

Significance
DNA replication accuracy is critical for genetic stability and is ensured by high-fidelity replicative DNA polymerases and the mismatch repair (MMR) system, both of which are regulated by the proliferating cell nuclear antigen (PCNA). Dysfunction of either system leads to genome instability and cancer development. Interestingly, many types of cancers have no obvious defects in either system, but they display increased mutation frequency during development. The underlying mechanism is unknown. We demonstrate here that PCNA tyrosine phosphorylation by epidermal growth factor receptor (EGFR) inhibits MMR and promotes misincorporation during DNA synthesis. This study therefore discovers a novel mechanism promoting genome instability and explains why progression of many cancer types is associated with EGFR overexpression and/or activation.
Keywords: mismatch repair, PCNA phosphorylation, EGFR, genome instability, cancer
Abstract
Proliferating cell nuclear antigen (PCNA) plays essential roles in eukaryotic cells during DNA replication, DNA mismatch repair (MMR), and other events at the replication fork. Earlier studies show that PCNA is regulated by posttranslational modifications, including phosphorylation of tyrosine 211 (Y211) by the epidermal growth factor receptor (EGFR). However, the functional significance of Y211-phosphorylated PCNA remains unknown. Here, we show that PCNA phosphorylation by EGFR alters its interaction with mismatch-recognition proteins MutSα and MutSβ and interferes with PCNA-dependent activation of MutLα endonuclease, thereby inhibiting MMR at the initiation step. Evidence is also provided that Y211-phosphorylated PCNA induces nucleotide misincorporation during DNA synthesis. These findings reveal a novel mechanism by which Y211-phosphorylated PCNA promotes cancer development and progression via facilitating error-prone DNA replication and suppressing the MMR function.
DNA mismatch repair (MMR) is an essential genome stability pathway that removes mismatches introduced by nucleotide misincorporation during DNA replication (1–4). MMR in eukaryotic cells is nick-directed and targeted specifically to the newly synthesized DNA strand (5, 6). MMR is carried out in three phases: initiation, excision, and resynthesis. The initiation phase involves mismatch recognition by MutSα or MutSβ; assembly of the initiation complex containing MutSα (or MutSβ), MutLα, and proliferating cell nuclear antigen (PCNA); and localization of the strand discrimination signal (a single-strand nick) by this complex (7–11) in a process that is incompletely understood. During the excision phase, exonuclease 1 (Exo1) removes nascent DNA exonucleolytically from a distal nick to the mismatch in a reaction that requires MutSα (or MutSβ), MutLα, and PCNA. Replication protein A (RPA) stimulates DNA excision by Exo1 and protects single-stranded DNA from cleavage by nucleases (12, 13). During the resynthesis phase of MMR, DNA polymerase δ fills in the single-strand DNA gap left by DNA excision in a concerted reaction that requires replication factor C (RFC), PCNA, and RPA, followed by DNA ligase I-catalyzed nick ligation.
Mutations or promoter hypermethylation in coding or regulatory regions of MMR genes MSH2, MSH6, MLH1, and PMS2 are linked to susceptibility to colorectal and other cancers in humans and other organisms (2, 14–16). The cellular phenotype associated with defects in MMR includes an elevated genome-wide mutation rate as well as frequent changes in DNA microsatellite repeats, a phenomenon called microsatellite instability (MSI). Thus, MSI is frequently used as a biomarker for (or hallmark of) MMR deficiency (1–4). However, a significant number of MSI-positive tumors do not carry mutations in known MMR genes (17). Recent studies have shown that mutations affecting the proofreading nuclease activity of DNA polymerases δ and ε are associated with some colorectal and/or endometrial cancers (18), and that defects in the gene encoding histone H3 Lys36 (H3K36) trimethyltransferase SETD2 result in MSI and loss of MMR function in vivo (19).
Although defects in MMR cause MSI and susceptibility to colorectal cancer, MSI-positive colorectal cancer cells are also closely associated with high levels of epidermal growth factor receptor (EGFR) (20), a transmembrane receptor protein kinase that promotes cell growth, tumor progression, and metastasis (21). EGFR overexpression in these cancer cells is mainly a result of its mRNA stability, which is caused by a one- or two-base deletion mutation within the A13/A14 repeat sequence in the 3′-untranslated region of the EGFR gene (20). EGFR can translocate to the nucleus (22) and phosphorylate PCNA on tyrosine 211 (Y211) to stimulate DNA synthesis and cell proliferation (23). Given that PCNA is required for the initiation and resynthesis steps of MMR (7, 9, 10), we hypothesized that EGFR-catalyzed PCNA phosphorylation might interfere with its role in MMR, leading to a mutator phenotype that promotes tumor progression.
In this study, we compared the MMR activities of PCNA and Y211-phosphorylated PCNA (PCNA-Y211p) and examined the correlation between expression of EGFR with abundance of PCNA-Y211p and MMR activity in several human cancer cell lines. Our results suggest that a high level of PCNA-Y211p inhibits the initiation step of MMR and that this inhibition is reversed by excess exogenous nonphosphorylated PCNA. Cells expressing a high level of PCNA-Y211p display significantly reduced fidelity of DNA synthesis because of an elevated rate of nucleotide misincorporation. The results support the conclusion that phosphorylation of PCNA on Y211 inhibits MMR activity and reduces the fidelity of DNA synthesis. This may contribute to the mechanism by which EGFR family tyrosine kinases promote tumor progression.
