

Abstract
The molecular motor kinesin moves along microtubules using energy from ATP hydrolysis in an initial step coupled with ADP release. In neurons, kinesin-1/KIF5C preferentially binds to the GTP-state microtubules over GDP-state microtubules to selectively enter an axon among many processes; however, because the atomic structure of nucleotide-free KIF5C is unavailable, its molecular mechanism remains unresolved. Here, the crystal structure of nucleotide-free KIF5C and the cryo-electron microscopic structure of nucleotide-free KIF5C complexed with the GTP-state microtubule are presented. The structures illustrate mutual conformational changes induced by interaction between the GTP-state microtubule and KIF5C. KIF5C acquires the ‘rigor conformation’, where mobile switches I and II are stabilized through L11 and the initial portion of the neck-linker, facilitating effective ADP release and the weak-to-strong transition of KIF5C microtubule affinity. Conformational changes to tubulin strengthen the longitudinal contacts of the GTP-state microtubule in a similar manner to GDP-taxol microtubules. These results and functional analyses provide the molecular mechanism of the preferential binding of KIF5C to GTP-state microtubules.
Keywords: cryo-electron microscopy, kinesin, microtubule, polarized transport in neuron, X-ray crystallography
Introduction
Kinesin superfamily proteins (KIFs) are microtubule (MT)-based molecular motors that play fundamental roles in intracellular transport. Kinesins use energy from ATP to move along the MTs (Hirokawa et al, 2009a,b). Structural studies to elucidate the molecular mechanism of kinesin motility suggest that the energy generated by the hydrolysis of ATP is used by kinesin for its active detachment from the MT track, whereas the energy produced by the binding to the MT, coupled with ADP release, allows the plus-end-directed movement of kinesin (Kull et al, 1996; Kozielski et al, 1997; Sablin et al, 1998; Kikkawa et al, 2001; Nitta et al, 2004, 2008; Hirose et al, 2006; Kikkawa & Hirokawa, 2006; Sindelar & Downing, 2010). Based on this alternating attachment and detachment of a kinesin catalytic head, the hand-over-hand model has been proposed to explain the processive, cooperative movement of dimeric kinesin. According to this model, the two catalytic heads alternately ‘step’ so that kinesin is always attached to the MT (Block et al, 1990; Asbury et al, 2003; Yildiz et al, 2004). This is accomplished by only one head adopting the detached (weak-binding) mode in the Mg-ADP state. This means that one head cannot hydrolyze ATP to detach from the MT until the other head has released ADP to attach strongly to the MT (Alonso et al, 2007). Hence, the mechanical process in which the catalytic head attaches to the MT, coupled with ADP release, defines the coordination between the two heads in the hand-over-hand mechanism.
To elucidate the molecular mechanisms of this fundamental process, kinesin structures before and after ADP release, that is the Mg-ADP state and the nucleotide-free state, are necessary. However, kinesin in the absence of MT is unstable and crystal structure determination of kinesin in the nucleotide-free state remains the missing link in the elucidation of the various possible structures. Instead, the cryo-EM structure of nucleotide-free kinesin complexed with MT was reported for two types of kinesins, KIF5 (kinesin-1/conventional kinesin) and Kar3 (kinesin-14) (Hirose et al, 2006; Sindelar & Downing, 2007; Atherton et al, 2014). Both forms, however, take very different conformations, especially at the switch II element located at their MT-binding surface. The switch II conformation of KIF5 is very similar to the crystal structure of kinesin in the Mg-ADP state (ADP-like conformation), whereas the interface of Kar3 differs from both the Mg-ATP form (ATP-like conformation) and the ADP-like conformation. The reason for the difference between KIF5 and Kar3 is still debated, and the high-resolution structure of kinesin in the nucleotide-free state in the presence and absence of MTs remains elusive.
Understanding this key mechanical step in kinesin motility has attracted the attention of cell biologists and neuroscientists. KIF5, the earliest discovered and a well studied KIF, transports several types of cargo to the axon among many processes in neurons (Nakata & Hirokawa, 2003; Jacobson et al, 2006). This polarized transport in neurons is fundamental to the formation of the neuronal circuitry and is possible because, in axons, GTP-state MTs (GTP-MTs) are enriched over GDP-state MTs (GDP-MTs) and are preferentially ‘searched for’ as landmarks by KIF5 (Nakata et al, 2011). This high-affinity binding of KIF5 to GTP-MTs guides the transport of the various cargoes to their correct neuronal destinations. Recently, the structure of GTP-MT was reported and the characteristic feature of its KIF5 binding site was suggested to account for the difference in the affinity between KIF5 and GDP-/GTP-MTs (Yajima et al, 2012). To further understand the molecular mechanism of this fundamental relationship between kinesins and MTs, elucidation of the structures of KIF5 at the beginning of kinesin stepping, in the nucleotide-free state and complexed with GTP-MT, is necessary.
Here, we describe the successful cryo-EM determination of the structure of nucleotide-free KIF5C complexed with MT in the GTP-state using a GTP analog, guanylyl 5′-α, β-methylenediphosphonate (GMPCPP), at 8.9 Å resolution. We also solved the crystal structure of nucleotide-free KIF5C in the absence of MTs. In silico docking of the crystal structure with the cryo-EM structure revealed the mutual conformational changes of KIF5C and GMPCPP-MT. Nucleotide-free KIF5C complexes with GMPCPP-MT to acquire a new conformation that we termed the ‘rigor conformation’, by analogy with myosin. This conformation of KIF5C not only provides an important missing link in the structural analysis of kinesin, but also elucidates the molecular mechanism of the preferential binding of KIF5C to the GTP-MT. Furthermore, this is the first observation of the conformational change in the GMPCPP-MT induced by KIF5C binding. MT predominantly changes its surface conformation of α- and β-tubulins with strengthening of the longitudinal contacts. These structural and functional analyses provide the molecular mechanism of the preferential binding of KIF5 to GTP-state microtubules.
Results
GMPCPP-MT is a better substrate for KIF5C than GDP-MT
The binding of KIF5C to GTP-MT is 3.7 times stronger than that to GDP-MT, which accounts for the preferential transport of KIF5C into axons and not into dendrites (Nakata et al, 2011). Considering that the MT acts as a nucleotide exchange factor and as a nucleotide-activating protein of KIFs, the nucleotide state of the MT might affect both motility and the ATPase kinetics of KIF5C on the MT. To examine this possibility, we first analyzed the MT-gliding velocities of monomeric KIF5C K345 using the different nucleotide states of MTs. As shown in Fig1A, K345 moved along the GMPCPP-MTs at a rate that was 36% faster than that of GDP-taxol-MTs at saturating ATP concentrations [GMPCPP-MT: 338 ± 48 nm/s (mean ± SD) (n = 100); GDP-taxol-MT: 248 ± 44 nm/s (n = 100)]. This difference was statistically significant (P < 0.001) and reproducible. The ratio of the difference is also conserved in the dimeric KIF5C K375 (Supplementary Fig S1) and a previous report (Vale et al, 1994). We then examined the MT-stimulated ATPase activities of K345 to test whether the faster velocity of this kinesin on GMPCPP-MT is related to a faster rate of ATP hydrolysis. Figure1B shows that the ATPase activity of K345 stimulated by GMPCPP-MTs was 32% faster than that simulated by GDP-taxol-MTs [kcat (GMPCPP-MT) = 14.3 ± 0.7/s; kcat (GDP-taxol-MT) = 10.8 ± 0.7/s]. This difference in ATPase rates matched the difference in the MT-gliding velocities and explained the faster transport of KIF5C on GMPCPP-MT. In addition, the Michaelis–Menten constants (KM, MT) were not significantly different between the two forms [KM, MT (GMPCPP-MT) = 3.2 ± 0.4 μM; KM, MT (GDP-taxol-MT) = 3.1 ± 0.6 μM], indicating the higher substrate specificity of GMPCPP-MT than GDP-taxol-MT for KIF5C. This suggests that although the conventional kinesin KIF5C could still move well on the GDP-MT stabilized with taxol, GMPCPP-MT is a better substrate for KIF5C.
Figure 1. Effect of nucleotide state of MT on KIF5C motility and kinetics.
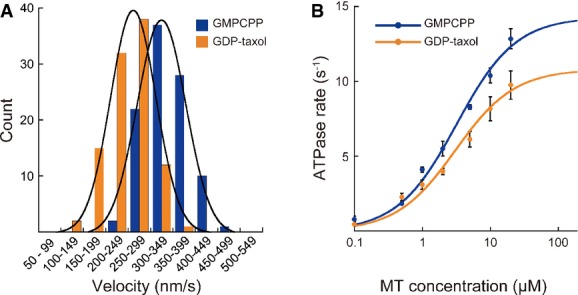
- A, B Gliding assays (A) and ATPase activity assays (B) of KIF5C on GMPCPP-MT (blue) and GDP-taxol-MT (orange). Error bars in (B) are the SEM of at least four independent measurements. See also Supplementary Fig S1.
Cryo-EM structure of the nucleotide-free KIF5C–GMPCPP-MT complex
As described above, compared with GDP-taxol-MT, GMPCPP-MT not only binds with high affinity to KIF5C but also results in faster transport because of the faster ATP hydrolysis cycle, both of which would be crucial for polarized axonal transport in neurons. However, the structural mechanisms explaining how KIF5C distinguishes GTP-MT from GDP-MT and the structural features that correspond to faster ATPase cycling are unknown. Therefore, we solved the cryo-EM structure of KIF5C complexed with GMPCPP-MT using the previously described real-space helical reconstruction (Fig2A and Supplementary Fig S2A) (Yajima et al, 2012; Ogura et al, 2014). We also confirmed the previously solved cryo-EM structure of the KIF5C–GDP-taxol-MT complex to validate the cryo-EM structures solved here (Fig2B, Supplementary Fig S2B and C) (Sindelar & Downing, 2007; Atherton et al, 2014). In these structural analyses, KIF5C was forced to release ADP, thereby yielding a nucleotide-free state that allowed us to analyze the structure in its initial high-affinity binding state to the MT. In the following, we refer to nucleotide-free KIF5C complexed with GMPCPP-MT or GDP-taxol-MT as KIF5(Ø)–CPP-MT and KIF5(Ø)–taxol-MT, respectively.
