

Abstract
Objectives:
To characterize phenotypes of T cells that accumulated in multiple sclerosis (MS) lesions, to compare the lesional T-cell receptor (TCR) repertoire of T-cell subsets to peripheral blood, and to identify paired α and β chains from single CD8+ T cells from an index patient who we followed for 18 years.
Methods:
We combined immunohistochemistry, laser microdissection, and single-cell multiplex PCR to characterize T-cell subtypes and identify paired TCRα and TCRβ chains from individual brain-infiltrating T cells in frozen brain sections. The lesional and peripheral TCR repertoires were analyzed by pyrosequencing.
Results:
We found that a TCR Vβ1+ T-cell population that was strikingly expanded in active brain lesions at clinical onset comprises several subclones expressing distinct yet closely related Vα7.2+ α chains, including a canonical Vα7.2-Jα33 chain of mucosal-associated invariant T (MAIT) cells. Three other α chains bear striking similarities in their antigen-recognizing, hypervariable complementarity determining region 3. Longitudinal repertoire studies revealed that the TCR chains that were massively expanded in brain at onset persisted for several years in blood or CSF but subsequently disappeared except for the canonical Vα7.2+ MAIT cell and a few other TCR sequences that were still detectable in blood after 18 years.
Conclusions:
Our observation that a massively expanded TCR Vβ1-Jβ2.3 chain paired with distinct yet closely related canonical or atypical MAIT cell–related α chains strongly points to an antigen-driven process in early active MS brain lesions.
CNS-invasive, presumably autoreactive T cells are believed to play a central role in the pathogenesis of multiple sclerosis (MS).1–3 In parenchymal MS brain infiltrates, CD8+ T cells are more frequent than CD4+ T cells.4–7 Furthermore, CNS-infiltrating CD8+ T cells are oligoclonal,6–10 whereas CD4+ T-cell infiltrates tend to be more polyclonal.6 Some CD8+ T-cell clones were shown to be expanded not only in the brain but also in CSF and blood, where they may persist for years.6
For technical reasons, most previous studies of the T-cell receptor (TCR) repertoire were limited to the β chain. However, the antigen-specific TCR is an αβ heterodimer, and both chains contribute to antigen recognition. Here we used immunohistochemistry, laser microdissection, and single-cell multiplex PCR11 to identify paired αβ TCRs from brain-infiltrating CD8+ T cells present in an early active lesion from “patient A,”6 whom we have followed for 18 years. We found that a clonally expanded and persisting Vβ1-Jβ2.3 chain pairs with several distinct yet closely related Vα7.2+ α chains. It is intriguing that one of the newly identified TCR α chains is characteristic for mucosal-associated invariant T (MAIT) cells, and 3 other α chains are highly homologous. MAIT cells are an innate-like T-cell subset with limited TCR variability12 that express the TCR Vα7.2 element and the natural killer cell marker CD16113–16 and are restricted by the MHC-related molecule 1 (MR1).17 MAIT cells are a heterogeneous, semi-invariant T-cell population, with most cells carrying a “canonical” TCR α chain defined by the usage of Vα7.2 and Jα33 and some cells carrying a “noncanonical” TCR α chain in which Jα33 is replaced by Jα12 or Jα20.15 Their development depends on gut microbiota, and they are thought to play a role in defense against various microorganisms.17–19 Because we found not only canonical MAIT α chains but also different, though homologous, α chains pairing with one β chain, our results illustrate the complexity of the CD8+ T-cell repertoire.
METHODS
Standard protocol approvals, registrations, and patient consents.
Written consent from patient A was obtained according to the Declaration of Helsinki. The study was approved by the ethics committee of the medical faculty of the LMU Munich.
Patient A.
The male patient A initially presented with left-sided hemianopia in 1996. His initial cranial MRI showed a large right temporo-occipital white matter lesion, raising suspicion of malignant glioma.5 Two weeks after onset of his clinical symptoms, the brain lesion was neurosurgically resected. Histopathology showed an inflammatory demyelinating lesion consistent with MS. Subsequently he had a typical relapsing-remitting course of MS. He has been continuously treated with interferon-β-1a IM from the time of his third relapse in 1998 until submission of this manuscript. Figure e-1 at Neurology.org/nn gives an overview of the course of experiments. Blood samples for this study were taken in 2003, 2005, 2013, and 2014. Written consent was obtained from the donor.
Immunohistochemistry.
The resected brain tissue was immediately frozen in liquid nitrogen and stored at −80°C until further use. Frozen sections of 10 µm were cut, mounted on positively charged slides (Superfrost Plus, Menzel, Braunschweig, Germany) for immunohistochemistry or on membrane-covered PET slides (Zeiss, Jena, Germany) for laser capture microscopy, and directly stored at −20°C or −80°C, respectively. Immunohistochemistry and microdissection was applied to tissue regions that contain lesions with high numbers of CD8+ T cells.
