

Abstract
The remodeling of axonal circuits after injury requires the formation of new synaptic contacts to enable functional recovery. Which molecular signals initiate such axonal and synaptic reorganisation in the adult central nervous system is currently unknown. Here, we identify FGF22 as a key regulator of circuit remodeling in the injured spinal cord. We show that FGF22 is produced by spinal relay neurons, while its main receptors FGFR1 and FGFR2 are expressed by cortical projection neurons. FGF22 deficiency or the targeted deletion of FGFR1 and FGFR2 in the hindlimb motor cortex limits the formation of new synapses between corticospinal collaterals and relay neurons, delays their molecular maturation, and impedes functional recovery in a mouse model of spinal cord injury. These results establish FGF22 as a synaptogenic mediator in the adult nervous system and a crucial regulator of synapse formation and maturation during post-injury remodeling in the spinal cord.
Keywords: axonal remodeling, fibroblast growth factor, functional recovery, spinal cord injury, synapse formation
Introduction
Incomplete lesions of the spinal cord can be followed by substantial functional recovery in both human patients and rodent models. This recovery is mediated by the remodeling of spinal and supraspinal axonal circuits (Weidner et al, 2001; Bareyre et al, 2004; Girgis et al, 2007; Courtine et al, 2008; van den Brand et al, 2012; Shah et al, 2013; Zörner et al, 2014). The hindlimb corticospinal tract (CST), for example, responds to a thoracic transection with the de novo formation of intraspinal detour circuits that circumvent the lesion site and re-establish a functional connection between the motor cortex and the lumbar spinal cord (Bareyre et al, 2004; Lang et al, 2012). A key step in the formation of these detour circuits is the establishment of synaptic contacts between newly formed CST collaterals that enter the cervical gray matter and long propriospinal neurons (LPSN) that are located in the cervical spinal cord and act as relays to lumbar motor circuits. Although the functional importance of this and similar detour circuits has been well established over the recent years (Bareyre et al, 2004; Courtine et al, 2008; van den Brand et al, 2012), it is currently unclear how the formation of these circuits is regulated and which molecular signals initiate the establishment of new synaptic contacts in the injured adult central nervous system (CNS).
In the developing nervous system, a number of molecules that can promote synapse formation have been identified (Sanes & Lichtman, 2001; Williams et al, 2010; Siddiqui & Craig, 2011). Among these, the family of the fibroblast growth factors and their receptors has emerged as important regulators of presynaptic differentiation (Umemori et al, 2004; Stevens et al, 2010; Terauchi et al, 2010). One member in particular, FGF22—a target-derived secreted factor that can act as a presynaptic organizer—is crucial for the establishment of excitatory synapses as shown for CA3 pyramidal cells in the developing hippocampus (Terauchi et al, 2010). Whether such developmental signaling pathways remain operational in adulthood is currently not fully understood.
Here, we now show that FGF22 and its receptors FGFR1 and FGFR2 are constitutively expressed in the adult CNS. Genetic interference with FGF22 signaling either on the level of the ligand or its receptors limits the formation and maturation of new synapses in the injured spinal cord and results in deficient detour circuit formation and attenuated functional recovery. These findings establish FGF22 as an important endogenous regulator of synaptic plasticity and circuit remodeling in the adult nervous system.
Results
Impaired post-injury synapse formation and circuit remodeling in FGF22-deficient mice
To investigate whether FGF22 signaling can regulate synapse formation during axonal remodeling after spinal cord injury, we first assessed whether and where FGF22 is expressed in the spinal cord of adult mice using in situ hybridization and single-cell laser microdissection followed by quantitative PCR analysis. Our results showed that FGF22 mRNA is present in spinal interneurons including in a large proportion of long propriospinal neurons both constitutively and at different timepoints after a mid-thoracic spinal cord injury (Fig1A–E). To investigate the role of spinal FGF22 expression during post-injury remodeling (Fig2A), we performed a bilateral dorsal hemisection of the spinal cord at T8 in FGF22-deficient mice (Terauchi et al, 2010) and age-matched FGF22-competent wild-type mice and traced the hindlimb portion of the corticospinal tract by stereotactic injection of the anterograde tracer biotinylated dextran amine (BDA). Unlesioned FGF22-deficient mice showed a normal density of cortical layer V neurons and an unaltered lumbar and cervical projection pattern of the hindlimb CST (Supplementary Fig S1). At 3 weeks after spinal cord injury, however, FGF22 deficiency diminished the density of boutons on newly formed CST collaterals that enter the gray matter of the cervical spinal cord as part of the detour circuit formation process (Fig2D and E). As a result, the proportion of long propriospinal relay neurons in the cervical spinal cord that are contacted by these CST collaterals was significantly reduced (Fig2F and G). In contrast, neither the overall number of CST collaterals that exited the main CST in the cervical spinal cord nor the length of these collaterals was altered in FGF22-deficient mice (Fig2B and C and Supplementary Fig S2).
Figure 1. FGF22 is expressed by interneurons in the healthy and injured adult spinal cord.
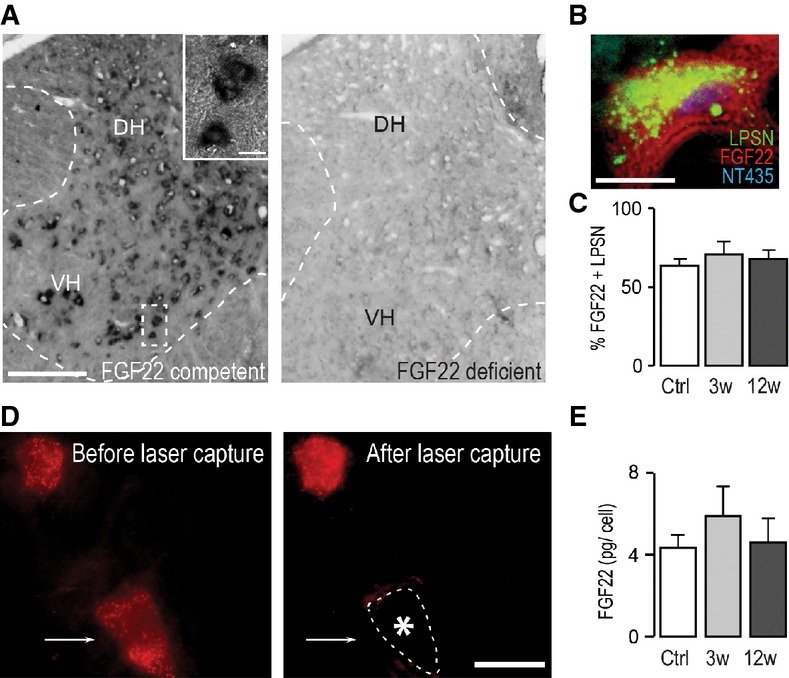
- In situ hybridization of FGF22 mRNA in the spinal cord of FGF22-competent (left panel) and FGF22-deficient (right panel) mice (DH: dorsal horn; VH: ventral horn). Scale bar equals 200 μm (10 μm in inset).
- In situ hybridization of a section of the cervical spinal cord at level C4/C5 showing presence of FGF22 mRNA in a long propriospinal neuron (LPSN) retrogradely traced from T12 (LPSN: green; FGF22 mRNA: red; NeuroTrace 435/455: blue). Scale bar equals 20 μm.
- Quantification of the percentage of LPSN showing a FGF22 in situ signal in unlesioned mice (‘Ctrl’) and in mice at 3 (‘3w’) and 12 (‘12w’) weeks after spinal cord injury (n > 30 LPSN from 2 to 3 animals per group). Mean ± SEM. No significant differences between the groups were detected (ANOVA followed by Tukey tests).
- Images of LPSN (red) before (left panel) and after (right panel) laser microdissection of a single neuron (arrow, asterisk and dotted line in right panel indicate previous location of the microdissected neuron). Scale bar equals 40 μm.