Figure 2. Cryo-EM structure of the KIF5C–MT complex.
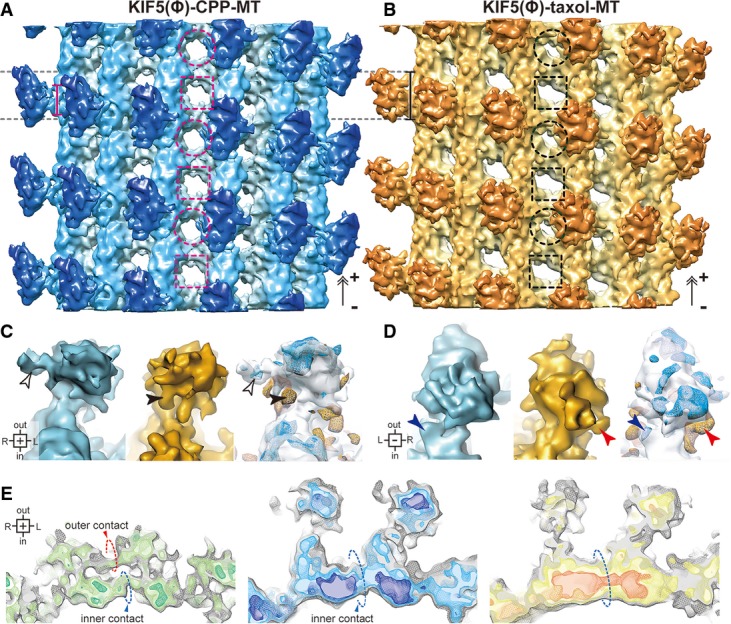
- A, B Cryo-EM structures of KIF5(Ø)–CPP-MT (A) and KIF5(Ø)–taxol-MT (B). The bars represent the length of the KIF5–MT interface. The dashed circles and squares indicate the holes surrounded by intra-tubulin dimers and inter-tubulin dimers, respectively.
- C, D Magnified views of KIF5C seen from the plus-end (C) and minus-end (D). KIF5(Ø)–CPP-MT (blue), KIF5(Ø)–taxol-MT (orange), and the statistical significance of their differences (t-map, n = 46). The blue and orange meshes of the t-map show the densities increase in maps of each color. P < 10−2.5 was defined as significant. Arrowheads indicate the regions differ significantly.
- E Cross-sectional views of CPP-MT (left), KIF5(Ø)–CPP-MT (middle) and KIF5(Ø)–taxol-MT (right), seen from the plus-end. Dashed circles indicate lateral contacts.
See also Supplementary Fig S2.
The resolutions of KIF5(Ø)–CPP-MT and KIF5(Ø)–taxol-MT were achieved at 8.9 and 9.8 Å, according to the 0.5 criteria of gold-standard FSC (Supplementary Fig S2D). In these cryo-EM maps, especially in KIF5(Ø)–CPP-MT, most of the α-helices were well resolved and the important flexible loops contributing to the KIF5C–MT interfaces, which are usually invisible in crystal structures, were readily observed.
Mutual conformational adaptation of KIF5C and MT upon their binding
The overall structure of KIF5(Ø)–CPP-MT was found to closely resemble that of KIF5(Ø)–taxol-MT, albeit with several striking differences in both the KIF5C and MT that were apparent in the statistical analysis. The shapes of the MT interface of KIF5C were dependent on the nucleotide state of the MT. KIF5C bound to GDP-taxol-MT with the longitudinally long interface, as previously reported (Sindelar & Downing, 2007; Atherton et al, 2014), whereas the binding interface for GMPCPP-MT was relatively short and concentrated around the intra-tubulin dimer groove (bars in Fig2A and B). The catalytic core of KIF5C was also farther from the surface of GMPCPP-MT than that of GDP-taxol-MT (Fig2C and D). On the MT side, several differences were also found among KIF5(Ø)–CPP-MT, KIF5(Ø)–taxol-MT and the previously solved GMPCPP-MT in the absence of KIF5C (CPP-MT) (Fig2E) (Yajima et al, 2012). If observed from the plus-end, the outer contact, reported to be one of the specific features of GMPCPP-MT, was clearly observed in CPP-MT, whereas it was not observed in KIF5(Ø)–CPP-MT or KIF5(Ø)–taxol-MT. The difference of size and shape of the MT holes at which four tubulin monomers meet were also observed. In the KIF5(Ø)–taxol-MT or the CPP-MT, the size and shape of the holes alternated between small and large along the protofilament, and is consistent with previous cryo-EM structures. In the KIF5(Ø)–CPP-MT structure, however, the holes were uniform (Fig2A and B and Supplementary Fig S2G). These findings indicate the distinct longitudinal and lateral contacts among them, suggesting the conformational change in GMPCPP-MT induced by KIF5C binding as well as the effects of the bound nucleotide and taxol to β-tubulins. This overview of the cryo-EM structures collectively illustrated that the binding of KIF5C to GMPCPP-MT induces mutual conformational changes, resulting in strong KIF5C–MT binding that accelerates the release of ADP from KIF5C.
Crystal structure of nucleotide-free KIF5C without MT
Examination of the cryo-EM structure of KIF5(Ø)–CPP-MT revealed the nature of the conformational differences of KIF5C and its track MT with respect to GMPCPP-MT and GDP-taxol-MT. To understand the underlying structural mechanism required, the atomic structure was docked onto the cryo-EM structure by a hybrid approach. Currently, the atomic structure of any of the KIFs without nucleotide has not been available, except for that of Kin I kinesin (Shipley et al, 2004). We therefore set out to solve the crystal structure of KIF5C without any bound nucleotide (Fig3A).
Figure 3. Crystal structure of nucleotide-free KIF5C.
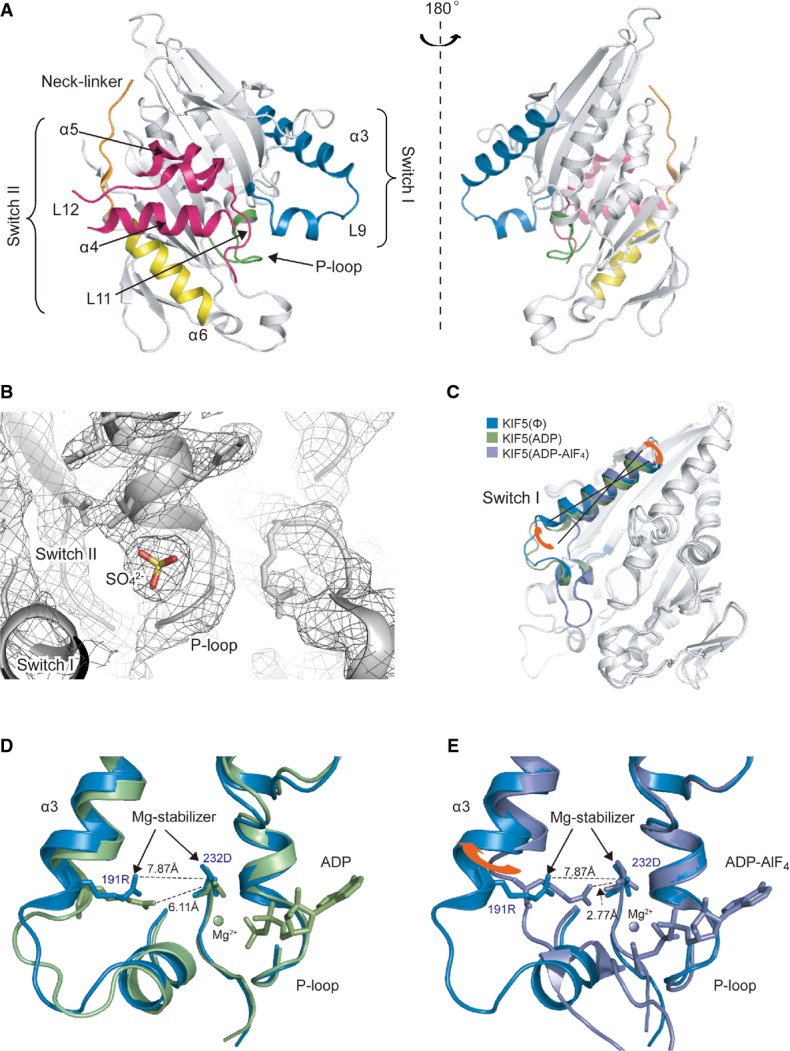
- A Overall structure of nucleotide-free KIF5C as seen from the MT-binding interface (left) and the opposite side (right).
- B Nucleotide-binding pocket of the electron density map of the nucleotide-free KIF5C. Meshes show the 2fo-fc map (σ = 1.0). The small round density of sulfate ion is surrounded by the P-loop.
- C Comparison of the nucleotide-free KIF5C and KIF5B in the preceding ADP state (PDB ID: 1BG2) and in the following ADP-AlF4 state (PDB ID:4HNA).
- D, E Close-up view of the nucleotide-binding pocket. Blue, nucleotide-free KIF5C; green, 1BG2; purple, 4HNA.
See also Supplementary Fig S3.