Results
Correlation Between EGFR Expression and PCNA-Y211p in Colorectal Cancer Cells.
Western blot analysis was used to examine the relationship between EGFR expression and PCNA-Y211p abundance in Lim2405, DLD-1, and HCT116 colorectal cancer cells and HeLa cervical cancer cells. EGFR and PCNA-Y211 were quantified, and the relative expression level was calculated, using expression values in HCT116 as a reference (i.e., values in HCT116 arbitrarily set to 1.0). This analysis revealed that relative EGFR expression was 3.7-fold or 3.5-fold higher in Lim2405 and DLD-1 cells than in HCT116 cells, respectively, and twofold lower in HeLa than in HCT116 cells (Fig. 1A), which is consistent with a previous report (20). As for EGFR, the abundance of PCNA-Y211p is threefold higher in Lim2405 and DLD-1 cells than in HCT116 cells and is comparable in HeLa and HCT116 cells. Interestingly, despite the fact that PCNA-Y211p is thought to enhance its stability (23), the amount of total (i.e., phosphorylated and unphosphorylated) PCNA is about the same in all four cell lines (Fig. 1B). This is likely because PCNA is very abundant in cells and only a small portion of PCNA is phosphorylated. Given the previous demonstration that EGFR phosphorylates PCNA on residue Y211 (23, 24), these data are consistent with the possibility that EGFR is involved in phosphorylating PCNA.
Fig. 1.
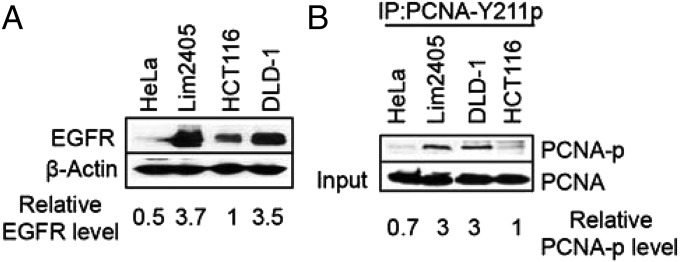
PCNA-Y211 phosphorylation in colorectal cancer cells overexpressing EGFR. (A) Western blot analysis of EGFR expression in HeLa and colorectal cancer cell lines HCT116, Lim2405, and DLD-1. (B) Coimmunoprecipitation-Western (IP-Western) analysis of PCNA-Y211p in HeLa, HCT116, Lim2405, and DLD-1. Cell lysates were incubated with a phospho-specific antibody to PCNA-Y211p. Precipitates were collected and analyzed by Western blot, using an antibody that detects phosphorylated and unphosphorylated PCNA.
Cells with High Levels of PCNA-Y211p Exhibit Reduced MMR Activity.
PCNA plays an essential role in MMR. Thus, genetic or posttranslational modifications that functionally impair this activity could manifest as a deficiency in MMR. We therefore investigated the possibility that PCNA-Y211p alters its function in MMR. A functional in vitro MMR assay (Fig. 2A and Fig. S1A) was used to determine MMR activities of nuclear extracts from MMR-competent HeLa (5), MutSα-deficient DLD-1, MutLα-deficient HCT116 (25, 26), and MutLα-deficient Lim2405 cells (Fig. S1B). As expected, repair products (arrows) were detected in assays containing HeLa nuclear extract (Fig. 2B, lane 1), but not in assays with extracts from Lim2405 (Fig. 2B, lane 3), HCT116 (Fig. 2B, lane 7), and DLD-1 (Fig. 2C, lane 2). Interestingly, despite both HCT116 and Lim2405 carrying the same MutLα defect, the addition of purified MutLα resulted in a repair of 47% in the HCT116 reaction (Fig. 2B, lane 8), but only 28% in the Lim2405 reaction (Fig. 2B, lane 4). Similarly, addition of purified MutSα only stimulated MMR from 5% to 22% in DLD-1 extracts (Fig. 2C, lane 3).
Fig. 2.

Inverse correlation between PCNA-Y211p and MMR activity. (A) DNA heteroduplex used in this study. (B and C) In vitro MMR assay in the presence of HeLa nuclear extract or nuclear extracts from Lim2405 (B) or DLD-1 (C) with or without exogenous PCNA. Arrows indicate repaired products. (D) Fluorescence confocal imaging showing EGF-dependent translocation of EGFR from cytosol to nucleus and colocalization of EGFR and PCNA in HeLa cells. (E) IP-Western analysis showing EGF-dependent interaction between EGFR and PCNA (Upper) and PCNA-Y211 phosphorylation (Lower) in HeLa cells. (F) In vitro MMR assay in control or EGF-treated HeLa cells. (G) Western blot analysis demonstrating increased expression of EGFR and PCNA-Y211 phosphorylation in HCT116 cells stably transfected with the EGFR gene. (H) In vitro MMR assay in HCT116 cells stably expressing high levels of EGFR.