- Quantification of the single-cell PCR analysis of FGF22 mRNA expression in LPSN dissected from unlesioned mice (‘Ctrl’) and from mice at 3 (‘3w’) and 12 (‘12w’) weeks after spinal cord injury (n = 7–8 LPSN per group). Mean ± SEM. No significant differences between the groups were detected (ANOVA followed by Tukey tests).
Figure 2. FGF22 deficiency impairs bouton formation and circuit remodeling after spinal cord injury.
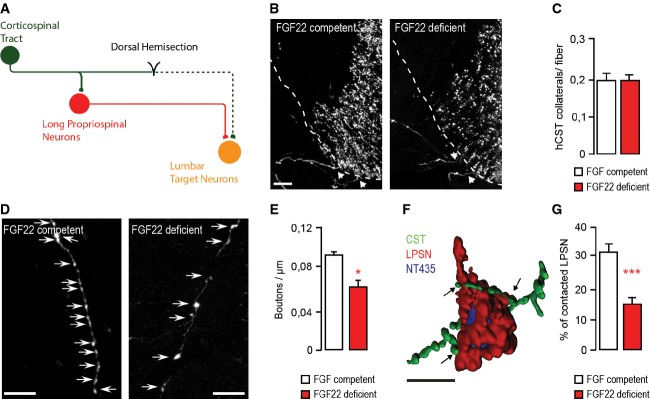
- Schematic representation of CST detour circuit formation following a mid-thoracic dorsal bilateral hemisection of the spinal cord.
- Confocal images of hindlimb CST collaterals exiting the main CST tract (arrows) in the cervical spinal cord 3 weeks following a T8 dorsal bilateral hemisection in FGF22-competent (left panel) and FGF22-deficient (right panel) mice. Scale bar equals 40 μm.
- Quantification of the number of exiting hindlimb CST collaterals per labeled hindlimb CST fiber at 3 weeks following T8 dorsal bilateral hemisection in FGF22-competent and FGF22-deficient mice (n = 8 animals per group). Mean ± SEM. No significant differences between the groups were detected (unpaired two-tailed t-test).
- Confocal images showing putative synaptic boutons (arrows) on newly formed cervical hindlimb CST collaterals at 3 weeks following spinal cord injury in FGF22-competent (left panel) and FGF22-deficient (right panel) mice. Scale bar equals 20 μm.
- Quantification of bouton density on newly formed cervical hindlimb CST collaterals in FGF22-competent and FGF22-deficient mice (n = 8 animals per group) at 3 weeks after injury. Mean ± SEM. *P = 0.0244 (unpaired two-tailed t-test).
- 3D Rendering of a confocal image stack that illustrates putative synaptic contacts between CST collaterals (green) and LPSN (red) counterstained with NeuroTrace 435/455 (blue). Scale bar equals 30 μm.
- Quantification of the percentage of LPSN contacted by cervical hindlimb CST collaterals in FGF22-competent and FGF22-deficient mice at 3 weeks after injury (n = 8 animals per group). Mean ± SEM. ***P = 0.0001 (unpaired two-tailed t-test).
FGFR1 and FGFR2 mediate the effects of FGF22 on synapse formation in the injured spinal cord
To better understand which receptors communicate FGF22 signaling to CST collaterals, we first established that the two main receptors of FGF22, FGFR1 and FGFR2 (Singh et al, 2012; Umemori et al, 2004; Zhang et al, 2006), were expressed by cortical projection neurons in adult mice (Fig3A and B). For this purpose, we performed in situ hybridization for FGFR1 and FGFR2 mRNA and retrogradely traced the cortical neurons projecting to the thoracic spinal cord (Fig3C). Our results showed that more than half of these cortical projection neurons express mRNA for FGFR1 and FGFR2 (Fig3D). We then conditionally deleted expression of the FGF22 receptors in the forebrain by crossing floxed FGFR1 and FGFR2 mouse strains (Umemori et al, 2004; Terauchi et al, 2010) to EMX1-Cre mice (Gorski et al, 2002; Bareyre et al, 2005). Deletion of either receptor did not affect mRNA expression of the other (Fig3E). Furthermore, forebrain deletion of FGFR1 or FGFR2 did not alter the density of layer V neurons in the cortex or the development of a mature hindlimb CST projection pattern in unlesioned mice (Supplementary Figs S3 and S4). However, when we performed a thoracic dorsal bilateral hemisection of the spinal cord in these mice, we observed that the deletion of either FGFR1 or FGFR2 reduced the density of boutons on newly formed CST collaterals in the cervical spinal cord at 3 weeks after injury (Fig4D and E). A similar reduction of CST bouton density in the cervical spinal cord was observed in double-floxed mice, in which both FGFR1 and FGFR2 were depleted in the hindlimb motor cortex by stereotactic injection of a recombinant adeno-associated virus expressing the Cre recombinase (rAAV-GFP-Ires-Cre, Fig4D and E). Notably, while the length of individual cervical CST collaterals was similar in all groups (Supplementary Fig S2), reduced synapse formation after injury was compensated by sprouting of additional CST collaterals, if either FGFR1 or FGFR2 alone was depleted but not if both receptors were missing (Fig4A–C). As a result, only mice, in which both FGFR1 and FGFR2 were co-deleted, showed an impaired formation of intraspinal detour circuits (Fig4F), similar to the one observed in FGF22-deficient mice (Fig2G).
Figure 3. FGFR1 and FGFR2 are expressed in adult cortical projection neurons.
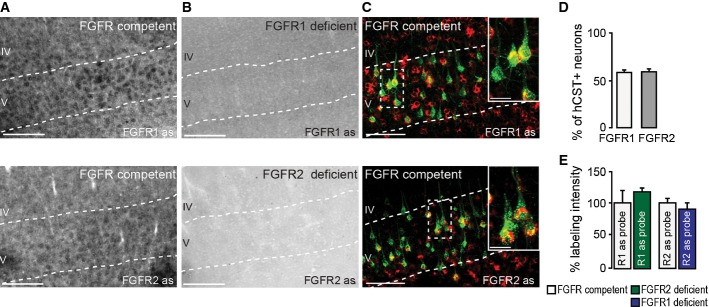
- In situ hybridization of FGFR1 (top) and FGFR2 (bottom) mRNA in FGFR-competent mice. Scale bar equals 200 μm.
- In situ hybridization of FGFR1 (top) and FGFR2 (bottom) mRNA in forebrain FGFR1 (top)- and FGFR2 (bottom)-deficient mice. Scale bar equals 200 μm.
- Retrograde labeling of CST projection neurons with dextran conjugated with Texas Red® (green) shows that many of these neurons express FGFR1 (red, top) and FGFR2 mRNA (red, bottom; insets in top and bottom panels are twofold magnification of boxed areas). Scale bar equals 100 μm (20 μm in insets).
- Quantification of the percentage of CST projection neurons in layer V of the cortex expressing FGFR1 (white bar) and FGFR2 mRNA (gray bar; n = 5 animals per group). Mean ± SEM.
- Quantification of the intensity of the in situ signal for FGFR1 mRNA in FGFR2-deficient mice (green bar) and FGFR2 mRNA in FGFR1-deficient mice (blue bar) normalized to the signal intensity measured in the respective FGFR-competent control group (white bars; n = 3 animals per group). Mean ± SEM. No significant differences between the groups were detected (unpaired two-tailed t-tests).
Figure 4. Deletion of forebrain FGFR1 and FGFR2 expression impairs bouton formation and circuit remodeling after spinal cord injury.

- Confocal images of hindlimb CST collaterals exiting the main CST tract (arrows) in the cervical spinal cord 3 weeks following T8 dorsal bilateral hemisection in FGFR-competent (left panel), forebrain FGFR1-deficient (middle panel), and forebrain FGFR2-deficient (right panel) mice. Scale bar equals 40 μm.
- Confocal images of hindlimb CST collaterals exiting the main CST tract (arrows) in the cervical spinal cord 3 weeks following T8 dorsal bilateral hemisection in FGFR-competent (left panel) and hindlimb motor cortex FGFR1/FGFR2 double-deficient (right panel) mice. Scale bar equals 40 μm.