We first crystallized KIF5C K334 in the final crystallization buffer. The construct used for the crystallization had a partially truncated neck-linker, but achieved the comparable ATPase activity with K345 that was used for the cryo-EM studies. It resulted in that the weakly bound ADP without Mg2+ was found in the nucleotide-binding pocket (average B-factor for ADP: 100.88 Å2) (Supplementary Fig S3A). We thus soaked several additives such as EDTA, apyrase or the C-terminal peptide of KIF5C to the weakly ADP-bound crystals to further accelerate the release of ADP. In the presence of the C-terminal peptide, then, a small round electron density was found in the nucleotide-binding pocket instead of ADP. The corresponding density for the peptide was not found in the electron density map possibly because of the peptide's high flexibility (Fig3B). Given the contents of the buffer used, this density was most likely the sulfate ion 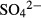 , as was the case in myosin (Himmel et al, 2002). This structure therefore represents nucleotide-free KIF5C, at the resolution of 2.9 Å (Table1), although there remains the possibility that the presence of the sulfate ion might prevent from taking the ‘full’ nucleotide-free conformation. It should be noted that the structures before and after the treatment of the peptide have very similar conformations with their RMSD being 0.309 Å, except that ADP was weakly bound to the pocket before the treatment. This suggests that the C-terminal peptide might help the release of ADP, that should be further confirmed by the future kinetic studies. The effect of ADP release for the overall conformation of KIF5C (from the weakly bound ADP state to the nucleotide-free state) was not clearly observed. We thus safely describe that since the KIF5C structures solved here detailed in the next paragraph share the structural features of switch I and switch II with the KIF1A in the weakly bound ADP in the absence of MT (Nitta et al, 2008), kinesins in the nucleotide-free state in the absence of MT will take this type of conformation. Especially, the switch I takes the favorable conformation to release Mg2+ and ADP, that has not been observed in the presence of MT or tubulin.
, as was the case in myosin (Himmel et al, 2002). This structure therefore represents nucleotide-free KIF5C, at the resolution of 2.9 Å (Table1), although there remains the possibility that the presence of the sulfate ion might prevent from taking the ‘full’ nucleotide-free conformation. It should be noted that the structures before and after the treatment of the peptide have very similar conformations with their RMSD being 0.309 Å, except that ADP was weakly bound to the pocket before the treatment. This suggests that the C-terminal peptide might help the release of ADP, that should be further confirmed by the future kinetic studies. The effect of ADP release for the overall conformation of KIF5C (from the weakly bound ADP state to the nucleotide-free state) was not clearly observed. We thus safely describe that since the KIF5C structures solved here detailed in the next paragraph share the structural features of switch I and switch II with the KIF1A in the weakly bound ADP in the absence of MT (Nitta et al, 2008), kinesins in the nucleotide-free state in the absence of MT will take this type of conformation. Especially, the switch I takes the favorable conformation to release Mg2+ and ADP, that has not been observed in the presence of MT or tubulin.
Table 1.
Data collection and refinement statistics (molecular replacement).
| KIF5C (3WRD) | KIF5C-ADP (3X2T) | |
|---|---|---|
| Data collection | ||
| Space group | P 212121 | P 212121 |
| Cell dimensions | ||
| a, b, c (Å) | 71.59, 71.32, 176.16 | 71.86, 71.83, 176.89 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50–2.86 (2.91–2.86) a | 50–2.70 (2.77–2.70) a |
| Rsym or Rmerge | 0.079 (0.942) | 0.049 (0.798) |
| I /σI | 17.0 (1.96) | 27.8 (1.66) |
| Completeness (%) | 99.7 (100.0) | 99.5 (98.4) |
| Redundancy | 7.2 (6.9) | 6.8 (6.3) |
| Refinement | ||
| Resolution (Å) | 20–2.86 | 20–2.70 |
| No. reflections | 21376 | 24347 |
| Rwork/Rfree | 0.237/0.299 | 0.224/0.298 |
| No. atoms | ||
| Protein | 4980 | 4996 |
| Ligand/ion | 10 | 54 |
| B-factors | 102.29 | 84.42 |
| R.m.s. deviation | ||
| Bond lengths (Å) | 0.012 | 0.011 |
| Bond angles (°) | 1.648 | 1.641 |
Values in parentheses are for highest resolution shell.
The crystal structure of the KIF5C motor domain without nucleotide adopts the ATP-like conformation. In comparison with the previously solved KIF5 (kinesin-1) structures, the conformations of switch II and the neck-linker in KIF5C are very similar to the previously solved ATP-like structures. On the other hand, the switch I conformation of KIF5C is slightly different from those in the other ATP-like structures (Fig3C–E and Supplementary Fig S3B–D). Helix α3 of KIF5C rotates so that the C-terminal region of α3 and the following L9 move away from the nucleotide-binding pocket. A comparison with the recently solved KIF5 structure in the ADP-AlF4 state complexed with tubulin dimers further highlights the conformational change of switch I (Fig3E). Switch I of KIF5 in the ADP-AlF4 state completely closes the nucleotide-binding pocket with the formation of the Mg-stabilizer between Arg191 in switch I and Asp232 in switch II, which plays a fundamental role in retaining the nucleotide in the pocket. The rotation of α3 observed in the nucleotide-free KIF5C (12 degrees from the KIF5-ADP-AlF4) breaks the Mg-stabilizer; thus, Mg-ADP is destabilized, which opens the pocket to facilitate the exchange of ADP with Mg-ATP (Nitta et al, 2008).
In silico docking of the atomic models into the cryo-EM map
To elucidate the structural mechanism by which KIF5C moves along the MT, the atomic structures of the nucleotide-free KIF5C, as solved here, and tubulin dimer (PDB ID: 1JFF) (Löwe et al, 2001) were docked onto the cryo-EM structure of KIF5(Ø)–CPP-MT (Fig4A, Supplementary Fig S4 and Supplementary Movie S1). The fitting trials were guided by several parameters, including the cross-correlation coefficient, the average map value, and the number of atoms inside the contour (Supplementary Fig S4A–D). In addition, the volume tracer tool in Sculptor (Birmanns et al, 2011) was also used to detect α-helices from the EM map as references (Supplementary Fig S4F–H).
Figure 4. Atomic model docking into KIF5(Ø)–CPP-MT map.

- Pseudo-atomic model fitted into the KIF5(Ø)–CPP-MT map for the orientation of (B) and (C).
- Cross section seen from the MT-binding side (left) and the comparison of the models (right). The gray model indicates the KIF5(Ø) crystal structures before the subdomain fittings.
- Cross sections seen from the minus-end. Dark gray models indicate the tubulin dimer (PDB ID: 1JFF) and KIF5(Ø) crystal structures before the subdomain fittings.
See also Supplementary Fig S4 and Supplementary Movie S1.
The crystal structure of the nucleotide-free KIF5C did not fit as a rigid body into the KIF5(Ø)–CPP-MT map (gray model in Fig4B). Thus, we rigidly fitted the atomic model of the β-sheet core into the map, which was followed by the fittings of the switch I helix α3 and switch II helix α4. For the fitting of tubulins, the atomic model of GMPCPP-MT and 1JFF were tested as the starting models. The reasonable fit of β-tubulin was achieved by rigid body fitting of 1JFF, although the atomic model of α-tubulin did not fit well (dark gray in Fig4C). Since most of the differences of α-tubulin might be explained by the relative movements among subdomains, we divided the atomic model as was the case in a previous report (Yajima et al, 2012), and rigidly docked each one separately. To avoid overfitting by introducing extra degree of freedom, we simulated several divisions (no division, divided into two, three, and four subdomains) and carefully checked the fitting parameters. Hence, α-tubulin divided into three subdomains, the N-domain, I-domain, and C-domain, was chosen as a final model, in which the N- and C-domains moved 6 Å toward the inside of the MT without any collision between subdomains, whereas I-domain did not show marked conformational change in comparison with that of 1JFF.
In this way, we finally created an atomic model of KIF5(Ø)–CPP-MT that fitted to our EM map. Note that the current pseudo-atomic model is still a conservative approximate model. Nonetheless, the positions of most secondary structures are supported by the local maxima of the map density such that we safely describe the conformational change at the secondary structure level. For further details, a higher resolution map is required.
Conformational change of MT upon KIF5C–MT binding
To clarify the conformational change to tubulin in the GMPCPP-MT, which is induced by the binding of KIF5C, the KIF5(Ø)–CPP-MT was compared with the previously solved CPP-MT (Yajima et al, 2012) (Fig5). Remarkable differences were also indicated by the difference map between them especially at the lateral contacts, at the longitudinal contacts, and at the MT surface (Supplementary Fig S5A and B).
Figure 5. KIF5C-induced conformational change of GMPCPP-MT.

- A, B The left side view of CPP-MT (A) and KIF5(Ø)–CPP-MT (B), with three different contour levels and at the same resolution (8.9 Å). Mesh, 1.3 σ; light color, 1.5 σ; dark color, 1.8 σ. The arrowheads indicate the major difference on the MT surface at the minus-end of α-tubulin.
- C KIF5C-induced movements of helices in N2- and C-domains seen from the left side.
- D, E CPP-MT (D) and KIF5(Ø)–CPP-MT (E), seen from the MT minus-end at the level of β-tubulin. OC, outer contact; IC, inner contact.
- F KIF5C-induced movements of helices in N2- and C-domain of β-tubulin seen from the MT minus-end.
See also Supplementary Fig S5.
KIF5C was supposed to target the strong intra-dimer interaction that was specifically observed in CPP-MT, between the H4-S5 loop of β-tubulin and H11′ and/or H3′ of α-tubulin (red broken line in Fig5A) (Yajima et al, 2012). In the KIF5(Ø)–CPP-MT structure solved here, KIF5C actually binds to the rear side of this interaction, the H11′ of α-tubulin. As a consequence, the intra-dimer interaction between H4-S5 loop and H11′ is broken and instead KIF5C bridges the intra-dimer groove by connecting the C-domains of α- and β-tubulin (red broken curve in Fig5B). The direct binding of KIF5C to H11′ further pushes the C- and N-domains of α-tubulin downward (Fig5C), decreasing the surface density at the intra-dimer whereas increasing the density around the nucleotide-binding pocket (Supplementary Fig S5A). This unique compact conformation of α-tubulin was observed in the KIF5(Ø)–CPP-MT structure for the first time.
The rupture of the strong intra-dimer interaction also triggers a conformational change to the lateral contact between neighboring β-tubulins. In the CPP-MT, helices H3 to H5 (N2-domain) of β-tubulin are raised and separated from H1 and H2 (N1-domain) to form double-layered lateral contacts, the outer contact and the inner contact (Fig5D). KIF5C binding then releases the H4-S5 loop of β-tubulin from H11′ of α-tubulin so that the N2-domain of β-tubulin becomes flexible (Fig5E). H3 to H5 of β-tubulin thus rotate downward to fuse these contacts into the single-layered canonical lateral contact (Fig5F and Supplementary Fig S5C).