Because both Lim2405 and DLD-1 cells contained high levels of PCNA-Y211p (Fig. 1B), we reasoned that the inhibitory factor in these cells could be the phosphorylated PCNA or an MMR inhibitor that is activated by PCNA phosphorylation. If this is the case, a repair activity comparable to that of HeLa extract should be seen in Lim2405 or DLD-1 extract supplemented with exogenous MutLα or MutSα and exogenous PCNA. The results shown in Fig. 2 confirm this hypothesis. Exogenous PCNA neither stimulates nor inhibits MMR in HeLa extract (Fig. 2B, lane 2) and HCT116 extract (Fig. 2B, lanes 8 and 10), but it greatly elevated MMR activity of Lim2405 extract supplemented with MutLα (Fig. 2B, lanes 4 and 6). An elevated MMR activity was also observed in DLD-1 extract complemented with MutSα and PCNA (Fig. 2C, lanes 3 and 5). These results are consistent with the notion that PCNA-Y211p by EGFR directly or indirectly inhibits MMR.
To further verify this assumption, two additional experiments were performed. First, HeLa cells were incubated with EGF, which is known to promote cell proliferation by activating the EGFR tyrosine kinase activity (21, 27). The results show that the vast majority (if not all) of EGFRs remained outside of the nucleus in the absence of EGF, but all EGFR molecules were found within the nucleus on EGF treatment (Fig. 2D and Fig. S1C), consistent with previous observations (22, 23). Interestingly, many EGFR foci colocalized with those of PCNA (Fig. 2D and Fig. S1C), suggesting a physical interaction between EGFR and PCNA in the nucleus. This postulation is supported by the fact that EGFR is efficiently coimmunoprecipitated with PCNA in EGF-treated but not untreated HeLa cells (Fig. 2E, Upper). Correspondingly, EGF-treated HeLa cells show higher abundance of PCNA-Y211p than untreated HeLa cells (Fig. 2E, Lower). Consistent with the hypothesis presented here, nuclear extracts isolated from EGF-treated HeLa cells have less MMR activity than untreated HeLa cells (Fig. 2F, lanes 1 and 2), and MMR activity in an extract of EGF-treated HeLa cells was stimulated by the addition of recombinant PCNA (Fig. 2F, lane 3).
Second, an HCT116 line containing an extra copy of the EGFR gene (H6-EGFR) was established. As expected, the H6-EGFR cell line possessed higher levels of EGFR and PCNA-Y211p than its parental line HCT116 (Fig. 2G). Similar to Lim2405, MMR activity of H6-EGFR could only be effectively complemented when both MutLα and nonphosphorylated PCNA were added to the reaction (Fig. 2H). Taken together, these results suggest that PCNA-Y211p acts as a direct or indirect inhibitor of MMR in cells lacking a genetic or epigenetic alteration in MMR genes.
PCNA-Y211p Inhibits MMR in Vitro.
A direct test of the hypothesis is to isolate and characterize homogenous PCNA-Y211p. Because of insurmountable technical challenges, it was not feasible to isolate PCNA-Y211p from cell extracts. An alternate approach was adopted instead; namely, site-directed mutagenesis was performed on the cloned PCNA gene, generating recombinant clones that express PCNA-Y211D (Y-D), PCNA-Y211E (Y-E), or PCNA-Y211F (Y-F). PCNA isoforms Y211D and Y211E mimic constitutively phosphorylated PCNA, whereas PCNA-Y211F mimics constitutively unphosphorylated PCNA. The mutant recombinant proteins were overexpressed, purified, and tested for their ability to influence MMR in vitro. The results show that PCNA-Y211F neither stimulates nor inhibits MMR activity in HeLa extracts (Fig. 3A, lane 3), but PCNA-Y211D (lane 4) and PCNA-Y211E (lane 5) inhibit MMR activity.
Fig. 3.

Phosphorylation-mimicking PCNA isoforms inhibit MMR. (A) MMR activity of HeLa nuclear extract in the presence of PCNA (WT), phosphorylation mimicking PCNA-Y211D (Y-D), PCNA-Y211E (Y-E), or nonphosphorylation-mimicking PCNA-Y211F (Y-F), as indicated. (B) As in A, except assays were carried out in the presence of p21c.
Wild-type (WT) and mutant PCNA isoforms were also used in MMR assays in the presence of HeLa nuclear extracts and the C-terminal peptide of p21 (p21c), previously shown to inhibit MMR (9, 10, 28) and DNA replication (29, 30). As expected, p21c inhibits MMR in HeLa extracts (Fig. 3B, lane 2). The effect was completely reversed by the addition of nonphosphorylated WT PCNA (Fig. 3B, lane 3) or PCNA-Y211F (Fig. 3B, lane 4), but not by PCNA-Y211D (Fig. 3B, lane 5) or PCNA-Y211E (Fig. 3B, lane 6). These results support the idea that PCNA-Y211p inhibits MMR.
