

Abstract
Altered stem cell homeostasis underlies functional tissue decline during muscle aging and disease progression. Janus kinase–signal transducer and activator of transcription (JAK-STAT) signal transduction is a crucial regulator of muscle regeneration, and targeting this pathway in mice relieves aspects of debilitating muscle wasting.
Skeletal muscle functional decline is an inevitable consequence of aging, which begins with loss of muscle mass in middle age and culminates in frailty and reduced quality of life in old age. When severe, this loss is termed sarcopenia and is associated with limited mobility and increased morbidity. The increased healthcare requirements associated with sarcopenia are a major concern as the global population ages. The mechanisms involved are multifaceted and probably include alterations in gene expression, proteostasis, metabolism and stem cell exhaustion1. Together, these influences antagonize muscle regeneration, leading to an inability to adequately replace lost or damaged muscle mass.
In this issue of Nature Medicine, Price et al.2 and Tierney et al.3 present complementary studies demonstrating a crucial role for JAK-STAT signal transduction during muscle regeneration. Using a combination of genetic and pharmacological approaches, the authors report that increases in JAK-STAT signaling impair muscle regeneration during aging in mice and further provide an avenue for therapy development for muscle-wasting diseases.
Skeletal muscle regeneration requires a population of resident stem cells to replace myonuclei (postmitotic muscle fiber nuclei) after an injury. Muscle stem cells, or satellite cells, lie juxtaposed between the plasma membrane of mature skeletal muscle myofibers and the basement membrane. In response to muscle damage, quiescent satellite cells activate and upregulate myogenic transcription factors, including Myf5 and MyoD, and then amplify to form a pool of myoblasts that fuse with each other and damaged myofibers to form new myofibers. Replenishment of the satellite cell pool requires self-renewal and a return to quiescence4,5. Satellite cell ablation and lineage-tracing experiments confirm that satellite cells are indispensable for proper muscle function6,7. As a consequence of aging, the myogenic potential of satellite cells is impaired. Factors responsible include cell-intrinsic defects, such as DNA damage repair deficits, and external influences such as alterations in signals from the satellite cell niche8–11. Understanding the interplay between cell intrinsic and extrinsic factors is important in pursuit of potential therapeutic targets for combating muscle wasting.
Price et al.2 report that in satellite cells isolated from juvenile mice (3 weeks old), young adult mice (12 weeks old) and elderly mice (~78 weeks old), there is an increase in JAK-STAT signal transduction in the muscle with age, as determined by gene expression analyses. JAKs phosphorylate STAT proteins, translocating STATs from the plasma membrane to the nucleus, where they activate gene transcription of their targets12. Alterations in JAK-STAT signaling are observed in adult hair-follicle stem cell populations, where age-associated external cytokine stimulation inhibits stem cell function13. Price et al.2 similarly report age-associated deficits in satellite cell function, prompting the authors to speculate that JAK-STAT signaling may suppress myogenic activity. In a series of in vitro and transplant-based siRNA and inhibitor studies targeted at JAK kinase (using inhibitor AG490) or Stat3 (using inhibitor 5,15 DPP) in mice, the authors demonstrate that inhibition of either JAK kinase or Stat3 promotes symmetric satellite cell expansion and rescues muscle regeneration defects in elderly and dystrophic mice (Fig. 1).
Figure 1.
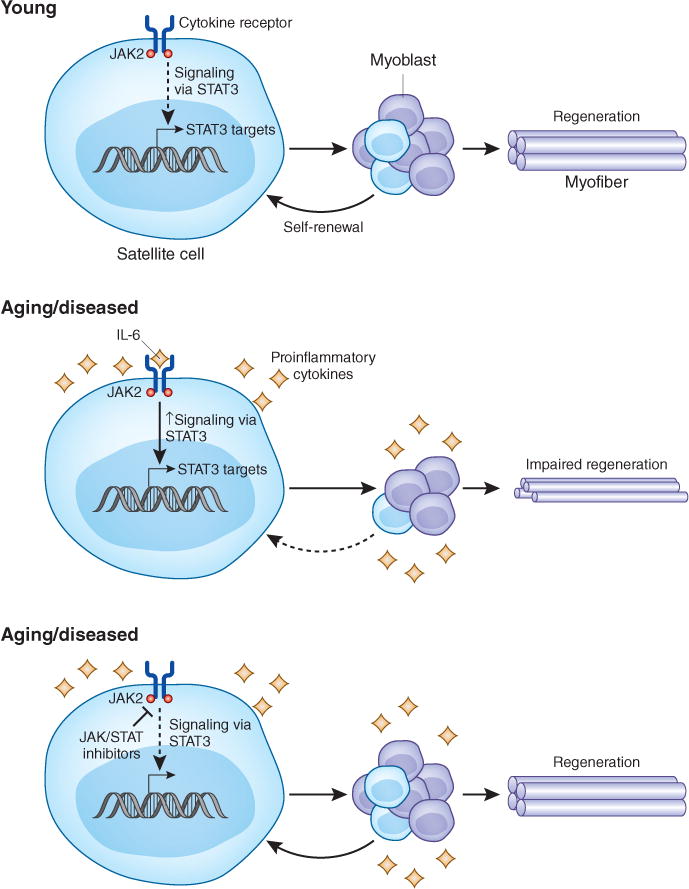
JAK-STAT signaling progressively increases with age and inhibits satellite cell function. In young mice, Price et al.2 and Tierney et al.3 find that JAK-STAT signaling is low and satellite cells are able to expand to form a proliferating myoblast pool, the majority of which differentiate and fuse to repair muscle fibers. A small fraction return to a quiescent satellite cell state in a process termed self-renewal. In mice in an aging or diseased environment, the authors find that hyperactive JAKSTAT signaling impairs myogenic proliferation, differentiation and self-renewal, leading to diminished regenerative capacity. In the presence of JAK or STAT inhibitors, satellite cell function is restored and muscle regeneration is rescued.