- Quantification of the number of exiting hindlimb CST collaterals at 3 weeks following T8 dorsal bilateral hemisection in forebrain FGFR single-deficient mice, hindlimb motor cortex FGFR1/FGFR2 double-deficient mice, and the corresponding FGFR-competent control mice (n = 6–15 animals per group). Mean ± SEM. *P < 0.05; ***P < 0.001 (ANOVA followed by Tukey tests for FGFR-competent versus FGFR single-deficient mice). No significant differences were found between FGFR-competent and FGFR1/FGFR2 double-deficient mice (unpaired two-tailed t-tests).
- Confocal images showing putative synaptic boutons (arrows) on newly formed cervical hindlimb CST collaterals at 3 weeks following spinal cord injury in FGFR-competent (left panel), forebrain FGFR1-deficient (second panel from left), forebrain FGFR2-deficient (second panel from right), and hindlimb motor cortex FGFR1/FGFR2 double-deficient (right panel) mice. Scale bar equals 20 μm.
- Quantification of the bouton density on newly formed cervical hindlimb CST collaterals in FGFR-competent, forebrain single FGFR-deficient, and hindlimb motor cortex FGFR1/FGFR2 double-deficient mice (n = 6–16 animals per group). Mean ± SEM. *P < 0.05, **P < 0.01 (ANOVA followed by Tukey tests in case of multiple group comparisons, e.g. FGFR-competent versus FGFR single-deficient mice). **P = 0.0028 (unpaired two-tailed t-tests for comparisons of FGFR-competent versus FGFR1/FGFR2 double-deficient mice).
- Quantification of the percentage of LPSN contacted by hindlimb CST collaterals in FGFR-competent, forebrain single FGFR-deficient, and hindlimb motor cortex FGFR1/FGFR2 double-deficient mice (n = 6–16 animals per group). ***P < 0.0001 (unpaired two-tailed t-tests for comparisons of controls versus FGFR1/FGFR2 double-deficient mice). No significant differences were found between FGFR-competent and FGFR single-deficient mice (ANOVA followed by Tukey tests).
Genetic disruption of FGF22 signaling delays molecular maturation of synapses after spinal cord injury
To determine whether FGF22 signaling regulates not only the number of new boutons that can be formed but also their molecular composition, we studied the expression of an active zone protein—bassoon—and a synaptic vesicle-associated protein—synapsin—as indicators of synapse maturation (Zhai et al, 2001; Shapira et al, 2003; Micheva et al, 2010; Lang et al, 2012). For this purpose, we used confocal microscopy to determine the proportion of boutons on newly formed cervical CST collaterals that were immunoreactive for synapsin and bassoon at different timepoints after a thoracic dorsal bilateral hemisection of the spinal cord. Our results showed that forebrain deletion of either FGFR1 or FGFR2 alone led to a moderate delay in synapse maturation that was most obvious at 3 weeks after injury and primarily altered the presence of synapsin (Fig5A–D). Furthermore, complete interruption of FGF22 signaling either by conditional deletion of both receptors in the hindlimb motor cortex or by constitutive FGF22 deficiency induced protracted synapse maturation defects that affected the recruitment of both active zone and synaptic vesicle-associated proteins and persisted for at least 12 weeks after injury (Fig5A–D).
Figure 5. Deletion of FGF22 or its receptors delays synapse maturation following spinal cord injury.

- Confocal image of synaptic contacts (arrows) between a CST collateral (green) and a LPSN (blue) that show bassoon immunoreactivity (red). Right images are magnification (two and a half-fold) of the area boxed on the left. Scale bar equals 25 μm.
- Quantification of the percentage of boutons on cervical hindlimb CST collaterals that are immunoreactive for bassoon at 3 weeks (left) and 12 weeks (right) after spinal cord injury in FGF22-deficient, forebrain FGFR1-deficient, forebrain FGFR2-deficient and hindlimb motor cortex FGFR1/FGFR2 double-deficient mice compared to the respective FGF22- and FGFR-competent control mice. A minimum of 100 boutons per mouse were evaluated for 3 mice per group. ***P < 0.0001 FGFR-competent versus FGFR1/FGFR2 double-deficient mice at 3 weeks (unpaired two-tailed t-tests), **P = 0.002 FGF-competent versus FGF22 deficient at 3 weeks (unpaired two-tailed t-tests), *P < 0.05 FGFR-competent versus FGFR2 single-deficient mice at 12 weeks (one-way ANOVA followed by Tukey tests), ***P = 0.001 FGFR-competent versus FGFR1/FGFR2 double-deficient mice at 12 weeks (unpaired two-tailed t-tests).
- Confocal image of synaptic contacts (arrows) between a CST collateral (green) and a LPSN (blue) that show synapsin I immunoreactivity (red). Right images are magnification (two and a half-fold) of the area boxed on the left. Scale bar equals 25 μm.
- Quantification of the percentage of boutons on cervical hindlimb CST collaterals that are immunoreactive for synapsin I at 3 weeks (left) and 12 weeks (right) after spinal cord injury in FGF22-deficient, forebrain FGFR1-deficient, forebrain FGFR2-deficient and hindlimb motor cortex FGFR1/FGFR2 double-deficient mice compared to the respective FGF22 and FGFR-competent control mice. A minimum of 100 boutons per mouse were evaluated for 3 mice per group. ***P < 0.001 FGFR-competent versus FGFR1 and FGFR2 single-deficient mice at 3 weeks (one-way ANOVA followed by Tukey tests). ***P = 0.0001 FGFR-competent mice versus FGFR1/FGFR2 double-deficient mice at 3 weeks (unpaired two-tailed t-tests), P = 0.00004 FGF-competent mice versus FGF22-deficient mice at 3 weeks (unpaired two-tailed t-tests). ***P < 0.001 FGFR-competent versus FGFR2 single-deficient mice (one-way ANOVA followed by Tukey tests). ***P = 0.0006 FGFR-competent mice versus FGFR1/FGFR2 double-deficient mice at 12 weeks (unpaired two-tailed t-tests), *P = 0.027 FGF-competent mice versus FGF22-deficient mice at 12 weeks (unpaired two-tailed t-tests).
Disturbed FGF22 signaling limits recovery of function after spinal cord injury
Finally, we wanted to understand whether the impaired formation and maturation of new synapses would interfere with the spontaneous recovery of CST function that follows an incomplete spinal cord injury (Bareyre et al, 2004). To assess this, we performed thoracic dorsal bilateral hemisections of the spinal cord and followed the recovery of CST function using behavioral testing paradigms such as the ‘ladder rung test’ (Metz & Whishaw, 2002) and the ‘catwalk analysis’ (Hamers et al, 2006) in FGF22-deficient mice and in mice in which FGFR1 and FGFR2 were conditionally deleted in the hindlimb motor cortex as well as in the respective FGF- and FGFR-competent control mice. While neither of these genetic manipulations altered the size of the thoracic lesions (0.37 ±0.047 mm3 in FGF22-deficient versus 0.37 ± 0.055 mm3 in FGF22-competent control mice and 0.35 ± 0.03 mm3 in hindlimb motor cortex FGFR1/FGFR2-deficient mice versus 0.32 ± 0.054 mm3 in the FGFR-competent controls; n = 5–7 mice per group), both deletion of FGF22 or co-deletion of FGFR1 and FGFR2 in the hindlimb motor cortex impeded functional recovery after injury. More pronounced deficits remained in mice with impaired FGF22 signaling for at least 3 weeks after injury in both the ‘regular walk’ and ‘irregular walk’ paradigms of the ladder rung test as well as measured by the ‘paw angle body axis’ parameter in the catwalk analysis (Fig6A–D). In contrast, deletion of either FGFR1 or FGFR2 alone did not alter functional recovery likely due to the compensatory increase in CST sprouting that prevented deficits in detour circuit formation under these conditions (Supplementary Fig S5). Taken together, the results of our behavioral analysis indicate that intact FGF22 signaling is required for the timely recovery of motor function after spinal cord injury.