This rotation further affects the inter-dimer contacts. Helices H11-H11’-H12 (C-domain) of β-tubulin, which locate over the N2-domain, also move downward mainly at their plus-end half because their minus-end interacts with KIF5C to suppress their movement (Fig5B and C). Thus, the surface density had decreased in the KIF5(Ø)–CPP-MT; instead, the density around the nucleotide-binding pocket of β-tubulin had increased (Supplementary Fig S5A). As a consequence of these conformational changes, β-tubulin in the KIF5(Ø)–CPP-MT takes the similar conformation with that in the GDP-taxol-MT with the essentially similar lateral and inter-dimer contacts because the I-domain conformation of α-tubulin that interacts with β-tubulin to make the inter-dimer contact also resembles that in the GDP-taxol-MT (Figs4C and 10A).
From these structural analyses, we expected that the strength of the MT lattice will be affected by the KIF5C-induced conformational changes to the GMPCPP-MT. Thus, we tested how KIF5C binding affected the stability of the GMPCPP-MT by examining the shrinking time of GMPCPP-MT with and without KIF5C (Fig6). As a result, GMPCPP-MT in the presence of KIF5C remained as a filamentous structure much longer than when KIF5C was absent. This suggests that the binding of KIF5C significantly stabilizes the GMPCPP-MT and also supports the conformational change induced by KIF5C binding.
Figure 6. Effect of KIF5C binding on GMPCPP-MT stability.
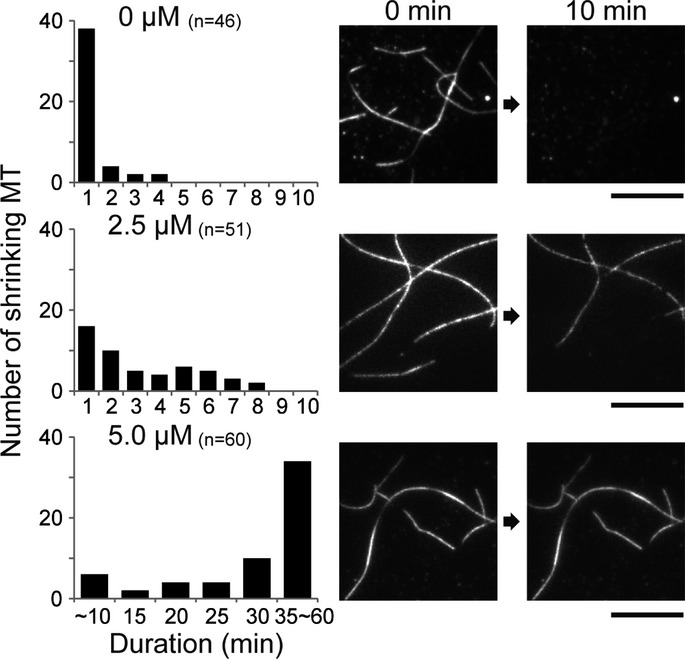
Shrinking rates of GMPCPP-MT with various concentrations (0, 2.5, and 5.0 μM) of KIF5C. The number of MTs which began to shrink in the duration is counted. Right panel depicts representative TIRF images at 0 min (left) and 10 min (right). Bar, 10 μm.
Conformational change of KIF5C upon KIF5C–MT binding
We next describe the conformational change of KIF5C induced by MT binding. As noted above, the conformations of switch I and switch II changed in the presence of GMPCPP-MT (Fig4B). In KIF5(Ø)–CPP-MT, switch I helix α3 adopted a similar conformation to the KIF5 in the ATP form, and switch II was a novel conformation distinct from both the ATP-like and ADP-like forms. Although the loops could not be modeled due to the resolution limit of our map, L5, L7, L8, L9, and the neck initial segment (NIS) of neck-linker may also change their conformation considering from the shape of the map.
The nucleotide-binding pocket in the crystal structure of nucleotide-free KIF5C consists of the shallow groove of the P-loop. In the KIF5(Ø)–CPP-MT EM map, the P-loop groove is clearly observed at the nucleotide-binding pocket and further deepened by two elements, L5 and L9 (switch I) that approach the pocket to form its top and left walls, respectively (Fig7A). Interestingly, this switch I conformation (α3-L9) is similar to the crystal structure of KIF5 in the ADP-AlF4 state complexed with tubulin (Gigant et al, 2013), suggesting that switch I had already acquired the proper conformation to welcome ATP into the pocket (Fig7B). This conformation is also conserved in the previously solved nucleotide-free KIF5C complexed with GDP-taxol-MT, indicating its functional importance for KIF5C motility (Supplementary Fig S6A) (Sindelar & Downing, 2007; Atherton et al, 2014).
Figure 7. Conformational changes of KIF5C on GMPCPP-MT.

- Cryo-EM map of KIF5(Ø)–CPP-MT, as seen from the left (left), minus-end (middle), and right (right).
- Comparison of the switch I conformation to that of KIF5(ADP) (PDB ID: 1BG2) (upper panel) and KIF5(ADP-AlF4) (PDB ID: 4HNA) (lower panel), as seen from the left. Blue, nucleotide-free KIF5C switch I; light green, 1BG2; dark cyan, 4HNA.
- Comparison of the switch II conformations as seen from the MT minus-end. Magenta, nucleotide-free KIF5C switch II; light green, 1BG2; dark cyan, 4HNA.
- Comparison of neck-linkers as seen from the right. Orange, nucleotide-free KIF5C neck-linker; light green, 1BG2; dark cyan, 4HNA.
See also Supplementary Fig S6.
On the other hand, switch II helix α4 of KIF5C on GMPCPP-MT exists distal from the core, both the nucleotide-binding pocket and switch I. This is markedly different from KIF5C of either the ADP-like or ATP-like conformation (Fig7C). KIF5(Ø)–CPP-MT therefore takes on a third, new type of switch II conformation, which we refer to as the ‘rigor’ conformation in analogy with myosin (Coureux et al, 2003). Since the helix α4 forms the bottom wall of the nucleotide-binding pocket just before the hydrolysis of ATP (Chang et al, 2013), the pocket in the nucleotide-free state lacks the bottom wall for the easy entry of ATP into the pocket. The helix α4 of the rigor conformation becomes shorter from that in the ADP-like conformation, coupled with the elongation of the adjacent L11. The elongated L11, then, squeezes into the space between α4 and α6. The NIS of the neck-linker docks to the core to support the raised position of the α4 (Fig7D). The main driving force for taking the characteristic conformation of α4 therefore might be the insertion of L11 and the NIS into the space between α4 and α6.
Interface between KIF5C and GMPCPP-MT
We then compared the KIF5–MT interfaces between KIF5(Ø)–CPP-MT and KIF5(Ø)–taxol-MT to examine the interactions between KIF5C and MT in detail (Fig8). Elements of KIF5 that contribute to the MT interface can be divided into five regions: L8 (plus), L11 (minus), α4-α5 (center), L12 (right), and L7 (left) (Supplementary Fig S7). The L8 in all the previously solved KIFs–MT complexes and our KIF5(Ø)–taxol-MT contributes to the interface at the plus-end side of KIF5 that interacts with helix H12 of β-tubulin (Fig8B). In the KIF5(Ø)–CPP-MT structure, however, the density corresponding to L8 is very weak because of the high flexibility of this loop, indicating it is far removed from β-tubulin and is not the main contributor to the KIF5C–GMPCPP-MT interaction. This high flexibility of L8 reflects the characteristic binding interface of KIF5C on the GMPCPP-MT (Fig2A).
Figure 8. Interface between KIF5C and GMPCPP-MT/GDP-taxol-MT.
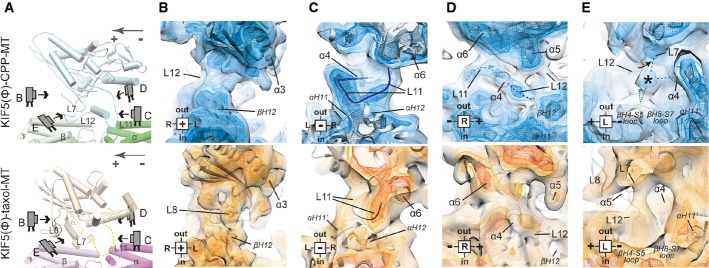
- A Cartoon representation of KIF5C complexed with GMPCPP-MT (upper panel) and GDP-taxol-MT (lower panel) for orientation in the following panels.
- B–E The differences of the L8 contribution to the KIF5C–MT interaction (B), the routes and the positions of L11 and α6 (C), the conformations of switch II and α6 (D), and the interaction among L7 and α4 of KIF5C and the loops of β-tubulin (E). Blue map, KIF5(Ø)–CPP-MT; orange map, KIF5(Ø)–taxol-MT. Four different contour levels are shown. Light color, 1.3 σ; light mesh, 1.35 σ; medium mesh, 1.43 σ; dark mesh, 1.5 σ.
See also Supplementary Fig S7.
The most striking difference is the conformation of L11. In KIF5(Ø)–taxol-MT, L11 comes from the left side of α6 and continues toward the plus-end to connect to α4 (Fig8C). In KIF5(Ø)–CPP-MT, however, L11 passes through the space between α6 and α4 and continues toward the left side to connect to the N-terminus of α4. The thick transverse route of L11 might be stabilized by the interaction with H11’ and H12 of α-tubulin, that triggers the mutual adaptation of KIF5C and α-tubulin as described. Loop L12, located on the C-terminal side of α4, also strongly contributes to KIF5C–MT binding. Our map clearly illustrates its route, which would include its ionic interaction with H12 of β-tubulin (Fig8D).
Another interesting difference at the left, plus-end sides of α4 is the strong connection between KIF5C and β-tubulin, which occurs only for GMPCPP-MT and not for GDP-taxol-MT (Fig8E). This connection might be made among L7, α4 of KIF5C and the loops (H4-S5 and/or H8-S7) at the minus-end side of β-tubulin, with a slight distortion of L7. A similar distortion was reported in the structural neighbor myosin, in which distortion of the β-sheet is necessary for the effective release of ADP (Coureux et al, 2003). Collectively, these observations suggest that the contact between the L7 and β-tubulin, aided by helix α4, will cause a twist of the β-sheet, which in turn will trigger the conformational change of switch I to release ADP from the nucleotide-binding pocket.