To characterize T-cell infiltrates in sections of MS brain, the following antibodies against cell surface molecules were used: mouse anti-human CD161 (1:5, 191B8, Miltenyi Biotec, Bergisch Gladbach, Germany), mouse anti-human Vα7.2 (1:5, 3C10, BioLegend, San Diego, CA), mouse anti-human CD8α (1:50, LT8, AbD Serotec, Kidlington, UK; labeled with the Cy3 MAb labeling kit, GE Healthcare, Freiburg, Germany), rabbit anti-human CCR7 (1:800, Y59, Abcam, Cambridge, UK), mouse anti-human CD45RA (1:250, HI100, BioLegend), mouse anti-human CD45RO (1:250, UCHL1, BioLegend), fluorescein isothiocyanate (FITC)-labeled mouse anti-human Vβ1 (1:100, BL37.2, Beckman Coulter, Brea, CA), rabbit anti-human CD3 (1:500, Dako, Glostrup, Denmark), and Alexa Fluor 488–conjugated mouse anti-human CD4 (1:50, RPA-T4, eBioscience, San Diego, CA). Frozen sections of CNS were thawed at room temperature, fixed in acetone, rehydrated in phosphate-buffered saline (PBS), and blocked with 2% bovine serum albumin in PBS. For staining with anti-Vα7.2 and anti-CD161 antibodies, samples were fixed in 4% paraformaldehyde for 10 minutes. Sections were then incubated for 1 hour with the primary antibodies at room temperature and subsequently—in cases where the primary antibody was not directly conjugated to a detectable fluorophore—the bound primary antibody was detected using a secondary Alexa Fluor 488–labeled goat anti-mouse IgG antibody (1:1,000, Life Technologies, Carlsbad, CA). For double staining of Vα7.2 and CD161, an Alexa Fluor 568–labeled goat anti-mouse IgG1 or an Alexa Fluor 488–labeled goat anti-mouse IgG2a antibody (1:1,000, Life Technologies) was used, respectively. If a FITC-labeled primary antibody was used, the signal was enhanced using an anti-FITC Alexa Fluor 488–conjugated secondary antibody (1:100, Life Technologies). For dual stainings with anti-CD8α, the Cy3-labeled anti-CD8α antibody was applied last. Slides were mounted using fluorescent mounting medium (Dako). Staining was visualized using an Olympus FV 1000 confocal microscope with a 40× water 1.15 NA objective or a Zeiss Axioplan 2 microscope.
Isolation of T cells from peripheral blood mononuclear cells.
Peripheral blood mononuclear cells (PBMCs) were isolated from ethylenediaminetetraacetic acid blood in 2003 by standard-density centrifugation using Percoll (Pan-Biotech, Aidenbach, Germany) and double stained with mouse anti-human Vβ1 (clone BL37.2) and CD8+ (clone LT8). CD8+Vβ1+ double-positive T cells were sorted using a BD FACSVantage SE flow cytometer and analyzed by single-cell PCR. Cells were either sorted directly into PCR tubes or obtained as bulk and then isolated by hand under a microscope. CD8+ T cells from 2003 to 2005 were analyzed by complementarity determining region (CDR) 3 spectratyping, as described previously.6
To isolate MAIT cells from PBMCs in 2013 and 2014, T cells were negatively isolated using the pan T-cell isolation kit (Miltenyi Biotec) and subsequently stained with the FITC-labeled mouse anti-human Vα7.2 (1:20, 3C10, BioLegend) and the allophycocyanin-labeled mouse anti-human CD161 (1:10, 191B8, Miltenyi Biotec) antibodies at 4°C for 30 minutes. Cells were then sorted using a BD FACSAriaTM2 flow cytometer. 5 × 102 CD161+Vα7.2+ cells, 6 × 103 CD161−Vα7.2+ cells, 1 × 105 CD161+ cells, and 2 × 105 CD161− cells were collected. CD4+ and CD8+ T cells were positively isolated from PBMCs in 2013 and 2014 using magnetic beads (Miltenyi Biotec).
Laser microdissection and single-cell PCR.
Single Vβ1+CD8α+ T cells were isolated by microdissection as described,7 with the exception that we replaced the anti-Vβ5 antibody with the anti-Vβ1 antibody BL37.2. TCRs of single Vβ1+CD8α+ cells obtained by microdissection or by flow cytometry were analyzed by reverse transcription PCR as described.11 PCR products were separated using a 2% agarose gel, and promising bands were excised and submitted to sequencing after isolation of DNA by the MinElute Gel Extraction Kit (Qiagen, Hilden, Germany). Throughout this manuscript, the nomenclature according to Arden et al.20 is used for the TCR V region.