Figure 6. Genetic disruption of FGF22 signaling impedes functional recovery following spinal cord injury.

- Image of a spinal cord injured mouse performing the irregular ladder rung test that assesses recovery of CST function. Scale bar equals 1 cm.
- Quantification of the functional recovery in the ladder rung test (regular walk, left panels; irregular walk, right panels) in FGF22-deficient (top panels, red bars), and hindlimb motor cortex FGFR1/FGFR2 double-deficient (bottom panels, blue bars) mice and the respective FGF22- and FGFR-competent control mice (white bars) before (‘Pre’) and 2 (‘2 wks’) and 3 (‘3 wks’) weeks after a spinal cord injury (n = 7–10 animals per group). *P < 0.05, **P < 0.01 (repeated-measure ANOVA followed by Bonferroni tests).
- Image of a spinal cord injured mouse walking on the catwalk that assesses locomotor recovery. Illumination of the paws from below allows to determine the paw angle body axis by relating the axis of the paw (line shown magnified in the right panel) to the axis of the body (line shown in left panel).
- Quantification of the paw angle body axis of the hindpaws in FGF22-deficient (top panel, red bars) and hindlimb motor cortex FGFR1/FGFR2 double-deficient (bottom panel, blue bars) mice and respective FGF22- and FGFR-competent control mice (white bars) before (‘Pre’) and 2 (‘2 wks’) and 3 (‘3 wks’) weeks after a spinal cord injury. Between 13 and 40 steps were analyzed per group and timepoint (n = 10–15 animals per group). **P < 0.01, ***P < 0.001 (two-way ANOVA followed by Bonferroni tests).
Discussion
Our study identifies FGF22 as an important contributor to circuit remodeling in the injured adult spinal cord. We show that FGF22 is constitutively expressed by a large proportion of spinal interneurons including those that can function as relays to lumbar motor circuits. FGF22 that is locally released in the spinal gray matter can then come into contact with growing axon collaterals, for example, those emerging from the transected corticospinal tract. The corresponding projection neurons located in layer V of the motor cortex express the mRNA for the main receptors of FGF22, FGFR1 and FGFR2, and the receptor proteins appear to be present along the CST axons as well as in the CST boutons (as we can show for FGFR2, Supplementary Fig S6). Presence of both of these receptors appears to be required for intact FGF22 signaling to cortical projection neurons as deletion of either one of them results in a decreased density of new CST synapses. However, the alterations induced by the loss of one receptor are mostly temporary and deficits of detour circuit formation and functional recovery can be prevented by compensatory CST sprouting. In contrast, the deletion of both receptors leads to a protracted impairment of synapse formation and maturation after injury that is not compensated by CST sprouting and results in deficient circuit remodeling and attenuated recovery of function. The finding that compensatory sprouting is only observed if either one of the FGF receptors is still present might indicate that FGF22 by itself can promote the formation of additional CST collaterals through either FGFR1 or FGFR2. Alternatively, it is also possible that the induction of this compensatory response requires the presence of at least some mature synapses that are mainly present on the corresponding CST collaterals in the single receptor-deficient mice but not on collaterals that lack both receptors or emerge in FGF22-deficient mice. Taken together, our observations after spinal cord injury indicate that while both FGFR1 and FGFR2 are required for normal FGF22 signaling, loss of one receptor can at least be partially compensated by the presence of its remaining partner. Finally, the finding that FGF22 deficiency and co-deletion of FGFR1 and FGFR2 result in essentially similar changes of synapse formation, circuit remodeling, and functional recovery supports the view that these are indeed the major receptors and ligands involved in this interaction.
It is interesting to note that genetic disruption of FGF22 signaling not only reduces the number of new synapses that can be formed in the injured spinal cord but also alters the molecular composition of those synapses that are formed. These findings are consistent with previous reports that analyzed the role of FGF22 signaling in the developing nervous system. For example, it was shown that genetic inactivation of FGFR2 or FGF22 inhibited presynaptic differentiation in the granule layer of the developing cerebellum as indicated by the reduced immunoreactivity for the vesicle-associated proteins, synapsin and synaptophysin, and the active zone protein, bassoon (Umemori et al, 2004). Likewise, the clustering of synaptic vesicles is significantly decreased in the developing hippocampus of FGF22-deficient mice (Terauchi et al, 2010). Similar deficits of developmental synapse formation have also been described as the result of impaired FGF22 signaling in the dorsal lateral geniculate nucleus (Singh et al, 2012) and at the neuromuscular junction (Fox et al, 2007). It is interesting to note that in contrast to these studies, we did not detect abnormalities of CST development in our analysis. This might indicate that the developmental effects of deficient FGF22 signaling are cell type-specific and thus not observed in CST projection neurons or that initial developmental alterations of CST projections are compensated over time and thus no longer apparent in adult mice that we investigate here. The latter seems to be the more plausible explanation as FGF22 signaling can clearly affect CST projection neurons in the injured adult nervous system as we show here, and our analysis of synapse maturation further suggests that FGF22-related synaptic defects can at least partially recover over time. Finally, it is of course possible that more subtle defects, for example, in the number of vesicles per synapse (Terauchi et al, 2010) still persist but are not captured by our morphological analysis of the CST projection pattern.
While the role of FGF22-FGFR signaling has thus been well characterized in the developing nervous system, we now show that neuronal expression of FGF22 and its receptors FGFR1 and FGFR2 persists into adulthood. This constitutive expression of FGF22 and its receptors in the adult CNS is in line with the findings of a recent more systematic analysis that shows that many developmental signals that regulate axon guidance and synapse formation are still present in the mature brain and spinal cord (Jacobi et al, 2014). Indeed, important roles in the injured adult nervous system are emerging for a number of these molecules. For example, molecules such as EphA4, Sema3A, and Sema6A have all been shown to limit axonal regeneration following adult spinal cord injury (Kaneko et al, 2006; Goldshmit et al, 2011; Shim et al, 2012), while Sema3A and Sema3F influence remyelination (Piaton et al, 2011). Taken together, the persistence of these developmental signaling pathways suggests that the molecular machinery that allows the initial formation of neuronal circuits stays in place in the adult nervous system and provides the blueprint for ongoing nervous system plasticity. Indeed, it is increasingly appreciated that axonal remodeling processes both at the lesion site but also involving distant spinal and supraspinal circuits take place in the injured adult CNS, where they enable functional recovery (Weidner et al, 2001; Bareyre et al, 2004; Girgis et al, 2007; Courtine et al, 2008; van den Brand et al, 2012; Zörner et al, 2014). While such circuit remodeling can be initiated spontaneously, its therapeutic success can be enhanced by manipulations that foster neuronal growth initiation (Lang et al, 2013; Yip et al, 2010), neutralize plasticity restrictions (García-Alías et al, 2009; Lindau et al, 2014), and stimulate activity-based rehabilitation (van den Brand et al, 2012; Shah et al, 2013). An improved understanding of the endogenous signaling networks that enable the reorganization of axonal connections is a key requirement both for the informed advancement of such therapeutic strategies and for the identification of novel targets and approaches. Here, we uncover an integral component of these signaling networks and identify FGF22 as a synaptogenic mediator in the adult nervous system that is required for de novo synapse formation and timely synapse maturation during post-injury remodeling of the CNS.