L11 is necessary for substrate recognition by KIF5C
The most prominent conformational change at the KIF5C–MT interface involves L11 of KIF5C, which not only serves as a MT interface but also supports the rigor conformation of switch II helix α4. This observation suggested that L11 might be the key player not only in discriminating GTP-MT from GDP-MT but also in determining the fast rate of ATP hydrolysis on GTP-MT. To test this hypothesis, we made the KIF5C swap mutant, KIF5C carrying L11 of KIF1A, and tested the binding affinity for GMPCPP- and GDP-MT (Fig9A and B). As previously reported, the affinity of the KIF5C catalytic head for GTP-MT was approximately three times higher (Kd = 190 ± 70 nM) than that for GDP-MT (Kd = 500 ± 170 nM) (Sweeney & Houdusse, 2010; Nakata et al, 2011). However, the L11 swap mutation completely abolished the substrate specificity for GTP-MT versus GDP-MT, reflected in the similar dissociation constants for GMPCPP- and GDP-MT (Kd = 590 ± 170 nM vs. Kd = 730 ± 210 nM, respectively). These results directly showed that the KIF5C motor head uses L11 to discriminate MT lattice structures differing in their guanine nucleotide content.
Figure 9. Importance of L11 to discriminate GTP-MT from GDP-MT.
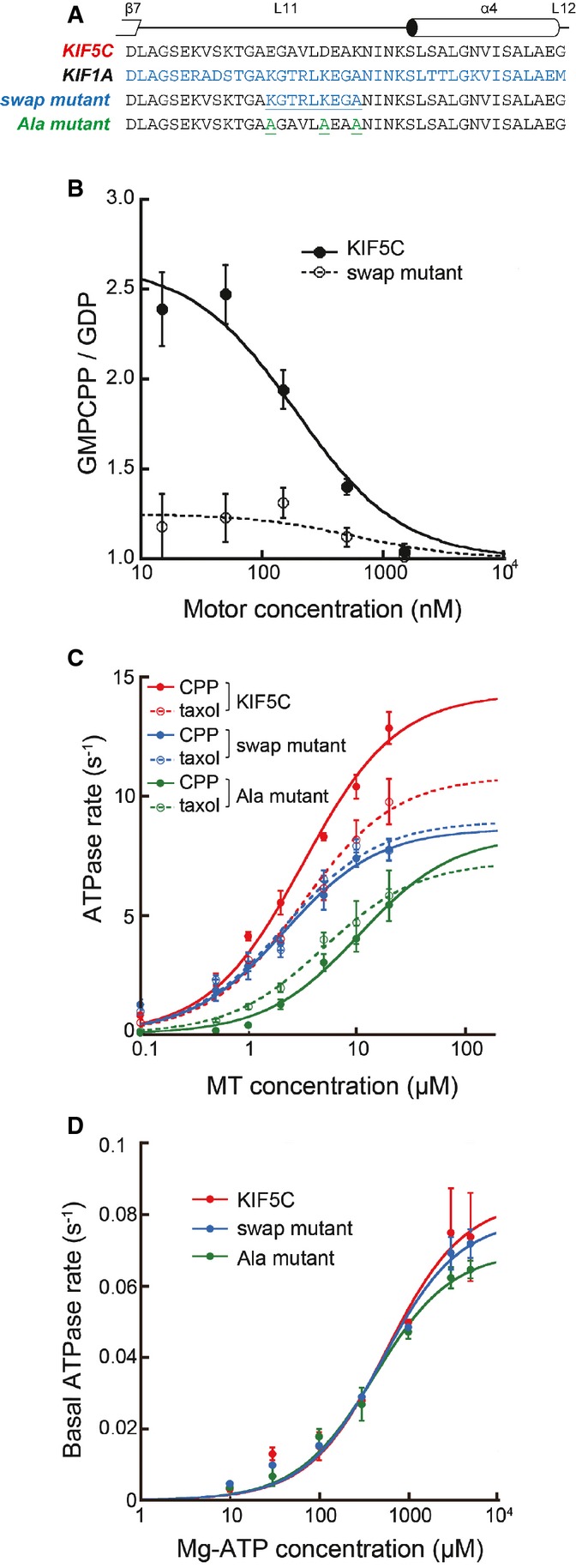
- Sequence alignments of KIF5C, KIF1A, and the L11 mutants.
- Binding assays of KIF5C and swap mutant in the nucleotide-free state. The ratio of MT-bound KIF5C vs. GMPCPP-MT and GDP-MT is plotted against the motor concentration. The mean and SEM were measured from 120 MTs at each point.
- ATPase activities of KIF5C (red) and the swap (blue) and Ala (green) mutants with GMPCPP-MT (solid lines) and GDP-taxol-MT (broken lines). Wild-type data are the same as in Fig1B.
- Basal ATPase activities. Error bars are the SEM of at least four independent experiments.
Next, the MT-activated ATPase activity of the L11 swap mutant was examined using GMPCPP- and GDP-taxol-MT, to elucidate the contribution of L11 to rapid ATPase cycling in the presence of GMPCPP-MT (Fig9C). The swap mutation again abolished the difference between GMPCPP- and GDP-taxol-MT. The substrate specificity of the swap mutant, as indicated by the lower MT-concentration area under the curve, was very similar to that of wild-type KIF5C for GDP-taxol-MTs, suggesting that the swapping area of L11 was responsible for the higher substrate specificity of KIF5C for GMPCPP-MT. To further examine whether the charged residues in this swapping area are the main contributor to the substrate specificity of KIF5C for GMPCPP-MTs, we introduced a triple-alanine mutation into the three charged regions in L11 and then measured the activity of its MT-activated ATPase. These triple mutations indeed lessened the difference between GMPCPP- and GDP-taxol-MT, suggesting that at least one of these residues contributes to substrate specificity. However, the alanine mutations also increased the KM, MT for both GMPCPP- and GDP-MT, indicating that one if not all of these residues is involved in the interface common to both GMPCPP- and GDP-taxol-MT. Thus, both the charged and the hydrophobic residues corresponding to those in the area of the swap mutation participate in defining the characteristic function of L11 of KIF5C. In addition, neither the swap mutation nor the alanine mutation affected the basal ATPase activity of KIF5C (Fig9D), suggesting that the L11 sequence of KIF5C is not involved in this function in the absence of MTs, but it is involved in the MT-activated pathway of the KIF5C ATPase.
Discussion
In this study, we solved both the crystal structure of nucleotide-free KIF5C and the cryo-EM structure of nucleotide-free KIF5C bound to GMPCPP-stabilized MT. These structures showed the first detailed structural evidence of mutual conformational changes to KIF5C and GMPCPP-MT upon their interaction.
Implications for conformational change of tubulin induced by KIF5C binding
There have been numerous cryo-EM studies of MT bound to the various MAPs including KIFs, albeit no or negligible conformational changes to MTs were reported. Some of the possible reasons for this might be the use of stabilizing reagents for MT, the effect of the bound nucleotide, or the technical limitation to solve the structure of the MT. In this study, we solved the GMPCPP-bound MT without any other MT-stabilizing reagent by the single-particle-based program (Ogura et al, 2014). Although clear evidence is shown here to support a different conformation of αβ-tubulin between MT with and without bound KIF5C, determining the nature of that new conformation requires higher resolution.
KIF5C binding on GMPCPP-MT generates the downward rotation of N- and C-domains of αβ-tubulin (Figs5 and 10A). These movements lead α- and β- tubulins to adopt compact conformations. Interestingly, the I-domain of α-tubulin and β-tubulin in the KIF5(Ø)–CPP-MT take very similar conformation with those in the GDP-taxol-MT despite the difference of the nucleotide states as well as the presence/absence of the MT-stabilizing agent (Fig10B). Considering that I-domain connects the top to the bottom of αβ-tubulin to stabilize or destabilize the longitudinal contact of MT, this I-domain conformation should be one of the energetically stable forms in the straight MT that could be induced by the binding of MT-stabilizing agent to the inside of the MT or the binding of some of the MT-associated proteins to the surface. On the other hand, conformations of N- and C-domains might be altered depending on the bound proteins/reagents, reflecting the distinct surface conformations of MT. In the recently solved KIF5–GMPCPP-MT complex, the tubulin dimer adopts a similar conformation with that observed in the GDP-taxol-MT (Alushin et al, 2014). Thus, the difference from our structure is the N- and C-domain conformations of α-tubulin. Possible explanations for this difference include the different nucleotide state of KIF5 (nucleotide-free versus a possible ATP-bound state) and the different symmetry of the microtubules (15 PFs four-start versus 13 PFs three-start MTs), albeit the latter is not likely because the lateral contacts did not differ between them.
Figure 10. Schematic representation of the conformational changes in GTP-MT and kinesin.

- Comparison of MT structures between KIF5(Ø)–CPP-MT (brown) and CPP-MT (green). KIF5C induces the downward movement of C/N-domains of α- and β-tubulin.
- Comparison of MT structures between KIF5(Ø)–CPP-MT and taxol-MT (PDB ID: 1JFF). KIF5C induces the downward movement of C/N-domains of α-tubulin. β-tubulin structures are very similar between them.
- Schematic model for the ATPase cycle in the dimeric kinesin motility on GTP-MT. In the Mg-ADP state, switch II acquires the ADP-like conformation with the flexible neck-linker, which represents the weak-binding state on the MT (i). Then, nucleotide-binding pocket opens to release Mg2+ and ADP (ii). After releasing Mg2+ and ADP, switch I adopts closed conformation to hold the next Mg-ATP (iii). Switch II takes the rigor conformation which is supported by the docked NIS and elongated L11. Switch II is distant from switch I; thus, Mg-ATP can enter the nucleotide-binding pocket. Mg-ATP binding allows neck-linker docking and produces the power stroke that carries the partner head (gray model) forward (iv).