PCNA-Y211p Inhibits Initiation of MMR.
Previous studies show that PCNA plays an important role during initiation of MMR by interacting with MutSα and activating MutLα endonuclease activity (7–11). Here, the influence of PCNA and PCNA-Y211p on MMR initiation was evaluated using a 3′ nick-directed in vitro MMR assay in the absence of exogenous deoxynucleotide triphosphates, which allows mismatch-provoked excision/incision but not DNA resynthesis to occur (28). As shown in Fig. 4B, MMR-proficient HeLa extracts generate multiple DNA incision/excision intermediates (lane 1); in contrast, Lim2405 nuclear extracts alone do not produce DNA incision/excision intermediates (lane 2), but this defect is partially corrected by addition of MutLα (Fig. 4B, lane 3) and corrected to a level comparable to that of HeLa extract by the addition of MutLα and PCNA (Fig. 4B, lane 6). When PCNA-Y211D (Fig. 4B, lane 7) was added, MMR initiation was greatly reduced (Fig. 4B, lane 7). These results suggest PCNA-Y211p fails to support initiation of MMR.
Fig. 4.

Initiation of MMR in the presence or absence of variant PCNA isoforms. (A) Diagram of Southern blot analysis to detect MMR initiation intermediates. In the absence of exogenous dNTPs, 3′ nick-directed MMR was performed. The reaction products were double-digested with restriction endonucleases PstI and BglI and separated by gel electrophoresis, followed by Southern blot analysis using a 32P-labeled probe, which anneals to the nicked strand near the BglI site. The distance from the BglI site to the mismatch or various restriction enzyme sites is shown. (B) Incision/excision products in HeLa extract or Lim2405 extract in the presence or absence of MutLα and the indicated PCNA isoforms. (C) DNA repair intermediates in HeLa extracts in the presence or absence of PCNA inhibitor, p21c, and the indicated PCNA isoforms. (D) Incision/excision intermediates generated by purified MMR components in the presence or absence of the indicated PCNA isoforms. Red stars indicate a nonspecific nick, which is present in untreated substrate (lane 2). Bands included in the blue bracket are incision/excision intermediates, although only those that are 5′ to the mismatch contribute to mismatch removal by a 5′–3′ exonuclease such as ExoI.
Assays for MMR initiation were also performed using HeLa cell nuclear extracts in the presence of PCNA inhibitor, p21c. As expected, p21c inhibits MMR initiation in HeLa extracts (Fig. 4C, compare lane 2 with lane 1). The inhibitory effect of p21c on MMR initiation was reversed by the addition of WT nonphosphorylated PCNA (Fig. 4C, lane 3) or PCNA-Y211F (Fig. 4C, lane 4) but was not reversed by the addition of PCNA-Y211D (Fig. 4C, lane 5). These results confirm that PCNA-Y211p either directly or indirectly inhibits the initiation of MMR in vitro.
PCNA-Y211p Inhibits MutLα Endonuclease Activity.
MMR incision/excision products were also analyzed in a minimal in vitro reconstituted MMR system containing MutSα, MutLα, RFC, RPA, and distinct isoforms of PCNA, as described earlier. Consistent with our hypothesis that PCNA-Y211p is not competent for MMR, the expected pattern of incision products was detected in reactions containing WT nonphosphorylated PCNA (Fig. 4D, lane 6) or PCNA-Y211F (Fig. 4D, lane 7), but DNA incision was largely inhibited in reactions containing PCNA-Y211D (Fig. 4D, lane 8) and PCNA-Y211E (Fig. 4D, lane 9). These results confirm that PCNA is required for activation of MutLα endonuclease, consistent with the idea that PCNA-Y211p, or its structural equivalents PCNA-Y211D and PCNA-Y211E, block MutLα endonuclease activity.
Influence of Y211 Phosphorylation on the Hydrophobic Pocket of PCNA and Its Interactions with MutSα and MutSβ.
PCNA interacts with MutSα, MutSβ (31–33), and MutLα (10, 34). Therefore, if phosphorylation of Y211 in PCNA alters one or more of these interactions, this could account for the fact that it fails to support MMR initiation. Far-Western analysis was used to study PCNA interactions with MutSα, MutSβ, and MutLα. The results were similar for WT PCNA and PCNA-Y211F, which bind all three factors (Fig. 5A). However, PCNA-Y211D displays significantly lower affinity for MutSα and MutSβ than PCNA WT and PCNA-Y211F, whereas all PCNA isoforms interact similarly with MutLα (Fig. 5A). These results suggest PCNA-Y211p may have reduced affinity for MutSα and MutSβ.
Fig. 5.

Virtual modeling of the hydrophobic pocket in PCNA isoforms and effect on interaction with MutSα and MutSβ. (A) Far-Western analysis for interactions between PCNA-WT, PCNA-Y211F, or PCNA-Y211D and MutSα or MutSβ. (B–E) Model to estimate conformation of the hydrophobic pocket in Y211, Y211F, Y211D, or Y211E isoforms. (F–I) Modeling the electrostatic charge distribution in the hydrophobic pocket (highlighted by a square in J) of the indicated PCNA isoforms.