Materials and Methods
Animals
Adult mice from 6 to 12 weeks of age were used in the study. To investigate the role of FGF22 during detour circuit formation after injury, we used FGF22 knockout mice (Terauchi et al, 2010). Age-matched wild-type C57Bl6j mice (Janvier, France) were used as FGF22-competent controls. To obtain forebrain FGFR1- or FGFR2-deficient mice, we crossed FGFR1fl/fl or FGFR2fl/fl mice (Pirvola et al, 2002; Yu et al, 2003), in which the FGFR1 or FGFR2 gene is flanked by loxP sites, to EMX1-Cre mice (Gorski et al, 2002; Bareyre et al, 2005), which express the Cre recombinase in the forebrain starting on embryonic day 10. Cre-negative littermates were used as FGFR-competent controls. For co-deletion of FGFR1 and FGFR2 in cortical projections, we injected a recombinant adeno-associated virus expressing the Cre recombinase (rAAV-GFP-Ires-Cre) in the hindlimb motor cortex of double-floxed FGFR1fl/fl/FGFR2fl/fl mice. Littermates injected in the same anatomical location with a rAAV-Ires-GFP served as FGFR-competent controls. All animal procedures were performed according to institutional guidelines and were approved by the local regulatory authorities.
In situ hybridization
Spinal cord tissue (cervical region C3–C5) and brain tissue (bregma −1.06 till −1.70) were sectioned coronally (20 μm thick for the spinal cord, 30 μm thick for the brain) using a cryostat (Leica CM1850) and processed as described previously (Jacobi et al, 2014). Briefly, all steps were carried out with DEPC-treated solutions to prevent degradation of target RNAs. Sections were washed in 2× SSC (from 20× stock solution containing 3 M NaCl and 0.3 M Na citrate), and before the pre-hybridization step, the sections were incubated in a 1:1 mixture of 2× SSC and hybridization buffer (50% formamide, 5× SSC, 5× Denhardt's solution, 250 μg/ml yeast tRNA, 500 μg/ml salmon sperm DNA) for 15 min at RT. Sections were then incubated for 1 h in hybridization buffer at the appropriate (pre-) hybridization temperature (65°C). For hybridization, the probe (200–400 ng/ml in hybridization buffer) was heated for 10 min at 80°C, applied to the tissue, and incubated overnight in an oven at 65°C. Sections were then rinsed at RT in 2× SSC and washed in decreasing concentration of SSC (2× to 0.1× SSC at hybridization temperature) before applying an alkaline-phosphatase-conjugated sheep anti-digoxigenin antibody, Fab fragments (1:2,000; Roche Diagnostics), in blocking buffer overnight at 4°C. Alkaline phosphatase activity was detected using nitroblue tetrazolium chloride (337.5 mg/ml) and 5-bromo-4-chloro-3-indolyl phosphate (175 mg/ml). The sections were washed in ddH2O after the staining procedure. The fluorescent Nissl stain NeuroTrace 435/455 (Life Technologies) was applied for 2 h at RT, and the sections were washed and mounted with Gel Mount (Sigma-Aldrich).
Laser microdissection and single-cell qPCR
Laser microdissection and single-cell qPCR was performed as follows (Hofbauer et al, 2003): Total RNA was isolated from LPSN derived from unlesioned animals and from lesioned animals at 3 weeks and 12 weeks post-injury. For this purpose, 20-μm-thick fresh frozen coronal sections of the cervical spinal cord (C3–C5) were collected on Membrane Slides 1.0 PET (Zeiss). Sections were covered with n-propanol to prevent drying and inhibit RNase activity, and transferred to a PALM Microbeam-Z microscope (Zeiss Axiovert 200 M). LPSN retrogradely labeled with dextran TexasRed® and located in the spinal laminae 6–9 at spinal level C4 were marked electronically. After evaporation of the n-propanol, neurons were laser (Crylas FTSS 355-50)-microdissected and pressure-catapulted into a reaction tube (AdhesiveCap 200 clear PCR tubes; Zeiss) and directly transferred on dry ice. For single-cell qPCR, we used the Single cell-to-CT kit (Life Technologies). Primers were as follows: FGF22 forward primer 5′-ACT TTT TCC TGC GTG TGG AC-3′, FGF22 reverse primer 5′-TCA TGG CCA CAT AGA AGC CT-3′; GAPDH forward primer 5′-TCA ACG ACC CCT TCA TTG-3′, GAPDH reverse primer 5′-ATG CAG GGA TGA TGT TCT G-3′. Amplification reaction was performed as follows: 95°C for 3 min, 95°C for 10 s, 56°C for 10 s (39 repeats), and a melting curve with 65–95°C increments.
Generation and production of AAV vectors
pAAV-CMV-GFP-Ires2-Cre (rAAV-GFP-Ires-Cre) was created by inserting an Ires sequence from pIres2-DsRed2 (BD Bioscience) at the HincII site of pAAV-CMV-MCS. The coding sequence for the Cre recombinase was excised from PBS185 (kind gift of Thomas Hughes, Montana State University) and inserted upstream of the Ires sequence. Green fluorescent protein (GFP) was excised from pEGFP-N1 and inserted downstream of the Ires sequence. The control pAAV-CMV-GFP (rAAV- GFP) used was a kind gift of Hildegard Büning (University of Cologne). Recombinant AAV chimeric virions containing a 1:1 ratio of AAV1 and AAV2 capsid proteins and the foreign gene were generated as previously described (Grimm et al, 2003; Klugmann et al, 2005). Genomic titers were as follows: rAAV-GFP-Ires-Cre, 1.2 × 1012 genome copies/ml; rAAV-GFP, 2.4 × 1012 genome copies/ml.
Surgical procedures
Mid-thoracic dorsal hemisection
Mice were anesthetized with i.p. injections of ketamine/xylazine (ketamine 100 mg/kg, xylazine 13 mg/kg). After a laminectomy to expose the dorsal spinal cord at thoracic level 8 (T8), a thoracic dorsal hemisection, which results in a bilateral transection of the main dorsal and minor dorsolateral CST component but leaves the ventral white matter intact, was performed with fine iridectomy scissors as previously described (Bareyre et al, 2004; Lang et al, 2013). Prior to and after surgery, animals were kept on a heating pad (38°C) until fully awake and treated with meloxicam (Metacam; Boehringer Ingelheim) twice per day for 48 h.
Stereotactic injection of rAAV into the hindlimb motor cortex
To study the effects of FGFR1 and FGFR2 deletion in cortical projection neurons, we pressure injected 0.7 μl of rAAV-GFP-Ires-Cre or control rAAV-GFP (concentration matched to 0.6 × 1012 genome copies/ml) 4 days prior to the spinal cord injury into the hindlimb motor cortex of double-floxed FGFR1fl/fl/FGFR2fl/fl mice using a finely pulled glass micropipette (coordinates from bregma: −1.3 mm, ± 1.0 mm lateral, 0.6 mm depth). The micropipette remained in place for 3 min following the injection. In order to verify that the virus remained confined to the hindlimb motor cortex and did not spread to the forelimb area, we amplified the GFP signal with an anti-GFP antibody (rabbit polyclonal anti-GFP; Life Technologies A11122), cut consecutive 50-μm-thick sections of the entire brain of all mice, and assessed the presence of GFP-labeled cells in layer V of the hindlimb and forelimb motor cortex (Supplementary Fig S7). Mice in which GFP-labeled cells were present in the forelimb motor cortex (coordinate from bregma starting at + 0.14 mm) were excluded from further analysis. To control for possible differences related to genetic or viral deletion of FGFRs, we deleted FGFR2 in the hindlimb cortex by injecting 0.7 μl of AAV-GFP-Ires-Cre at the following coordinates (bregma: −1.3 mm caudal, ± 1.0 mm lateral, 0.6 mm depth) in single-floxed FGFR2fl/fl mice. This deletion strategy induced similar changes of CST collateral and bouton formation (Supplementary Fig S8) as the genetic deletion of FGFR2 induced by crossing FGFR2fl/fl mice to EMX1-Cre mice (Fig4A–F).
Labeling of the hindlimb CST (hCST) fibers
The hindlimb CST of forebrain FGFR1- or FGFR2-deficient mice was traced by pressure injecting 1.5 μl of a 10% (in 0.1 M PB) solution of biotinylated dextran amine (BDA, 10,000 MW; Life Technologies) into the hindlimb motor cortex using a finely pulled glass micropipette 2 weeks prior to sacrifice (coordinates: −1.3 mm to bregma, 1 mm lateral to bregma, 0.6 mm depth). The micropipette remained in place 3 min following the injection.