In the companion report, Tierney et al.3 explore the hypothesis that Stat3, a downstream effector of the proinflammatory cytokine interleukin-6, which is involved in muscle regeneration and in muscle atrophy, regulates tissue regeneration during aging and disease progression. The authors conditionally ablate Stat3 in satellite cells in mice and implicate Stat3 signaling in satellite cell maintenance and muscle regeneration. Tierney et al.3 provide evidence that Stat3 regulates expression of MyoD, a transcription factor that converts satellite cells to proliferating myoblasts, via a conserved STAT3 enhancer element. Additionally, Stat3 inhibitor studies in aged and dystrophic mice mirrored observations made by Price et al.2 demonstrating that inhibition of JAK-STAT signaling improves skeletal muscle regeneration.
In both reports, the authors conclude that elevated JAK-STAT signaling inhibits satellite cell function. Tierney et al.3 further suggest that inhibiting Stat3 reduces MyoD transcription, thus inhibiting differentiation and promoting satellite cell self-renewal. Price et al.2 confirm recent reports demonstrating a reduction in satellite cell self-renewal with advanced age8,14 and further show that JAK-STAT inhibition promotes symmetric satellite cell expansion with a reduction in commitment to myogenesis. Thus, the enhanced production of satellite cells by symmetric expansion reported by Price et al.2 and the inhibition of differentiation reported by Tierney et al.3 probably work together to improve muscle regeneration in aged and diseased mice.
The aged muscle environment is known to negatively affect satellite cell function10, yet recent studies identify age-related, cell-intrinsic changes to satellite cells that include alterations in extracellular signal–regulated kinase–mitogen-activated protein kinase (MAPK) signaling9 and p38 MAPK signaling8,14—indicative of increased cellular stress—that also impair satellite cell regenerative function. These new studies further solidify the link between aging, stress signaling and satellite cell defects previously reported8,14. Tierney et al.3 suggest that as one source of age-associated stress, interleukin-6, a proinflammatory cytokine that signals via JAK2-STAT3, may be a contributing STAT3 stimulus. Candidate stress signal sources include infiltrating immune cells or senescent cells, the latter contributing to stress signaling via a senescence-associated secretory phenotype composed of numerous proinflammatory cytokines and chemokines15. A comprehensive study of these possibilities during muscle aging would be extremely informative.
The clinical use of JAK-STAT inhibitors to combat diverse pathologies is well documented. Indeed, the JAK inhibitors ruxolitinib (JAK1 and JAK2 inhibitor) and tofacitinib (JAK3 inhibitor), are approved for inflammatory disorders including psoriasis, myelofibrosis and rheumatoid arthritis. Additional JAK-STAT inhibitors are in late-stage clinical trials for an even broader range of diseases16. Price et al.2 used AG490, a potent inhibitor of JAK2, JAK3 and epidermal growth factor receptor, and the STAT3 inhibitor 5,15 DPP to provide convincing evidence that the inflammatory stimulus is probably mediated by JAK2. Rigorously identifying the distinct contributions of individual JAKs to muscle regeneration will further refine the pathways involved and possibly help identify the culpable inflammatory signals. Notably, Price et al.2 demonstrate that transient exposure to JAK-STAT inhibitors before satellite cell transplantation improves engraftment efficiency, raising the therapeutic potential of JAK-STAT inhibitors for improving muscle regeneration by stem cell transplantation, thus circumventing potential problems arising from systemic administration of anti-inflammatory agents. An important next step would be to determine whether human myoblasts behave similarly when treated with JAK-STAT inhibitors before in vivo transplantation.
An aging world population highlights the importance of identifying interventions to maintain skeletal muscle mass and function. The results from Price et al.2 and Tierney et al.3 identify JAK-STAT signaling as a potential therapeutic target for the treatment of age-related muscle wasting. Given the proven safety and utility of using JAK inhibitors in the clinic, these studies provide new information supporting satellite cell–centered therapeutic interventions to improve quality of life and attenuate the loss of muscle function occurring in progressive muscle disorders and as a consequence of aging.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Piccirillo R, Demontis F, Perrimon N, Goldberg AL. Dev Dyn. 2014;243:201–215. doi: 10.1002/dvdy.24036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Price FD, et al. Nat Med. 2014;20:1174–1181. doi: 10.1038/nm.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tierney MT, et al. Nat Med. 2014;20:1182–1186. doi: 10.1038/nm.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Troy A, et al. Cell Stem Cell. 2012;11:541–553. doi: 10.1016/j.stem.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Development. 2011;138:3625–3637. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sambasivan R, et al. Development. 2011;138:3647–3656. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- 8.Bernet JD, et al. Nat Med. 2014;20:265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakkalakal JV, Jones KM, Basson MA, Brack AS. Nature. 2012;490:355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conboy IM, et al. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 11.Sousa-Victor P, et al. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- 12.Rawlings JS, Rosler KM, Harrison DA. J Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 13.Doles J, Storer M, Cozzuto L, Roma G, Keyes WM. Genes Dev. 2012;26:2144–2153. doi: 10.1101/gad.192294.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cosgrove BD, et al. Nat Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freund A, Orjalo AV, Desprez PY, Campisi J. Trends Mol Med. 2010;16:238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pardanani A, et al. Leukemia. 2011;25:218–225. doi: 10.1038/leu.2010.269. [DOI] [PubMed] [Google Scholar]