To understand the molecular basis by which PCNA phosphorylation blocks its interactions with MutSα and MutSβ, virtual protein structure modeling of PCNA-Y211F, PCNA-Y211D, and PCNA-Y211E was performed, using the published crystal structure of PCNA as a starting point (35, 36). Y211 is located in the A2 α-helix and oriented to the central loop (residues 41–45), which is underneath the hydrophobic pocket. Both the central loop and the hydrophobic pocket of PCNA are important for its interactions with PCNA-interacting protein (PIP) box-containing proteins (37, 38). Under the nonphosphorylated condition, Y211 is 3.2 Å away from D41 of the central loop (Fig. 5B). Tyrosine substitution mutations were modeled into this structure using PyMol. This analysis reveals that although substitution of Y211 with phenylalanine slightly increases the distance to 3.8 Å (Fig. 5C), substitution to aspartic acid (Fig. 5D) or glutamic acid (Fig. 5E) changes the distance to 4.9 or 4.5 Å, respectively. These large-distance alterations (13 and 17 Å) can be rationalized by the expected repulsion between negatively charged D/E residues and D41. Negative charge at position 211 also changes the electrostatic balance status around the hydrophobic pocket from neutral in PCNA WT (Fig. 5F) and PCNA-YF (Fig. 5G) to negative in PCNA YD (Fig. 5H) and PCNA YE (Fig. 5I). These putative structural changes could explain the influence of Y211p, Y211D, or Y211E on interactions with MutSα or MutSβ.
Phosphorylated PCNA Is Mutagenic During DNA Synthesis.
PCNA is an essential cofactor for processive DNA synthesis during MMR and DNA replication. Therefore, an in vitro assay for efficiency and fidelity of DNA synthesis was used to estimate the relatively functional competence of PCNA and PCNA-Y211p during DNA gap-filling synthesis. For this experiment, a standard “blue-white” plaque-screening assay (39) was used to quantify forward mutagenesis in the β-galactosidase α-fragment (Fig. 6A). The results show ∼threefold higher mutation frequency during gap-filling DNA synthesis in Lim2405 nuclear extracts than in HeLa nuclear extracts (Fig. 6B, reactions 1 and 6). Increase in mutation frequency was also observed when gap-filling synthesis was conducted with HeLa extracts in the presence of PCNA-Y211D (reaction 3) or PCNA-Y211E (reaction 4) in comparison with HeLa extract alone (reaction 1), but a slightly reduced mutation frequency was seen when PCNA-WT or PCNA-Y211F was added to HeLa extracts (reactions 2 and 5). The addition of PCNA-Y211F to Lim2405 nuclear extracts reduced the mutation frequency (reaction 10), but the addition of PCNA-Y211D or PCNA-Y211E did not result in reduced mutability in Lim2405 (reactions 8 and 9). Unexpectedly, WT nonphosphorylated PCNA (PCNA-WT, reaction 7) was found to induce a mutation frequency that is higher than that induced by PCNA-Y211F. We attributed this to the possibility that PCNA-WT can be phosphorylated at Y211 in Lim2405 extract during the synthesis reaction, leading to the observed increase in mutation frequency. To test this hypothesis, in vitro phosphorylation experiments were performed. First, activated EGFR was able to phosphorylate PCNA-WT, but not PCNA-Y211F (Fig. 6C). Second, PCNA-WT and PCNA-Y211F were incubated with nuclear extract of Lim2405 or HeLa, followed by Ni-NTA agarose pull-down (all recombinant PCNAs contained a His-tag) and Western blot analysis to detect PCNA phosphorylation. Regardless of extracts used, there was a slow-migrated band (indicated by a red arrow) in reactions with PCNA-WT (Fig. 6D, lanes 3 and 5), but not in those with PCNA-Y211F (Fig. 6D, lanes 4 and 6), and this band is much more abundant in the Lim2405 reaction than in the HeLa reaction. Because this band appears to have the same mobility as the EGFR-phosphorylated PCNA (Fig. 6D, lane 1), it is likely the Y211-phosphorylated PCNA, supporting the idea that PCNA-WT can be phosphorylated in Lim2405 extracts during gap-filling DNA synthesis. However, a fast-migrated band (blue star) was found in all nuclear extract reactions, but not in EGFR-catalyzed reaction. The fast-migrated species is probably a tyrosine-phosphorylated factor from extracts that interacts with PCNA.
Fig. 6.

Fidelity of gap-filling DNA synthesis in the presence of the indicated PCNA isoforms. (A) Schematic diagram of DNA gap-filling synthesis and the “blue-white” plaque mutagenesis screening. (B) Mutation frequencies during gap filling by HeLa or Lim2405 nuclear extracts in the presence or absence of the indicated PCNA isoforms. (C) In vitro phosphorylation of PCNA by EGFR. (D) In vitro phosphorylation of PCNA by HeLa or Lim2405 extracts.