Labeling of long propriospinal neurons
Long propriospinal neurons were retrogradely labeled by pressure injections of 0.5 μl of 2.5% dextran conjugated with Texas Red® (3,000 MW; Life Technologies D-3328). Briefly, a laminectomy was performed at thoracic level 12 as previously described (Lang et al, 2013) and 0.5 μl of 2.5% dextran conjugated with Texas Red® was injected into each side of the spinal cord using a thin glass capillary (coordinates from central vein: ± 0.6 mm, depth: 0.9 mm). The capillary was maintained in place for 3 min following the injection. The number of labeled long propriospinal neurons in the cervical spinal cord (spinal level C4) did not differ significantly between lesioned and unlesioned mice, indicating that the thoracic dorsal bilateral hemisection did not transect the propriospinal projections that run in the ventral white matter (118.6 ± 8.41 labeled propriospinal neurons in lesioned mice versus 111.3 ± 3.8 labeled propriospinal neurons in unlesioned mice; n = 20 sections per mouse, 3 mice per group analyzed).
Labeling of cortical projection neurons
Co-localization of the in situ hybridization (ISH) signal with the cortical projection neurons of the transected CST was assessed after retrogradely labeling these neurons as shown previously (Lang et al, 2013; Jacobi et al, 2014). Briefly, 7 days before sacrifice, a laminectomy at thoracic level 8 of the spinal cord was performed and 0.5 μl of dextran conjugated with Texas Red® (5% in 0.1 M PB; Life Technologies) was stereotactically injected rostral to the lesion with a glass capillary into each side of the spinal cord (± 0.2 mm lateral from spinal midline, depth 0.3 mm). The micropipette remained in place for 3 min after completing the injection to avoid backflow. After retrograde labeling, mice were kept on a heating pad (38°C) until fully awake and treated with meloxicam (Metacam; Boehringer Ingelheim) for two more days.
Tissue processing and histological analysis
Mice were deeply anesthetized with isoflurane and perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB). Brains and spinal cords were dissected and post-fixed overnight in 4% PFA. The tissue was then cryoprotected in 30% sucrose (Sigma) for at least 3 days. Coronal sections (50 μm thick) were cut on a cryostat. To visualize CST collaterals, BDA detection was performed as follows: Sections were incubated in ABC complex (Vector Laboratories) overnight at 4°C. After a 20-min tyramide amplification (Biotin-XX, TSA Kit #21; Life Technologies), sections were incubated overnight with streptavidin conjugated to FITC (1:500; Life Technologies). To visualize CST collaterals in rAAV-injected mice, an anti-GFP (rabbit polyclonal anti-GFP; Life Technologies A11122) staining was performed to amplify the GFP signal. For this purpose, an anti-GFP antibody (Life Technologies) diluted 1:500 in PBS containing 0.1% Triton X-100 and 2.5% goat serum (Life Technologies) was applied and incubated overnight at 4°C. On day 2, the corresponding secondary antibody was applied for at least 4 h (goat anti-rabbit conjugated with Alexa 488).
For immunohistochemical analysis of synapse maturation, 20-μm-thick sections were cut and blocked for 1 h with 5% goat serum and 0.3% Triton X-100 diluted in 1× PBS. Sections were incubated with ABC (Vector Laboratories) and a rabbit polyclonal antibody reactive against synapsin I (Millipore AB1543; dilution 1:500) or a mouse monoclonal antibody reactive against bassoon (ENZO Life Science SAP7F407, dilution 1:200) in 0.1 M Tris buffer containing 2% horse serum overnight at 4°C. The following day, after a 20-min tyramide amplification (Biotin-XX, TSA Kit #21; Life Technologies) to detect BDA, sections were then incubated together with streptavidin–FITC (1:500; Life Technologies) and the appropriate secondary antibodies for the synaptic markers (donkey anti-rabbit conjugated with Alexa Fluor 647 or goat anti-rabbit conjugated with Alexa Fluor 635) overnight at 4°C. For the analysis of rAAV-injected animals, the sections were first incubated with an anti-GFP antibody (see above) to amplify the GFP signal together with the primary antibodies against synapsin I or bassoon (concentrations as above) in 2.5% goat serum and 0.1% Triton X-100 in 1× PBS overnight at 4°C. On the next day, the appropriate secondary antibodies for the anti-GFP (goat anti-rabbit conjugated with Alexa Flour 488) and anti-synapsin I and anti-bassoon primary antibodies (donkey anti-rabbit conjugated with Alexa Fluor 647 or goat anti-rabbit conjugated with Alexa Fluor 635) were applied overnight at 4°C. For FGFR2 immunohistochemistry, the above-described protocol was used, but sections were pre-treated with trypsin for 10 min at 37°C for antigen retrieval. Then, sections were incubated with an antibody reactive against FGFR2 (1:500, ABCAM ab10648) and a goat anti-rabbit Alexa 488 secondary antibody (1:500). The counterstaining was performed with NeuroTrace 435/455, and sections were mounted in Vectashield (Vector Laboratories).
Quantifications
Quantification of exiting hindlimb CST fibers
To evaluate axonal remodeling following a mid-thoracic dorsal bilateral hemisection of the spinal cord, CST collaterals entering the gray matter at cervical levels C4 were counted on 30 consecutive coronal sections per animal using a light microscope (Olympus IX471) with a ×40/0.65 air objective. To correct for differences in inter-animal tracing efficiency, the number of collaterals was divided by the number of labeled fibers in the main CST tract and expressed as the ratio of exiting collaterals per main CST fiber (Bareyre et al, 2004). All quantifications were performed by an observer blinded with respect to injury status and genotype/treatment.
Quantification of contacts onto LPSN
For quantifying the proportion of contacts of LSPN contacted by CST collaterals, a total amount of 30 sections of the cervical spinal cord (level C3–C5) were evaluated using a fluorescent microscope (Olympus IX471) with a ×40/0.65 air objective. Collaterals were visualized as mentioned above (tyramide amplification or GFP amplification); the number of LPSN contacted by CST collaterals and the total number of LPSN labeled were counted. The proportion of LPSN contacted by CST collaterals was then calculated as the ratio of all LPSN contacted by collaterals over the total number of LPSN. Values were further normalized according to the number of labeled fibers in the main CST tract. All quantifications were performed by an observer blinded with respect to injury status and genotype/treatment.
Quantification of the length of the collaterals and density of boutons
To determine the length of the collaterals and the number of bouton per μm collateral, 10 sections spanning the C3 to C5 area of the cervical spinal cord (50 μm thickness, sections randomly taken) were acquired with an Olympus FV1000 confocal microscope equipped with standard filter sets and a ×60/1.45 oil immersion objective. Image stacks obtained with confocal microscopy were processed using ImageJ software to generate maximum intensity projections. The lengths of all individual collaterals in those sections were measured with the help of the measurement tool of ImageJ and averaged to obtain the average collateral length per section. This value was then further averaged across all (10) sections evaluated for a given animal. All boutons on CST collaterals in the cervical cord were counted in 30 sections under a fluorescent microscope. A bouton was defined as a thick varicosity along a comparably thin CST collateral in the cervical spinal cord. The total number of boutons per animal was divided by the total length in micrometers of collaterals per animal to calculate the density in boutons/μm. All quantifications were performed by an observer blinded with respect to injury status and genotype/treatment.