A novel conformation of KIF5C bound with GTP-state microtubule
The structural features of nucleotide-free KIF5C in the presence of GMPCPP-MT can be summarized as follows: (i) Switch I (α3-L9) is in the closed conformation. (ii) Switch II helix α4 assumes the rigor conformation, in which switch II is distal from the nucleotide-binding pocket. (iii) The L11 and NIS are inserted between α4 and α6.
The C-terminus of the helix α3 rotates toward the nucleotide-binding pocket, and the elongated L9 covers the pocket (Fig10C (iii)). This closed switch I conformation is similar to that of KIF5 holding Mg-ATP in the nucleotide-binding pocket. This means that KIF5C switch I is ready for ATP binding and hydrolysis in the nucleotide-free condition. The rigor conformation of switch II contains the clockwise rotation of α4 around the MT axis, with the elongation of L11 caused by melting of 2.5 turns of the N-terminus of α4. Elongated L11 and NIS support the rotation of α4 by squeezing into the space between α4 and α6. NIS is stabilized and points downward, albeit more than two-thirds of the neck-linker remains flexible. The catalytic core floats above α4 with the help of L11 and NIS. Switch II helices are thus separated from the nucleotide-binding pocket and from switch I, thereby ensuring that the entrance of the nucleotide-binding pocket is open for ATP entry.
On the GTP-MT, KIF5 tends to adopt the rigor conformation, which enables the effective ATP cycling with the recognition of the specific surface structures of GTP-MT, H4-S5 loop of β-tubulin and H11’ of α-tubulin, by the L11 of KIF5. This specific binding accomplishes a higher specificity of KIF5C to the GTP-MT than to the GDP-MT, and explains a preferential binding to the GTP-MT in the polarized transport of KIF5 to the axon over the dendrites (Nakata et al, 2011).
Conserved structural mechanism for ADP release from KIFs
Among the three features above, only the closed switch I conformation is common between the nucleotide-free kinesin-1 on GMPCPP-MT, GDP-taxol-MT, or GDP-tubulin (Atherton et al, 2014; Cao et al, 2014; Shang et al, 2014). Since kinesin-1 moves well both on the GMPCPP- and GDP-taxol-MTs, albeit with different motility speeds, common structural features between kinesin-1 on two distinct types of MTs may reveal important aspects that describe the fundamental functionality of kinesin-1 motility. Considering that the α4/L11 serves as the common interface for both types of MTs, there might be two possibilities about how the information of MT binding is transmitted from the MT to switch I. The first possibility is that the closed switch I might be triggered by MT binding through the common interface α4/L11. A matter of concern for this is that no contact between switch I and α4/L11 (and even switch II) was observed in KIF5C on the GMPCPP-MT; thus, how information of MT binding is transmitted to switch I is currently unaccountable. The second possibility is that the closed switch I might be triggered through the L7-involved pathway. Kinesin–tubulin contact through the L7 was observed in several KIFs, including kinesin-1, kinesin-3, and kinesin-14 (Hirose et al, 2006; Nitta et al, 2008; Gigant et al, 2013). Further structural studies are required to solve the question.
Conformational variations of KIFs regulated by the MT
In the previous structural studies and in this work, three conformational variations of each switch I, switch II, and neck-linker were determined. Switch I can assume three conformations, trapped, open, and closed. The trapped conformation always includes ADP in the nucleotide-binding pocket, which is surrounded by the Mg-water cap (Nitta et al, 2008). The open conformation has weakly bound ADP without Mg2+ or nothing in the pocket. In the closed conformation, Mg-ATP is in the pocket or the pocket is empty only in the presence of MT. Switch II can also assume three conformations, ADP-like, rigor, and ATP-like, that are completely coupled with three conformations of neck-linker, undocked, NIS-docked, and docked, respectively. Currently, seven variations of switch I = switch II = neck-linker combinations have been reported: (i) trapped = ADP-like = undocked, (ii) open = ADP-like = undocked, (iii) open = rigor = NIS-docked, (iv) open = ATP-like = docked, (v) closed = ADP-like = undocked, (vi) closed = rigor = NIS-docked, and (vii) closed = ATP-like = docked.
Here, nucleotide-free KIF5 on the GDP-taxol-MT or GDP-tubulin takes the closed = ADP-like = undocked conformation, whereas nucleotide-free KIF5 on the GTP-MT takes the closed = rigor = NIS-docked conformation (Rice et al, 1999; Atherton et al, 2014; Cao et al, 2014; Shang et al, 2014). Since the switch I is responsible for the chemical cycle of kinesins, its conformation might be conserved even on the different type of MTs. Switch II, however, is one of the major interface for the microtubule so that switch II as well as the neck-linker conformations would be altered depending on the difference of the MT conformation. Interestingly, GDP-taxol-MT induces the nucleotide-free Kar3 to take the closed = rigor conformation, suggesting that the favorable conformation of MT varies with the type of kinesin motors (Supplementary Fig S6C). The difference in preference between KIF5 and Kar3 would reflect the biochemical results, which showed that KIF5 tends to stabilize (GTP-)MT (Fig6), whereas Kar3 tends to destabilize (GDP-)MT (Endow et al, 1994).
Finally, we assume the structural model of KIF5C during ADP release cooperates with rebinding (or initial binding) to the GTP-MT and during the ATP binding cooperates with the power stroke in dimeric motility (Rice et al, 1999) (Fig10C). In the Mg-ADP state, KIF5C acquires the trapped = ADP-like = undocked conformation, which represents its weak binding or detached state on/above the MT (i). MT-sensing through L7 and/or α4/L11 might trigger the transient opening of the nucleotide-binding pocket to release Mg2+ and ADP (switch I: open) (ii), presidential to taking the closed conformation (Nitta et al, 2008) (iii). This switch I movement cooperates with the conformational change of switch II from the ADP-like to the rigor conformation and thus a transition from weak to strong MT binding. With ATP entry and binding, conformational changes from the rigor to the ATP-like conformation occur and produce the power stroke that carries the partner head forward (iv). Since the KIF5C structure in the AMPPNP state on the GMPCPP-MT is still missing, further structural studies are required to clarify this structural model.
Materials and Methods
X-ray crystallography
The KIF5C motor domain K334 (mouse KIF5C residues 1–334 with a 7 × His-tag C-terminal) purified by immobilized metal affinity chromatography and cation exchange chromatography (AKTA Explorer 10S, RESOURCE S column; GE Healthcare) was dialyzed against crystallization buffer (10 mM PIPES, pH 7.4, 10 mM HEPES, pH 7.4, 100 mM NaCl, 1 mM MgCl2, 1 mM EGTA, 20% (w/v) sucrose and 100 μM ADP, pH 7.0) and then concentrated to ∽25 mg/ml using an Amicon Ultrafree 30K filter (Millipore). Crystallization was done using the hanging-drop vapor diffusion method. The reservoir buffer consisted of 2 M ammonium sulfate, 100 mM sodium citric acid, pH 5.5 and 16% (v/v) glycerol. One microliter of protein solution and 1 μl of reservoir buffer were mixed to form a drop, with crystals appearing within a week in samples kept at 20°C. Subsequently, the C-terminal tail peptide solution of KIF5C (EAVRAKNMARRAHSAQIAKPIRPG) was added to the drop to release bound ADP, such that the final concentration of the tail peptide was 1–2 mM. A few minutes after the addition of the tail peptide, the crystals were frozen in liquid nitrogen. X-ray diffraction experiments were performed at −178°C on a NW12A beam line (wavelength λ = 1.00 Å) at the Photon Factory Advanced Ring (KEK, Tsukuba, Japan) and on the BL41XU (wavelength λ = 1.00 Å) at SPring-8 (Hyogo, Japan), and final data were obtained on a NW12A beam line. The data were processed using the program HKL2000 (Otwinowski & Minor, 1997). The structures were determined using molecular replacement methods, with the structure of KIF5 in the ADP state (PDB ID: 2KIN) as the initial model. Subsequent rounds of model building and refinement were performed using the program Coot (Emsley et al, 2010) and the program Refmac5 (Murshudov et al, 2011). Finally, the model of the nucleotide-free KIF5C was refined to R and R-free values of 23.7 and 29.9%, respectively. Data collection and the refinement statistics are shown in Table1.
Preparation of MT and KIF5C for cryo-EM
Tubulin was purified from porcine brains by six cycles of polymerization and depolymerization. A high-molarity PIPES buffer was used to remove contaminating MT-associated proteins (Castoldi & Popov, 2003). For GDP-taxol-MTs, 18 μM tubulin was incubated in a polymerization buffer (100 mM PIPES, pH 6.8 adjusted with KOH, 1 mM EGTA, 1 mM MgCl2, 1 mM GTP, and 6–9% DMSO) at 4°C for 15 min and then clarified by centrifugation at 4°C for 30 min at 100,000 g using a TLA-100.3 rotor (Beckman Coulter) in a TLX Ultracentrifuge (Beckman Coulter). The supernatant was polymerized at 37°C for 60 min. Taxol was added in a stepwise fashion to a final concentration of 20 μM. The MTs were collected by centrifugation through a 20% (v/v) glycerol cushion at 37°C for 15 min at 20,000 g and resuspended in PEM-DMSO buffer (100 mM PIPES, pH 6.8 adjusted with KOH, 1 mM EGTA, 1 mM MgCl2, and 6–9% DMSO) containing 20 μM taxol. GMPCPP-MTs were prepared as described previously (Yajima et al, 2012). The KIF5C motor domain construct K345 [mouse KIF5C residues 1–345 and a 7 × His-tag; the same construct as K351, used in our previous report (Okada & Hirokawa, 1999)] was expressed in Escherichia coli BL21 (DE3) cells (Novagen) and purified from the cell pellet by immobilized metal affinity chromatography. The eluted fractions were purified further by cation exchange chromatography (AKTA Explorer 10S, RESOURCE S column; GE Healthcare). The purified protein was concentrated in an IMEKOAc buffer (50 mM imidazole, 5 mM Mg-acetate, 1 mM EGTA, 50 mM K-acetate, pH 7.2 adjusted with HCl), frozen in liquid N2, and stored at −80°C.