To determine the mutagenic nature induced by Y211-phosphorylated PCNA, we sequenced a lacZα fragment of at least 20 white plaques derived from gap-filling synthesis by nuclear extracts of Lim2405 and HeLa or HeLa extract supplemented with PCNA-Y211D or PCNA-Y211E. The results reveal various base-substitutions, nucleotide deletions, and clustered mutations in all reactions (Figs. S2 and S3). However, much more clustered mutations were observed in Lim2405 extract-catalyzed reactions than in HeLa extract-catalyzed reactions (Fig. S2), suggesting Y211-phosphorylated PCNA promotes clustered mutations during DNA synthesis. Interestingly, although both PCNA-Y211D and PCNA-Y211E enhanced mutagenic activity of HeLa extract during gap-filling synthesis (Fig. 6B), PCNA-Y211E appeared to induce more clustered mutations than PCNA-Y211D (Fig. S3). The molecular mechanism underlying this discrepancy remains to be investigated.
It is worth mentioning that we detected a number of clustered mutations near the PstI site, the boundary of nick ligation and strand displacement synthesis after gap-filling, and that the mutation frequency of these clustered mutations in HeLa extracts is at least twofold lower than in Lime2405 extract or HeLa extract supplemented with PCNA-Y211E (Figs. S2 and S3). There was essentially no difference in ligation efficiency after gap-filling between reactions with HeLa and Lim2405 extracts (Fig. S4). Whether or not these clustered mutations are caused by abnormal strand-displacement synthesis directly or indirectly related to PCNA phosphorylation remains to be studied. Nevertheless, the results shown here suggest that PCNA-Y211p supports in vitro high-fidelity DNA synthesis less well than unphosphorylated PCNA.
Discussion
PCNA orchestrates essentially all metabolic reactions at the replication fork, including DNA replication, MMR, and DNA translesion synthesis (37, 40). PCNA roles in these reactions appear to be regulated through posttranslational modifications such as ubiquitination, sumoylation, and phosphorylation. Although the role of PCNA ubiquitination and sumoylation is relatively well understood (40), little is known about the functional significance of PCNA phosphorylation.
In this study, we show that PCNA-Y211p inhibits MMR and induces nucleotide misincorporation during DNA synthesis. It is therefore predicted that tumor cells with a high level of PCNA-Y211p are likely displaying a mutator phenotype, despite that fact that they normally express all MMR proteins. Given that previous studies have always linked mutations in MMR genes and sequences encoding the proofreading activity of DNA polymerases with mutator phenotype and cancer predisposition (2, 14, 18), the present study has identified a novel mechanism, that is, posttranslational modifications of MMR proteins, for promoting genome instability and cancer progression.
Overexpression and/or constitutive activation of receptor family tyrosine kinases, including EGFR (21), are highly correlated with cancer progression. Phosphorylation of PCNA by EGFR stimulates cell proliferation, possibly by stabilizing PCNA (23). However, whether or not EGFR-catalyzed PCNA phosphorylation contributes to EGFR-dependent tumorigenesis is unknown. Surprisingly, findings presented here suggest that DNA synthesis in cells that overexpress EGFR may be error-prone (Fig. 6). This is consistent with observations that cancer cells often display a mutator phenotype, and this further promotes oncogenesis, leading to tumor progression and metastasis. The findings presented here also provide evidence that posttranslational modification of DNA repair proteins or accessory factors may frequently contribute to genetic instability in tumor cells.
We provide strong evidence that PCNA-Y211p inhibits MMR at the initiation step, possibly by altering the structure of the hydrophobic pocket of PCNA (Fig. 5) and weakening the interaction between PCNA and MutSα/MutSβ. It remains unclear how PCNA-Y211p promotes misincorporation during DNA synthesis. It is interesting to note that monoubiquitinated PCNA recruits error-prone translesion synthesis (TLS) DNA polymerases (41). This suggests that PCNA-Y211p might have enhanced affinity for one or more TLS DNA polymerases, relative to nonphosphorylated PCNA. Recent studies revealed that DNA polymerase ζ promotes clustered mutations (42), a phenomenon observed in nuclear extracts containing high levels of PCNA-Y211p or its equivalents (Figs. S2 and S3). However, whether or not polymerase ζ and/or other TLS polymerases are involved in PCNA-Y211p-catalyzed mutagenesis remains to be investigated.
It is interesting to note that only ∼15–20% of PCNA molecules are phosphorylated in cells with high levels of EGFR. This raises a question as to whether all three PCNA subunits or only one of the three subunits is phosphorylated. Our data support the latter. First, the limited amount of phosphorylated PCNA is capable of inhibiting MMR activity by 50% in Lim2405, DLD-1, and EGF-treated HeLa cells (Fig. 2); if all three subunits of a PCNA complex are phosphorylated, phosphorylated PCNA could only account for, at most, ∼7% of PCNA trimers. If this were the case, it is unlikely that phosphorylated trimers would compete effectively with nonphosphorylated PCNA trimers to inhibit MMR. However, if only one subunit of a PCNA trimer needs to be phosphorylated to impair its function in MMR, we estimate that 45–60% of PCNA trimers might fail to support MMR. Second, ∼30% of PCNA monomers were phosphorylated after incubation with EGFR in vitro (Fig. 6C). This is consistent with the idea that one phosphorylated PCNA monomer per trimer is sufficient to inhibit the function of the trimer in MMR. Therefore, it will be important to perform detailed structure/function studies of posttranslationally modified PCNA and to determine the stoichiometry of PCNA phosphorylation in the trimeric state to fully understand the implications and significance of the present study.