Quantification of synaptic marker expression
To determine the proportion of boutons that express the active zone protein, bassoon, and the synaptic vesicle-associated protein, synapsin I, about 20 sections spanning the C3 to C5 area of the cervical spinal cord (20 μm thickness, with every 5th section taken) were stained with anti-bassoon and anti-synapsin I antibodies as described above. Image stacks of CST collaterals (labeled as described above) were then acquired with an Olympus FV1000 confocal microscope equipped with standard filter sets and a ×60/1.45 oil immersion objective and processed using ImageJ software. Image stacks were used for analysis, and maximum intensity projections were generated for figure representation. The percentages of synapsin I- or bassoon-positive boutons were determined in confocal image stacks (all single planes from the image stacks were analyzed) upon the following criteria: A bouton was defined as a thick varicosity along a comparably thin CST collateral in the cervical spinal cord. Such a bouton was considered synapsin I or bassoon positive when the respective synapsin I or bassoon immunosignal covered the contour of the bouton but did not extend beyond it. The number of boutons positive for synapsin I or bassoon was expressed as a percentage of all CST boutons evaluated in the cervical spinal cord. A minimum of 300 boutons per group was counted. All quantifications were performed by an observer blinded with respect to injury status and genotype/treatment.
Cortical neuronal density
To determine whether genetic deletion of FGFR1 or FGFR2 at embryonic day 10 or constitutive FGF22 deficiency alters cortical lamination and cortical neuronal density, we cut 50-μm brain sections and performed NeuN immunohistochemistry (using an anti-NeuN antibody, Millipore, dilution 1:500, stained at 4°C overnight). Counterstaining was performed with NeuroTrace 500/525. The density of cells in layer V of the motor cortex and somatosensory cortex was quantified by counting the number of NeuN-positive cells in a defined box of 37.5 mm2 that was positioned on layer V on a total of 5 sections spanning the cortex at the following coordinates from bregma 1.5, 0.5, −0.22, −1.06, −1.3. Data were expressed as a number of neurons per 37.5 mm2. All quantifications were performed by an observer blinded with respect to injury status and genotype.
Quantification of retrogradely labeled CST neurons co-labeled with ISH
The co-labeling of retrogradely labeled cortical projection neurons with an ISH signal was assessed under the fluorescent microscope (Olympus IX71) by alternating between fluorescence and bright field illumination. To determine the proportion of cortical projection neurons that express FGFR1 or FGFR2, we counted all retrogradely labeled neurons on every third section of the cortex (n = 3 mice). Sections were assessed from anterior to posterior starting with the first section in which retrogradely labeled CST neurons appeared. Results were expressed as a ratio of the number of double-labeled neurons divided by the total number of retrogradely labeled neurons. All counts were performed by an observer blinded with respect to injury status and genotype/treatment.
Lesion volume
We assessed the extent of the spinal cord lesion in FGF22-deficient and hindlimb motor cortex FGFR1/FGFR2 double-deficient mice as well as the respective FGF22- and FGFR-competent control animals by measuring the lesion volume on longitudinal 50-μm-thick sections of the thoracic spinal cord that spanned the entire lesion extent. Following staining with a fluorescent Nissl dye (NT435; Life Technologies N-21479, dilution 1:500), the sections were imaged using an Olympus IX71 microscope. Images were then processed with ImageJ and the lesion area, including both the cavity and surrounding damaged tissue, was outlined. To calculate the total lesion volume, the measured lesion area of each section was multiplied by the section thickness (50 μm) and the results of all consecutive sections spanning the entire lesion extension were summed up for each animal.
Behavioral analysis
The following behavioral tests were used to assess locomotor recovery after spinal cord injury.
Ladder rung test
For assessment of the CST function following spinal cord injury, we used the ladder rung test (also called grid walk test) as previously described (Metz & Whishaw, 2002). Briefly, mice were scored for their ability to cross a 1-m-long horizontal metal-rung runway with either regular gaps of 1 cm (regular walk) or varying gaps of 1–2 cm (irregular walk) between the rungs. All mice underwent a couple of familiarization sessions with the task prior to preoperative baseline testing. Following familiarization, sessions were videotaped and scored to determine baseline performance. Preoperative scores as well as postoperative performance at 2 and 3 weeks post-injury were assessed. A hindlimb foot error was defined as a complete miss or slip from the rung at the moment of the placement of the paw onto the rung. Baseline and postoperative testing sessions consisted of 3 runway crossings. The total number of errors of the hindlimbs in each session was counted.
Catwalk analysis
To further evaluate unforced locomotion and gait patterns in FGF22-deficient as well as hindlimb motor cortex FGFR1/FGFR2 double-deficient mice, the Catwalk XT™ (Hamers et al, 2006; Noldus) was used. For data collection, all animals were familiarized with the system at least 3 times prior to preoperative baseline acquisition and the system was calibrated each day prior to acquisition. Three valid runs for each timepoint (−2 days, 2 weeks, 3 weeks) and each animal were recorded. A run was considered valid if it fulfilled the requirements pre-set in the system, a minimum run duration of 0.5 s, a maximum run duration of 4.0 s, and a maximum speed variation of 60% to ensure that the animal constantly walks on the runway without pausing. The body axis and paw angle was calculated by the software and the paw angle body axis was calculated as the deviation (in °) of the paw angle from the body axis.
Image processing
Image stacks obtained with confocal microscopy were processed using ImageJ software to generate maximum intensity projections. To obtain final images, these maximum intensity projections were processed in Adobe Photoshop using gamma adjustments to enhance visibility of intermediate gray values and median filtering to suppress noise when necessary. For the 3D rendering of contacts between CST collaterals and LPSN (Fig2F), Imaris software (Bitplane) was used. For visualizing the co-localization of the FGF22 in situ signal with long propriospinal neurons (LPSN) labeling (Fig1B), the ISH signal was inverted and pseudocolored (red) before being overlaid with the fluorescent LPSN label. For imaging retrogradely labeled CST neurons (Fig3C), we first imaged the fluorescence signals using a confocal microscope (FV1000; Olympus) using standard filter settings before we unmounted the sections, performed ISH, and re-imaged the sections as previously described (Jacobi et al, 2014). Again, the ISH signal was inverted and pseudocolored (red) before being overlaid with the retrogradely labeled CST neurons.
Statistical evaluation
Results are given as mean ± SEM. GraphPad Prism 5.01 for Windows (GraphPad Software) was used to perform statistical analysis. Student's t-tests were used for comparisons between two groups. For multiple comparisons, a one-way ANOVA followed by a Tukey test was performed. For behavioral analysis over time, a repeated one-way ANOVA followed by a Bonferroni post hoc test was used for the ladder rung test. A two-way ANOVA followed by Bonferroni tests was used for analyzing the catwalk data (as the number of suitable steps for analysis differs for each timepoint). Significance levels are indicated as follows: *P < 0.05; **P < 0.01; ***P < 0.001.
Acknowledgments
We would like to thank Martin Adrian and Geraldine Heitmann, for excellent technical assistance, Dana Matzek for animal husbandry, Klaus Dornmair and Kathrin Held for their help with single-cell laser microdissection and quantitative PCR, and Alexander Gun for his help analyzing CST maturation in mutant mice. We thank Daniel Kerschensteiner for critical reading of the manuscript. Work in F.M.B.'s laboratory is supported by grants from the Deutsche Forschungsgemeinschaft (DFG, SFB 870) and the German Federal Ministry of Education and Research (BMBF). Work in M.K.'s laboratory is financed through grants from the DFG (Transregio 128, SPP1710), the BMBF (Competence Network Multiple Sclerosis), the European Research Council under the European Union's Seventh Framework Program (FP/2007–2013; ERC Grant Agreement n. 310932), the Hertie-Foundation, and the ‘Verein Therapieforschung für MS-Kranke e.V.’. F.M.B. and M.K. are further supported by the Munich Center for Systems Neurology (SyNergy; EXC 1010). Work in H.U.'s laboratory is funded by NIH Grant NS070005.
