboj.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall J, et al. Oct4 and LIF/Stat3 additively induce Krüppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell. 2009;5(6):597–609. doi: 10.1016/j.stem.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 34.de Waard H, et al. Cell-type-specific consequences of nucleotide excision repair deficiencies: Embryonic stem cells versus fibroblasts. DNA Repair (Amst) 2008;7(10):1659–1669. doi: 10.1016/j.dnarep.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 35.Prabhu VV, et al. Therapeutic targeting of the p53 pathway in cancer stem cells. Expert Opin Ther Targets. 2012;16(12):1161–1174. doi: 10.1517/14728222.2012.726985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matoba R, et al. Dissecting Oct3/4-regulated gene networks in embryonic stem cells by expression profiling. PLoS ONE. 2006;1:e26. doi: 10.1371/journal.pone.0000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ziani S, et al. Sequential and ordered assembly of a large DNA repair complex on undamaged chromatin. J Cell Biol. 2014;206(5):589–598. doi: 10.1083/jcb.201403096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yasuda G, et al. In vivo destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation. Mol Cell Biol. 2007;27(19):6606–6614. doi: 10.1128/MCB.02166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Araújo SJ, Nigg EA, Wood RD. Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol. 2001;21(7):2281–2291. doi: 10.1128/MCB.21.7.2281-2291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koch F, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol. 2011;18(8):956–963. doi: 10.1038/nsmb.2085. [DOI] [PubMed] [Google Scholar]
- 41.Ito S, Yamane M, Ohtsuka S, Niwa H. The C-terminal region of Xpc is dispensable for the transcriptional activity of Oct3/4 in mouse embryonic stem cells. FEBS Lett. 2014;588(7):1128–1135. doi: 10.1016/j.febslet.2014.02.033. [DOI] [PubMed] [Google Scholar]
- 42.Sands AT, Abuin A, Sanchez A, Conti CJ, Bradley A. High susceptibility to ultraviolet-induced carcinogenesis in mice lacking XPC. Nature. 1995;377(6545):162–165. doi: 10.1038/377162a0. [DOI] [PubMed] [Google Scholar]
- 43.Cheo DL, et al. Characterization of defective nucleotide excision repair in XPC mutant mice. Mutat Res. 1997;374(1):1–9. doi: 10.1016/s0027-5107(97)00046-8. [DOI] [PubMed] [Google Scholar]
- 44.Dantuma NP, Heinen C, Hoogstraten D. The ubiquitin receptor Rad23: At the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair (Amst) 2009;8(4):449–460. doi: 10.1016/j.dnarep.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Durairaj G, Kaiser P. The 26S proteasome and initiation of gene transcription. Biomolecules. 2014;4(3):827–847. doi: 10.3390/biom4030827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szutorisz H, Georgiou A, Tora L, Dillon N. The proteasome restricts permissive transcription at tissue-specific gene loci in embryonic stem cells. Cell. 2006;127(7):1375–1388. doi: 10.1016/j.cell.2006.10.045. [DOI] [PubMed] [Google Scholar]
- 47.Wade SL, Poorey K, Bekiranov S, Auble DT. The Snf1 kinase and proteasome-associated Rad23 regulate UV-responsive gene expression. EMBO J. 2009;28(19):2919–2931. doi: 10.1038/emboj.2009.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ng JM, et al. Developmental defects and male sterility in mice lacking the ubiquitin-like DNA repair gene mHR23B. Mol Cell Biol. 2002;22(4):1233–1245. doi: 10.1128/MCB.22.4.1233-1245.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergink S, et al. Erythropoietic defect associated with reduced cell proliferation in mice lacking the 26S proteasome shuttling factor Rad23b. Mol Cell Biol. 2013;33(19):3879–3892. doi: 10.1128/MCB.05772-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ng SY, Bogu GK, Soh BS, Stanton LW. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol Cell. 2013;51(3):349–359. doi: 10.1016/j.molcel.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 51.Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 52.Schwartz JC, Wang X, Podell ER, Cech TR. RNA seeds higher-order assembly of FUS protein. Cell Reports. 2013;5(4):918–925. doi: 10.1016/j.celrep.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kwon I, et al. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155(5):1049–1060. doi: 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davidovich C, et al. Toward a consensus on the binding specificity and promiscuity of PRC2 for RNA. Mol Cell. 2015;57(3):552–558. doi: 10.1016/j.molcel.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fazzio TG, Huff JT, Panning B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008;134(1):162–174. doi: 10.1016/j.cell.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ang YS, et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145(2):183–197. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ding L, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell. 2009;4(5):403–415. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 58.Thompson NE, Steinberg TH, Aronson DB, Burgess RR. Inhibition of in vivo and in vitro transcription by monoclonal antibodies prepared against wheat germ RNA polymerase II that react with the heptapeptide repeat of eukaryotic RNA polymerase II. J Biol Chem. 1989;264(19):11511–11520. [PubMed] [Google Scholar]
- 59.Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 61.Blankenberg D, et al. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;Chapter 19:Unit 19.10.11–21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giardine B, et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goecks J, Nekrutenko A, Taylor J. Galaxy Team Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dull T, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72(11):8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Amendola M, Venneri MA, Biffi A, Vigna E, Naldini L. Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat Biotechnol. 2005;23(1):108–116. doi: 10.1038/nbt1049. [DOI] [PubMed] [Google Scholar]
- 66.Lombardo A, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25(11):1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 67.Ortolan TG, Chen L, Tongaonkar P, Madura K. Rad23 stabilizes Rad4 from degradation by the Ub/proteasome pathway. Nucleic Acids Res. 2004;32(22):6490–6500. doi: 10.1093/nar/gkh987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133(6):1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 71.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449(7162):570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.