Cryo-EM
A 5-μl drop of the polymerized MT was placed onto a glow-discharged EM grid (Maxtaform HF35; Pyser-SGI) coated with a perforated carbon film. After 30 s, the solution was wicked away with a piece of Whatman No. 1 filter paper and a 5-μl drop of the KIF5C-containing solution (1 μM) was quickly applied. After another 30 s, this solution was replaced with 2 U apyrase/ml and the mixture was allowed to stand for 60 s to obtain a nucleotide-free state. The sample was then washed with IMEKOAc buffer containing 0.1% Triton X-100. Immediately after absorbing the drop by the filter paper, the grid was frozen by plunging it into liquid ethane. The specimens were observed using 200-kV field emission cryo-EM (JEM-2010F; JEOL) with a 626 cryotransfer holder (Gatan, Inc.). Images were recorded at 40,000-fold magnification on SO163 film (Eastman Kodak Co.), with a defocus of 1,200–2,600 nm, and developed with D19 (Eastman Kodak Co.) for 10 min at 20°C.
Image processing and statistical analysis
The 15-protofilament and 2-start helix MTs (15-protofilament and 4-start for tubulin monomers) were chosen by their moiré pattern. Selected films were digitized with a charge-coupled device film scanner (Scitex Leafscan45; Leaf Systems) to obtain a final pixel size in the digitized images of 2.5 Å. Image analysis was carried out as previously described (Ogura & Sato, 2006; Yajima et al, 2012; Ogura et al, 2014), with a slight modification in that the high-pass-filtered image was cropped into square pieces of 64 × 64 nm instead of 60 × 60 nm to ensure that the size was sufficient to visualize the kinesin–MT complex. For KIF5(Ø)–CPP-MT and KIF5(Ø)–taxol-MT, >302,000 and >271,000 tubulin dimers were averaged in total, respectively, to achieve the claimed resolution. For estimating resolution, data sets were split in half to make two independent reconstructions and the FSC function was calculated for the whole map (32.5 × 32.5 × 32.5 nm). The statistical significance of differences between KIF5(Ø)–CPP-MT and KIF5(Ø)–taxol-MT maps was examined by Student's t-test as described previously (Yajima et al, 2012).
Atomic model fitting
The atomic models were fitted in silico to clarify the regions where the major conformational changes occur upon the KIF5C–MT binding. As detailed below, we only performed the rigid body fitting of subdomains of KIF5C or tubulin due to the resolution limit except for the fitting of the helix α4 of KIF5C whose density was well separated as a tubular density and also was detected as a helix by the volume tracer tool in Sculptor (Birmanns et al, 2011).
The electron crystallography structure of tubulin [PDB ID: 1JFF (Löwe et al, 2001)] was used as the initial model for tubulin fitting of our KIF5–GMPCPP-MT map. 1JFF (dimer) was fitted rigidly into the map using Fit in Map tool in UCSF Chimera (Pettersen et al, 2004). However, the dimer model did not fit into the map; thus, we separated the 1JFF model into monomers and fitted these two monomers as rigid bodies. As a result, the fitting scores of the average map value (AMV) increased from 160.0 to 161.1, the number of atoms inside the contour increased from 3,499 to 3,509, and the cross-correlation coefficient (ccc) did not change with a value of 0.699 (Supplementary Fig S4B). To achieve better fitting, we divided tubulin into four domains pieces, the N1-domain (aa 1–102), N2-domain (aa 103–203), I-domain (aa 204–382) and C-domain (aa 383–C-terminal). Based on this division, we examined several subdomain fittings: two-subdomain fittings (N/IC, N/IC, and NI/C), three-subdomain fitting (N/I/C), and four-subdomain fitting (N1/N2/I/C). Those subdomains were rigidly docked separately and reconnected using MODELLER to satisfy the spatial restraints (Sali & Blundell, 1993). The fitting scores improved to reach a plateau at the three-subdomain fitting of α-tubulin (Supplementary Fig S4A). Consequently, we decided to take this docking as a pseudo-atomic model of MT (AMV = 163.5, atoms inside the contour = 3,776, and ccc = 0.722). Fitting of α-tubulin via the monomer model to the three-subdomain model gave AMV, atoms inside the contour, and ccc varied from 160.7 to 164.8, from 1,697 to 1,920, and from 0.706 to 0.746, respectively. On the other hand, subdividing β-tubulin gave the little additional improvement of fittings with the little alteration of the fitting parameters; those scores of β-tubulin (monomer fitting through to four-subdomain fitting) increased from 160.9 to 162.7 (AMV), from 1,802 to 1,856 (atoms inside the contour), and from 0.708 to 0.718 (ccc) (Supplementary Fig S4A). The helices H11 and H12 in the newly created atomic model of α-tubulin matched the helical densities detected by the volume tracer tool in Sculptor, also supporting the validity of this fitting (Supplementary Fig S4F and G).
For the density of KIF5C, the X-ray crystal structure of nucleotide-free KIF5C solved in this work was used as the initial model (AMV = 154.0, atoms inside the contour = 882, and ccc = 0.713) (Supplementary Fig S4D). Since the map corresponding to the switch I region (aa 177–205), in which the nucleotide-free KIF5C crystal model did not fit, was preferably similar to the map of KIF5(ADP-Mg-AlFx)–MT, we rigidly fit the switch I region by referring to the 4HNA structure. This fitting of switch I enabled us to obtain better fitting scores of α3. The AMV increased from 146.6 to 152.6, atoms inside the contour from 39 to 40, and the ccc from 0.322 to 0.330 (Supplementary Fig S4C). To fit switch II (aa 233–296), especially helix α4, which was different between the crystal structure and the cryo-EM structure, the volume tracer tool in Sculptor was used to find a possible helical density corresponding to α4. We then moved and fit the switch II α4 to the detected helical density using the Fit in Map tool (Supplementary Fig S4H). Through these fittings, the AMV of the α4 increased from 147.6 to 158.7, atoms inside the contour increased from 24 to 34, and the ccc from 0.274 to 0.302 (Supplementary Fig S4C). Fragmented models were reconnected, and finally, the KIF5 atomic model was found to fit well to the map and was set as the model of the KIF5 rigor model (AMV = 156.8, atoms inside contour = 948, ccc = 0.740) (Supplementary Fig S4D). We finally confirmed that the model had no steric clashes by the ADIT deposition tool. As a consequence, we got a pseudo-atomic model of KIF5(Ø)–CPP-MT, which fit in the map in the similar level as previously published model (Supplementary Fig S4E). Further high-resolution cryo-EM study is necessary to validate this model.
Preparation of kinesin L11 mutants
Mutated K345 with a C-terminal reactive cysteine tag (-RKRCR-) was generated by overlapping PCR using the K345 expression vector as the template. The final PCR product was checked by DNA sequencing before it was cloned into the pET21b expression vector (Novagen). The protein was expressed in E. coli BL21 (DE3) cells and purified by immobilized metal affinity chromatography. For the binding assay, wild-type K345 and its mutants were labeled with Alexa 594 maleimide (Invitrogen).
Binding assay
Purified tubulin was labeled with Alexa Fluor 488 or 647 succinimidyl ester (Invitrogen) as described previously (Desai & Mitchison, 1998). Labeled tubulin was mixed with unlabeled tubulin such that 10% of the tubulin was labeled with the Alexa Fluor dye. After 15-min incubation on ice, the mixture was clarified and polymerized to yield GMPCPP-MT and GDP-MT. Labeled GMPCPP-MT and GDP-MT were then mixed at a ratio of 1:1 in assay buffer (80 mM PIPES, pH 6.8, adjusted with KOH, 0.8 mM EGTA, 0.8 mM MgCl2, 20% glycerol, 2 mM AMP-PNP, 1 mM glucose, 0.2 mg glucose oxidase/ml, 0.04 mg catalase/ml, and 5 mM cysteamine). The Alexa Fluor 594-labeled kinesin or the mutants were then mixed into the MT solution to varying final concentrations (15–1,500 nM). For the nucleotide-free state, 2 U apyrase/ml was added to the buffer. After incubation at 37°C, the kinesin-MT mixture was spread onto the cover glass and glutaraldehyde was added to a final concentration of 2.5% to stop MT depolymerization. The specimen was observed using the ELYRA P.1 system (Carl Zeiss, Jena, Germany) in the TIRF mode.
Gliding assay
MT gliding assays were performed as described previously (Carter & Cross, 2001), with a slight modification. In brief, a flow chamber assembled from a glass slide and a coverslip was coated with a penta-His antibody (Qiagen) for 3 min and washed with PEM buffer (100 mM PIPES, pH 6.8 adjusted with KOH, 1 mM EGTA, and 1 mM MgCl2). Then, 0.1 mg of kinesin/ml was introduced to allow its immobilization at the C-terminal His-tag. The chamber was then washed and blocked with casein-containing PEM buffer (1 mM DTT, 20 μM taxol for GDP-taxol-MT and 2 mg casein/ml in PEM buffer). Subsequently, the MT solution was injected into the chamber together with the motility buffer (1 mM DTT, 20 μM taxol for GDP-taxol-MT, 2 mg casein/ml, 5 mM Mg-ATP, and 5 mg glucose/ml in PEM buffer) and an oxygen scavenging system consisting of 0.05 mg glucose oxidase/ml and 0.1 mg catalase/ml. Time-lapse images were acquired using the ELYRA P.1 system in the TIRF mode at 37°C.
ATPase assay
ATPase activity was assayed at 25°C with the EnzChek phosphate assay kit (Molecular Probes) using a spectrophotometer (V630-Bio; JASCO). MT-activated ATPase activity was measured in PEM buffer containing 20 nM K345, varying concentrations of MT (0–20 μM) and 2 mM Mg-ATP. Basal ATPase activity was determined in PEM buffer containing 2 μM K345 and 0–5 mM Mg-ATP.