Author contributions
FMB, HU, and MK conceived the experiments. AJ performed and analyzed spinal surgeries and tracing. AJ and AMS performed and analyzed in situ hybridizations and single-cell PCR. AJ, KL, MH, and AMS performed anatomical and immunohistochemical analysis. HU characterized mutant mouse strains. AJ, FMB, and KL performed and analyzed behavioral tests. FMB, AJ, and MK wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figures S1–S8
Review Process File
References
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Misgeld T, Sanes JR. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat Med. 2005;11:1355–1360. doi: 10.1038/nm1331. [DOI] [PubMed] [Google Scholar]
- van den Brand R, Heutschi J, Barraud Q, DiGiovanna J, Bartholdi K, Huerlimann M, Friedli L, Vollenweider I, Moraud EM, Duis S, Dominici N, Micera S, Musienko P, Courtine G. Restoring voluntary control of locomotion after paralyzing spinal cord injury. Science. 2012;336:1182–1185. doi: 10.1126/science.1217416. [DOI] [PubMed] [Google Scholar]
- Courtine G, Song B, Roy RR, Zhong H, Herrmann JE, Ao Y, Qi J, Edgerton VR, Sofroniew MV. Recovery of supraspinal control of stepping via indirect propriospinal relay connections after spinal cord injury. Nat Med. 2008;14:69–74. doi: 10.1038/nm1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MA, Sanes JR, Borza DB, Eswarakumar VP, Fassler R, Hudson BG, John SW, Ninomiya Y, Pedchenko V, Pfaff SL, Rheault MN, Sado Y, Segal Y, Werle MJ, Umemori H. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–193. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- García-Alías G, Barkhuysen S, Buckle M, Fawcett JW. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat Neurosci. 2009;12:1145–1151. doi: 10.1038/nn.2377. [DOI] [PubMed] [Google Scholar]
- Girgis J, Merrett D, Kirkland S, Metz GA, Verge V, Fouad K. Reaching training in rats with spinal cord injury promotes plasticity and task specific recovery. Brain. 2007;130:2993–3003. doi: 10.1093/brain/awm245. [DOI] [PubMed] [Google Scholar]
- Goldshmit Y, Spanevello MD, Tajouri S, Li L, Rogers F, Pearse M, Galea M, Bartlett PF, Boyd AW, Turnley AM. EphA4 blockers promote axonal regeneration and functional recovery following spinal cord injury in mice. PLoS One. 2011;6:e24636. doi: 10.1371/journal.pone.0024636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Kay MA, Kleinschmidt JA. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol Ther. 2003;7:839–850. doi: 10.1016/s1525-0016(03)00095-9. [DOI] [PubMed] [Google Scholar]
- Hamers FP, Koopmans GC, Joosten EA. CatWalk-assisted gait analysis in the assessment of spinal cord injury. J Neurotrauma. 2006;23:537–548. doi: 10.1089/neu.2006.23.537. [DOI] [PubMed] [Google Scholar]
- Hofbauer M, Wiesener S, Babbe H, Roers A, Wekerle H, Dornmair K, Hohlfeld R, Goebels N. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc Natl Acad Sci USA. 2003;100:4090–4095. doi: 10.1073/pnas.0236183100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi A, Schmalz A, Bareyre FM. Abundant expression of guidance and synaptogenic molecules in the injured spinal cord. PLoS One. 2014;9:e88449. doi: 10.1371/journal.pone.0088449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Iwanami A, Nakamura M, Kishino A, Kikuchi K, Shibata S, Okano HJ, Ikegami T, Moriya A, Konishi O, Nakayama C, Kumagai K, Kimura T, Sato Y, Goshima Y, Taniguchi M, Ito M, He Z, Toyama Y, Okano H. A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med. 2006;12:1380–1389. doi: 10.1038/nm1505. [DOI] [PubMed] [Google Scholar]
- Klugmann M, Symes CW, Leichtlein CB, Klaussner BK, Dunning J, Fong D, Young D, During MJ. AAV-mediated hippocampal expression of short and long Homer 1 proteins differentially affect cognition and seizure activity in adult rats. Mol Cell Neurosci. 2005;28:347–360. doi: 10.1016/j.mcn.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Lang C, Guo X, Kerschensteiner M, Bareyre FM. Single collateral reconstructions reveal distinct phases of corticospinal remodeling after spinal cord injury. PLoS One. 2012;7:e30461. doi: 10.1371/journal.pone.0030461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang C, Bradley PM, Jacobi A, Kerschensteiner M, Bareyre FM. STAT3 promotes corticospinal remodelling and functional recovery after spinal cord injury. EMBO Rep. 2013;14:931–937. doi: 10.1038/embor.2013.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau NT, Bänninger BJ, Gullo M, Good NA, Bachmann LC, Starkey ML, Schwab ME. Rewiring of the corticospinal tract in the adult rat after unilateral stroke and anti-Nogo-A therapy. Brain. 2014;137:739–756. doi: 10.1093/brain/awt336. [DOI] [PubMed] [Google Scholar]
- Metz GA, Whishaw IQ. Cortical and subcortical lesions impair skilled walking in the ladder rung walking test: a new task to evaluate fore- and hindlimb stepping, placing, and co-ordination. J Neurosci Methods. 2002;115:169–179. doi: 10.1016/s0165-0270(02)00012-2. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O'Rourke N, Smith SJ. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68:639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piaton G, Aigrot MS, Williams A, Moyon S, Tepavcevic V, Moutkine I, Gras J, Matho KS, Schmitt A, Soellner H, Huber AB, Ravassard P, Lubetzki C. Class 3 semaphorins influence oligodendrocyte precursor recruitment and remyelination in adult central nervous system. Brain. 2011;134:1156–1167. doi: 10.1093/brain/awr022. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hébert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Shah PK, Garcia-Alias G, Choe J, Gad P, Gerasimenko Y, Tillakaratne N, Zhong H, Roy RR, Edgerton VR. Use of quadrupedal step training to re-engage spinal interneuronal networks and improve locomotor function after spinal cord injury. Brain. 2013;136:3362–3377. doi: 10.1093/brain/awt265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger E, Ziv N, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron. 2003;38:237–252. doi: 10.1016/s0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Shim SO, Cafferty WB, Schmidt EC, Kim BG, Fujisawa H, Strittmatter SM. PlexinA2 limits recovery from corticospinal axotomy by mediating oligodendrocyte-derived Sema6A growth inhibition. Mol Cell Neurosci. 2012;50:193–200. doi: 10.1016/j.mcn.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui TJ, Craig AM. Synaptic organizing complexes. Curr Opin Neurobiol. 2011;21:132–143. doi: 10.1016/j.conb.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Su J, Brooks J, Terauchi A, Umemori H, Fox MA. Fibroblast growth factor 22 contributes to the development of retinal nerve terminals in the dorsal lateral geniculate nucleus. Front Mol Neurosci. 2012;4:61. doi: 10.3389/fnmol.2011.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens HE, Smith KM, Maragnoli ME, Fagel D, Borok E, Shanabrough M, Horvath TL, Vaccarino FM. Fgfr2 is required for the development of the medial prefrontal cortex and its connections with limbic circuits. J Neurosci. 2010;30:5590–5602. doi: 10.1523/JNEUROSCI.5837-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H. Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature. 2010;465:783–787. doi: 10.1038/nature09041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemori H, Linhoff MW, Ornitz DM, Sanes JR. FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell. 2004;118:257–270. doi: 10.1016/j.cell.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH. Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc Natl Acad Sci USA. 2001;98:3513–3518. doi: 10.1073/pnas.051626798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, de Wit J, Ghosh A. Molecular mechanisms of synaptic specificity in developing neural circuits. Neuron. 2010;68:9–18. doi: 10.1016/j.neuron.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip PK, Wong LF, Sears TA, Yáñez-Muñoz RJ, McMahon SB. Cortical overexpression of neuronal calcium sensor-1 induces functional plasticity in spinal cord following unilateral pyramidal tract injury in rat. PLoS Biol. 2010;8:e1000399. doi: 10.1371/journal.pbio.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active zone precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zörner B, Bachmann LC, Filli L, Kapitza S, Gullo M, Bolliger M, Starkey ML, Röthlisberger M, Gonzenbach RR, Schwab ME. Chasing central nervous system plasticity: the brainstem's contribution to locomotor recovery in rats with spinal cord injury. Brain. 2014;137:1716–1732. doi: 10.1093/brain/awu078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1–S8
Review Process File