Next-generation sequencing.
For the analysis of the Vα7.2 chains, RNA was isolated from sorted cells using Trizol (Life Technologies), and complementary DNA was transcribed using the Qiagen One Step RT-PCR Kit (Qiagen). Two rounds of nested PCR were performed using the primers TRAV1-2-for-out, Cα-rev-out, TRAV1-2-for-in, and Cα-rev-in.11 The inner primers were extended for tags CS1 and CS2, barcodes MIDXX, and 454Ti-A and 454Ti-B adaptors according to the Access Array System 454 user guide (Fluidigm, San Francisco, CA). Samples were pooled in equimolar concentrations.
The library for sequencing all TCR β chains was prepared using 5′-RACE (Clontech, Mountain View, CA), with the gene-specific primers in the constant region CβRT11 (20 µM) and the 5′ adaptor (10 µM; 5′-AAGCAGTGGTATCAACGCAGAGTACTCTT(rG)53′). This was followed by 2 nested PCRs for 20 cycles, each for 20 seconds at 94°C, 20 seconds at 60°C, and 50 seconds at 72°C using gene-specific reverse primers Cβ-mid and Cβ-in.21 Cβ-in was extended for barcodes and the adaptor 454Ti-B. As forward primers we used the step-out primers sa1: CTAATACGACTCACTATAGGGCAAGCAGTGGTATCAACGCAG; sa2: 454TiA_MIDXX_CTAATACGACTCACTATAGGGC. PCR products were excised from agarose gels, purified by the MinElute Kit (Qiagen), and pooled in equimolar concentrations. Quality control of the libraries and sequencing on a Roche 454 GS FLX+ platform (Roche, Basel, Switzerland) was then carried out at IMGM Laboratories GmbH, Martinsried, Germany.
We obtained a total of 183,291 TCRα reads and 182,245 TCRβ reads, which were analyzed using the IMGT/HighV QUEST platform.22 We selected for in-frame sequences that contained the complete CDR3 region with clearly distinguishable V and J genes only. Thereby we obtained 46,682 and 46,133 readable TCRα and TCRβ chains, respectively.
RESULTS
Vβ1+ T cells accumulate in inflammatory foci and pair with MAIT cell–related α chains.
It was known that in patient A a particular T-cell clone expressing the Vβ1-Jβ2.3 TCRβ chain was expanded in brain lesions and persisted in peripheral blood and CSF between 1996 and 2003.5,6 Immunohistochemistry revealed clusters of Vβ1+CD8+ cells within the CNS (figure 1A). In some of these clusters, up to 38% of all CD8+ T cells coexpressed the Vβ1 chain together with CD8α. This observation links the clonal T-cell expansions initially detected by CDR3 spectratyping to morphologically identifiable accumulations of T cells.
Figure 1. Expanded Vβ1 chain pairs with MAIT cell–related α chains.
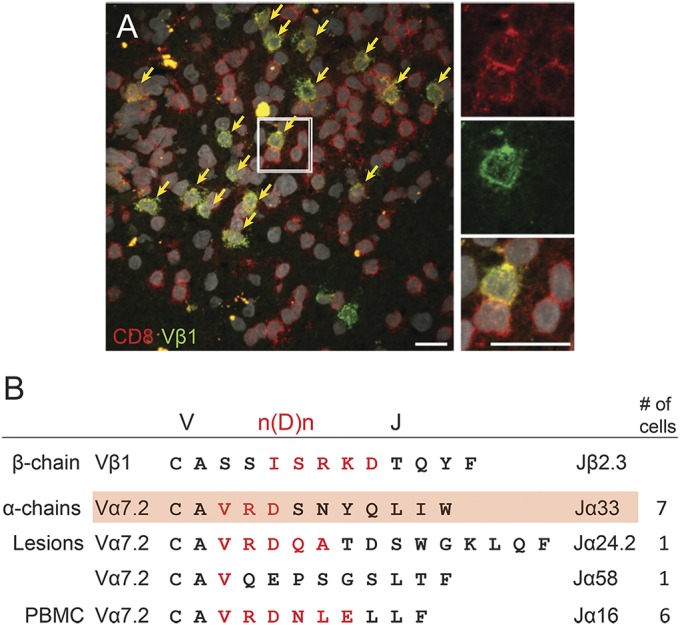
(A) Cluster of expanded Vβ1+ T cells within multiple sclerosis (MS) lesions. Immunohistochemistry for the expanded and persisting Vβ1 clone (green) in clusters of CD8+ T cells (red) in parenchymal MS lesions. Several such clusters were observed. Only very few scattered CD8−Vβ1+ T cells could be identified in the brain lesion. Nuclei are visualized with 4',6-diamidino-2-phenylindole (white). Scale bar 20 µm. (B) Sequences of paired T-cell receptor (TCR) α and β chains. Single sorted or laser microdissected Vβ1+CD8+ T cells from peripheral blood or brain sections were submitted to single-cell TCR PCR to identify Vβ1 chains and all possible matching α chains. The V, n(D)n, and J regions are indicated. Amino acids encoded by n(D)n nucleotides are printed in red. The expanded Vβ1-Jβ2.3 β chain (upper line) was found to pair with 4 different α chains. Three α chains were identified from brain lesions, and 1 α chain was found in blood. The α chains expressing the Jα33 (second line) and Jα16 (fifth line) elements were identified in 7 and 6 