MT shrinking assay
Labeled tubulin was mixed in a 1:5 molar ratio to unlabeled tubulin and incubated in polymerization buffer (100 mM PIPES, pH 6.8 adjusted with NaOH, 1 mM EGTA, 1 mM MgCl2, 6% DMSO, and 1 mM GMPCPP) at 37°C for 2 h. Labeled GMPCPP-MT and K345 were mixed with 5 mM AMPPNP, and the mixture was spread onto a coverslip. Time-lapse observations were performed at room temperature using TIRF as described in the sections above and measured the duration until depolymerization began. GMPCPP-MTs polymerized with PIPES adjusted with KOH were also tested and showed similar tendencies as was observed for NaOH (Fig6), albeit spending much longer periods.
Accession numbers
The crystal structures, the cryo-EM maps, and the coordinates we described and used in this study have been deposited: nucleotide-free KIF5C (PDB ID 3WRD); KIF5-ADP (PDB ID 3X2T); nucleotide-free KIF5C complexed with GMPCPP microtubule (EMD-5916); GMPCPP microtubule (EMD-2697, PDB ID 3J7I).
Acknowledgments
We are grateful to Y. Okada for helpful discussions. We thank K. Hirose for providing us with the cryo-EM maps of the Kar3–MT complex. We thank members of the Hirokawa Lab for assistance and discussions. This work was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), as a Grant-in-Aid for Specially Promoted Research to N.H., and by grants to T.O. and C.S. from MEXT, the Japan New Energy and Industrial Development Organization (NEDO), and Japan Science and Technology Corporation (JST).
Author contributions
MM and HY collected the cryo-EM data; TO and CS processed the cryo-EM data; SI solved the crystal structures; RN, MM, and HY performed the molecular docking; MM performed the biochemical experiments; MM, HY, SI, RN, and NH discussed the data and wrote the manuscript; NH conceived and directed the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Supplementary Movie S1
Review Process File
References
- Alonso MC, Drummond DR, Kain S, Hoeng J, Amos L, Cross RA. An ATP gate controls tubulin binding by the tethered head of kinesin-1. Science. 2007;316:120–123. doi: 10.1126/science.1136985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. High-resolution microtubule structures reveal the structural transitions in αβ-Tubulin upon GTP hydrolysis. Cell. 2014;157:1117–1129. doi: 10.1016/j.cell.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asbury CL, Fehr AN, Block SM. Kinesin moves by an asymmetric hand-over-hand mechanism. Science. 2003;302:2130–2134. doi: 10.1126/science.1092985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton J, Farabella I, Yu IM, Rosenfeld SS, Houdusse A, Topf M, Moores CA. Conserved mechanisms of microtubule-stimulated ADP release, ATP binding, and force generation in transport kinesins. Elife. 2014;3:e03680. doi: 10.7554/eLife.03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmanns S, Rusu M, Wriggers W. Using Sculptor and Situs for simultaneous assembly of atomic components into low-resolution shapes. J Struct Biol. 2011;173:428–435. doi: 10.1016/j.jsb.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block SM, Goldstein LS, Schnapp BJ. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- Cao L, Wang W, Jiang Q, Wang C, Knossow M, Gigant B. The structure of apo-kinesin bound to tubulin links the nucleotide cycle to movement. Nat Commun. 2014;5:5364. doi: 10.1038/ncomms6364. [DOI] [PubMed] [Google Scholar]
- Carter N, Cross R. An improved microscope for bead and surface-based motility assays. Methods Mol Biol. 2001;164:73–89. doi: 10.1385/1-59259-069-1:73. [DOI] [PubMed] [Google Scholar]
- Castoldi M, Popov AV. Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expr Purif. 2003;32:83–88. doi: 10.1016/S1046-5928(03)00218-3. [DOI] [PubMed] [Google Scholar]
- Chang Q, Nitta R, Inoue S, Hirokawa N. Structural basis for the ATP-induced isomerization of kinesin. J Mol Biol. 2013;425:1869–1880. doi: 10.1016/j.jmb.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Coureux PD, Wells AL, Ménétrey J, Yengo CM, Morris CA, Sweeney HL, Houdusse A. A structural state of the myosin V motor without bound nucleotide. Nature. 2003;425:419–423. doi: 10.1038/nature01927. [DOI] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ. Preparation and characterization of caged fluorescein tubulin. Methods Enzymol. 1998;298:125–132. doi: 10.1016/s0076-6879(98)98014-4. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endow SA, Kang SJ, Satterwhite LL, Rose MD, Skeen VP, Salmon ED. Yeast Kar3 is a minus-end microtubule motor protein that destabilizes microtubules preferentially at the minus ends. EMBO J. 1994;13:2708–2713. doi: 10.1002/j.1460-2075.1994.tb06561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigant B, Wang W, Dreier B, Jiang Q, Pecqueur L, Plückthun A, Wang C, Knossow M. Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat Struct Mol Biol. 2013;20:1001–1007. doi: 10.1038/nsmb.2624. [DOI] [PubMed] [Google Scholar]
- Himmel DM, Gourinath S, Reshetnikova L, Shen Y, Szent-Györgyi AG, Cohen C. Crystallographic findings on the internally uncoupled and near-rigor states of myosin: further insights into the mechanics of the motor. Proc Natl Acad Sci USA. 2002;99:12645–12650. doi: 10.1073/pnas.202476799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Nitta R, Okada Y. The mechanisms of kinesin motor motility: lessons from the monomeric motor KIF1A. Nat Rev Mol Cell Biol. 2009a;10:877–884. doi: 10.1038/nrm2807. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009b;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Hirose K, Akimaru E, Akiba T, Endow SA, Amos LA. Large conformational changes in a kinesin motor catalyzed by interaction with microtubules. Mol Cell. 2006;23:913–923. doi: 10.1016/j.molcel.2006.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson C, Schnapp B, Banker GA. A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron. 2006;49:797–804. doi: 10.1016/j.neuron.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Kikkawa M, Sablin EP, Okada Y, Yajima H, Fletterick RJ, Hirokawa N. Switch-based mechanism of kinesin motors. Nature. 2001;411:439–445. doi: 10.1038/35078000. [DOI] [PubMed] [Google Scholar]
- Kikkawa M, Hirokawa N. High-resolution cryo-EM maps show the nucleotide binding pocket of KIF1A in open and closed conformations. EMBO J. 2006;25:4187–4194. doi: 10.1038/sj.emboj.7601299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozielski F, Sack S, Marx A, Thormählen M, Schönbrunn E, Biou V, Thompson A, Mandelkow EM, Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwe J, Li H, Downing KH, Nogales E. Refined structure of alpha beta-tubulin at 3.5 A resolution. J Mol Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J Cell Biol. 2003;162:1045–1055. doi: 10.1083/jcb.200302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Niwa S, Okada Y, Perez F, Hirokawa N. Preferential binding of a kinesin-1 motor to GTP-tubulin-rich microtubules underlies polarized vesicle transport. J Cell Biol. 2011;194:245–255. doi: 10.1083/jcb.201104034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta R, Kikkawa M, Okada Y, Hirokawa N. KIF1A alternately uses two loops to bind microtubules. Science. 2004;305:678–683. doi: 10.1126/science.1096621. [DOI] [PubMed] [Google Scholar]
- Nitta R, Okada Y, Hirokawa N. Structural model for strain-dependent microtubule activation of Mg-ADP release from kinesin. Nat Struct Mol Biol. 2008;15:1067–1075. doi: 10.1038/nsmb.1487. [DOI] [PubMed] [Google Scholar]
- Ogura T, Sato C. A fully automatic 3D reconstruction method using simulated annealing enables accurate posterioric angular assignment of protein projections. J Struct Biol. 2006;156:371–386. doi: 10.1016/j.jsb.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Ogura T, Yajima H, Nitta R, Hirokawa N, Sato C. New simulated annealing approach considering helix bending applied to determine the 8.8Å structure of 15-protofilament microtubules. J Struct Biol. 2014;188:165–176. doi: 10.1016/j.jsb.2014.08.009. [DOI] [PubMed] [Google Scholar]
- Okada Y, Hirokawa N. A processive single-headed motor: kinesin superfamily protein KIF1A. Science. 1999;283:1152–1157. doi: 10.1126/science.283.5405.1152. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- Sablin EP, Case RB, Dai SC, Hart CL, Ruby A, Vale RD, Fletterick RJ. Direction determination in the minus-end-directed kinesin motor ncd. Nature. 1998;395:813–816. doi: 10.1038/27463. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Shang Z, Zhou K, Xu C, Csencsits R, Cochran JC, Sindelar CV. High-resolution structures of kinesin on microtubules provide a basis for nucleotide-gated force-generation. Elife. 2014;3:e04686. doi: 10.7554/eLife.04686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley K, Hekmat-Nejad M, Turner J, Moores C, Anderson R, Milligan R, Sakowicz R, Fletterick R. Structure of a kinesin microtubule depolymerization machine. EMBO J. 2004;23:1422–1432. doi: 10.1038/sj.emboj.7600165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindelar CV, Downing KH. The beginning of kinesin's force-generating cycle visualized at 9-A resolution. J Cell Biol. 2007;177:377–385. doi: 10.1083/jcb.200612090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindelar CV, Downing KH. An atomic-level mechanism for activation of the kinesin molecular motors. Proc Natl Acad Sci USA. 2010;107:4111–4116. doi: 10.1073/pnas.0911208107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney HL, Houdusse A. Structural and functional insights into the Myosin motor mechanism. Annu Rev Biophys. 2010;39:539–557. doi: 10.1146/annurev.biophys.050708.133751. [DOI] [PubMed] [Google Scholar]
- Vale RD, Coppin CM, Malik F, Kull FJ, Milligan RA. Tubulin GTP hydrolysis influences the structure, mechanical properties, and kinesin-driven transport of microtubules. J Biol Chem. 1994;269:23769–23775. [PubMed] [Google Scholar]
- Yajima H, Ogura T, Nitta R, Okada Y, Sato C, Hirokawa N. Conformational changes in tubulin in GMPCPP and GDP-taxol microtubules observed by cryoelectron microscopy. J Cell Biol. 2012;198:315–322. doi: 10.1083/jcb.201201161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz A, Tomishige M, Vale RD, Selvin PR. Kinesin walks hand-over-hand. Science. 2004;303:676–678. doi: 10.1126/science.1093753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Movie S1
Review Process File