Materials and Methods
Cell Culture.
Unless specified otherwise, cells used in this study were grown in Roswell Park Memorial Institute 1640 with 5% (vol/vol) FBS and 4 mM l-glutamine at 37 °C in a humidified atmosphere with 5% (vol/vol) CO2. For Lim2405 cells, 25 U/L insulin, 10 µM α-thioglycerol, and 1 μg/mL hydrocortisone were added in the culture medium. To obtain a HCT116 line stably expressing exogenous EGFR, HCT116 cells were infected with a human 293T cell-derived retrovirus carrying the EGFR gene (43), and clones expressing the exogenous EGFR, designated H6-EGFR, were selected by virtue of their tolerance of 10 µg/mL puromycin.
MMR Assay and Analysis of Incision/Excision Intermediates.
The MMR assay was performed in a 20-μL reaction containing 30 fmol mismatched DNA containing a 3′ nick, 75 μg nuclear extract in the presence or absence of 1 µM individual PCNA isoforms, 1 µM p21c, or 100 nM MutLα. DNA samples were recovered and digested with restriction enzymes to score for mismatch removal, as described (12, 25). Mismatch-provoked incision/excision reactions were conducted by omitting dNTPs from the standard MMR assay. For reconstituted reactions, minimum required proteins MutSα (100 nM), MutLα (100 nM), RPA (40 nM), RFC (10 nM), and PCNA (330 nM homotrimer) were used. DNA samples recovered were digested with PstI and BglI and fractionated by 6% (wt/vol) denaturing polyacrylamide gel electrophoresis, followed by Southern blot analysis using a 32P-labeled oligonucleotide probe (12). Reaction products were visualized by a phosphor imager.
Microscopy and Immunofluorescence Analysis.
Immunofluorescence analysis was performed as described previously (19). Cells were fixed using 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 in PBS. Fixed cells were blocked with 5% (wt/vol) BSA and incubated with primary antibody overnight at 4 °C, followed by incubation with a secondary antibody for 1 h at room temperature. Immunofluorescence images were obtained using an FV-1000 Olympus confocal scanner laser microscopy system.
DNA Gap-Filling Synthesis and Mutagenesis Assays.
The mutagenesis assay was performed essentially as described previously (39). A gapped DNA substrate (283-nt gap) was derived from bacterial phage M13mp18, with the gap carrying a segment of the lacZ gene sequence coding for the α fragment of β-galactosidase. Thus, misincorporation during DNA synthesis leads to a dysfunctional α fragment, which fails to complement β-galactosidase in the host strain on transfection. The resulting mutant plaques were determined by a blue-white color screening (39).
Coimmunoprecipitation, Western Blot, and Far-Western Blot Analyses.
Whole lysates or nuclear extracts were incubated with a PCNA Y211-phosphorylation-specific antibody (Bethyl Laboratories), and the precipitates were subjected to Western blot analysis using a PCNA antibody (Santa Cruz). For far-Western blot analysis, purified nonphosphorylated and phosphorylation-mimicking PCNAs were spotted onto a nitrocellulose membrane and incubated with 1 µg MutSα, MutSβ, or MutLα. The membrane was immunoblotted with antibodies against MSH2, MSH3, and MLH1 (BD Pharmingen). The proteins were detected using enhanced chemiluminescence, followed by autoradiography.
In Vitro Kinase Assay.
PCNA was incubated at room temperature with 50 ng GST-EGFR (Millipore) or 200 µg nuclear extracts in kinase buffer containing 60 mM Hepes (pH 7.5), 5 mM MgCl2, 5 mM MnCl2, 3 µM Na3VO4, 1.25 mM DTT, and 200 µM ATP. Reactions were resolved by SDS PAGE, followed Western blot analysis using an antibody specific to phospho-tyrosine (ECM Bioscience).
Supplementary Material
Acknowledgments
We thank Wei Yang and Hong Ling for help in PCNA structure analysis and Mark Ensor for comments on the manuscript. This work is supported in part by grants from the National Cancer Institute (CA167181), the National Natural Science Foundation of China (31370766 and 31461143005), and National Cancer Institute Training Grant T32 CA165990 (to J.O.). G.-M.L. holds the James-Gardner Chair in Cancer Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 5556.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417711112/-/DCSupplemental.
References
- 1.Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev. 1996;10(12):1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- 2.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 3.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 4.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 5.Holmes J, Jr, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci USA. 1990;87(15):5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowen N, et al. Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins. Proc Natl Acad Sci USA. 2013;110(46):18472–18477. doi: 10.1073/pnas.1318971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126(2):297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 8.Lau PJ, Kolodner RD. Transfer of the MSH2.MSH6 complex from proliferating cell nuclear antigen to mispaired bases in DNA. J Biol Chem. 2003;278(1):14–17. doi: 10.1074/jbc.C200627200. [DOI] [PubMed] [Google Scholar]
- 9.Gu L, Hong Y, McCulloch S, Watanabe H, Li GM. ATP-dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res. 1998;26(5):1173–1178. doi: 10.1093/nar/26.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Umar A, et al. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996;87(1):65–73. doi: 10.1016/s0092-8674(00)81323-9. [DOI] [PubMed] [Google Scholar]
- 11.Goellner EM, et al. PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol Cell. 2014;55(2):291–304. doi: 10.1016/j.molcel.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, et al. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122(5):693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 13.Ramilo C, et al. Partial reconstitution of human DNA mismatch repair in vitro: Characterization of the role of human replication protein A. Mol Cell Biol. 2002;22(7):2037–2046. doi: 10.1128/MCB.22.7.2037-2046.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev. 1995;5(3):382–395. doi: 10.1016/0959-437x(95)80055-7. [DOI] [PubMed] [Google Scholar]
- 15.Herman JG, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95(12):6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kane MF, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57(5):808–811. [PubMed] [Google Scholar]
- 17.Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21(6):1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 18.Palles C, et al. CORGI Consortium WGS500 Consortium Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45(2):136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell. 2013;153(3):590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan Z, et al. An A13 repeat within the 3′-untranslated region of epidermal growth factor receptor (EGFR) is frequently mutated in microsatellite instability colon cancers and is associated with increased EGFR expression. Cancer Res. 2009;69(19):7811–7818. doi: 10.1158/0008-5472.CAN-09-0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–S15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 22.Lin SY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3(9):802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 23.Wang SC, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8(12):1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- 24.Zhao H, et al. Targeting tyrosine phosphorylation of PCNA inhibits prostate cancer growth. Mol Cancer Ther. 2011;10(1):29–36. doi: 10.1158/1535-7163.MCT-10-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li GM, Modrich P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci USA. 1995;92(6):1950–1954. doi: 10.1073/pnas.92.6.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drummond JT, Li GM, Longley MJ, Modrich P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268(5219):1909–1912. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- 27.Carpenter G, Cohen S. Epidermal growth factor. Annu Rev Biochem. 1979;48:193–216. doi: 10.1146/annurev.bi.48.070179.001205. [DOI] [PubMed] [Google Scholar]
- 28.Guo S, et al. Differential requirement for proliferating cell nuclear antigen in 5′ and 3′ nick-directed excision in human mismatch repair. J Biol Chem. 2004;279(17):16912–16917. doi: 10.1074/jbc.M313213200. [DOI] [PubMed] [Google Scholar]
- 29.Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371(6497):534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- 30.Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369(6481):574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 31.Clark AB, Valle F, Drotschmann K, Gary RK, Kunkel TA. Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J Biol Chem. 2000;275(47):36498–36501. doi: 10.1074/jbc.C000513200. [DOI] [PubMed] [Google Scholar]
- 32.Flores-Rozas H, Clark D, Kolodner RD. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet. 2000;26(3):375–378. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- 33.Kleczkowska HE, Marra G, Lettieri T, Jiricny J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001;15(6):724–736. doi: 10.1101/gad.191201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SD, Alani E. Analysis of interactions between mismatch repair initiation factors and the replication processivity factor PCNA. J Mol Biol. 2006;355(2):175–184. doi: 10.1016/j.jmb.2005.10.059. [DOI] [PubMed] [Google Scholar]
- 35.Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79(7):1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 36.Kontopidis G, et al. Structural and biochemical studies of human proliferating cell nuclear antigen complexes provide a rationale for cyclin association and inhibitor design. Proc Natl Acad Sci USA. 2005;102(6):1871–1876. doi: 10.1073/pnas.0406540102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129(4):665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Zhang G, Gibbs E, Kelman Z, O’Donnell M, Hurwitz J. Studies on the interactions between human replication factor C and human proliferating cell nuclear antigen. Proc Natl Acad Sci USA. 1999;96(5):1869–1874. doi: 10.1073/pnas.96.5.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kunkel TA. The mutational specificity of DNA polymerase-beta during in vitro DNA synthesis. Production of frameshift, base substitution, and deletion mutations. J Biol Chem. 1985;260(9):5787–5796. [PubMed] [Google Scholar]
- 40.Mailand N, Gibbs-Seymour I, Bekker-Jensen S. Regulation of PCNA-protein interactions for genome stability. Nat Rev Mol Cell Biol. 2013;14(5):269–282. doi: 10.1038/nrm3562. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann AR. Ubiquitin-family modifications in the replication of DNA damage. FEBS Lett. 2011;585(18):2772–2779. doi: 10.1016/j.febslet.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Stone JE, Lujan SA, Kunkel TA, Kunkel TA. DNA polymerase zeta generates clustered mutations during bypass of endogenous DNA lesions in Saccharomyces cerevisiae. Environ Mol Mutagen. 2012;53(9):777–786. doi: 10.1002/em.21728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ory DS, Neugeboren BA, Mulligan RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA. 1996;93(21):11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.