independent cells, respectively. All α chains share the Vα7.2 element, and even though they do not share the same Jα element, they all show homologous complementarity determining region 3α regions with a conserved valine (V) followed by a positively charged arginine (R) (with only one clone showing a glutamine [Q]), a negatively charged amino acid (D/E), and a relatively large hydrophilic amino acid. One of the α chains (highlighted in red) is the mucosal-associated invariant T (MAIT) cell canonical TCR Vα7.2-CAXXDSNYQLIW-Jα33 chain with 2 N nucleotide–encoded amino acids between Vα7.2 and Jα33 (here VR). The other clones with Jα16, Jα24.1, and Jα58 chains are atypical for MAIT cells, which usually carry Jα33, Jα20, or Jα12. PBMC = peripheral blood mononuclear cell.
To identify the TCRα chain associated with the expanded Vβ1-Jβ2.3 chain, we double stained brain sections of the 1996 tissue for CD8α and Vβ1 and isolated single CD8α+Vβ1+ T cells by laser microdissection from brain sections (figure e-2A). In parallel, we isolated single peripheral blood CD8α+Vβ1+ T cells obtained in 2003 by flow cytometry (figure e-2B). To distinguish probable antigen-experienced Vβ1-Jβ2.3+ T cells from bystanders, we tested all Vβ1+ cells for their n(D)n-J sequences. For cells that expressed the expanded and persisting sequence Vβ1-IGRKD-Jβ2.3 (figure 1B), we analyzed the matching α chain by multiplex PCR.11 We identified 15 α chains that paired with the Vβ1-Jβ2.3 chain. One T-cell clone was found 7 times in brain, and another clone was found 6 times in blood (figure 1B). All α chains share the Vα7.2 element, and even though they do not share the same Jα element, they all show homologous CDR3α regions with the consensus sequence valine, arginine (one clone: glutamine), aspartic or glutamic acid, and a medium size hydrophilic amino acid. Of note, 5 of 6 CDR loops of the 4 αβ heterodimers are identical because all clones share the identical β chain and the Vα7.2 variable element. The common motif in the CDR3 loop of the α chains therefore hints at related antigens. Strikingly, one of the α chains identified from microdissected brain-infiltrating CD8+ T cells represents a canonical MAIT cell α chain that contains 2 N amino acids (X) in the CDR3α loop (CAXXDSNYQLIW) and the Jα33 element.12,15 Thus, the expanded, dominant, and persisting Vβ1 chain found in earlier studies5,6 pairs with several distinct yet related Vα7.2+ α chains.
Tissue-invading cells in early active lesions mostly show a CD8+ memory phenotype.
The immune cell infiltrates in lesions were dominated by CD8+ T cells. Four hundred twenty of 503 (84%) examined tissue-infiltrating parenchymal CD3+ T cells expressed CD8α (figure 2A). The parenchymal CD4:CD8 ratio was 1:8 (figure 2B). Expression of the naive T-cell marker CD45RA was found on only 11% of tissue-infiltrating CD8α+ cells (figure 2C), whereas 84% expressed the memory marker CD45RO (figure 2D). Of all parenchymal CD8α+ cells, 60% coexpressed the homing receptor CCR7. Double staining of CCR7 together with CD45RA or CD45RO revealed the following phenotypes in decreasing frequency: central memory (CD45RO+CCR7+), effector memory (CD45RO+CCR7−), and naive T cells (CD45RA+CCR7+), which accounted for only 6% of all CCR7+ cells (figure 2E). Sixty-five percent of CCR7+ cells coexpress CD45RO, and 62% of all CD45RO+ cells express CCR7 (figure 2F). Thus, the brain-infiltrating T cells predominantly show a CD8α+CD45RO+CCR7+ central memory or CD8α+CD45RO+CCR7− effector memory phenotype.
Figure 2. Brain-infiltrating immune cells mainly consist of CD8+ memory T cells.
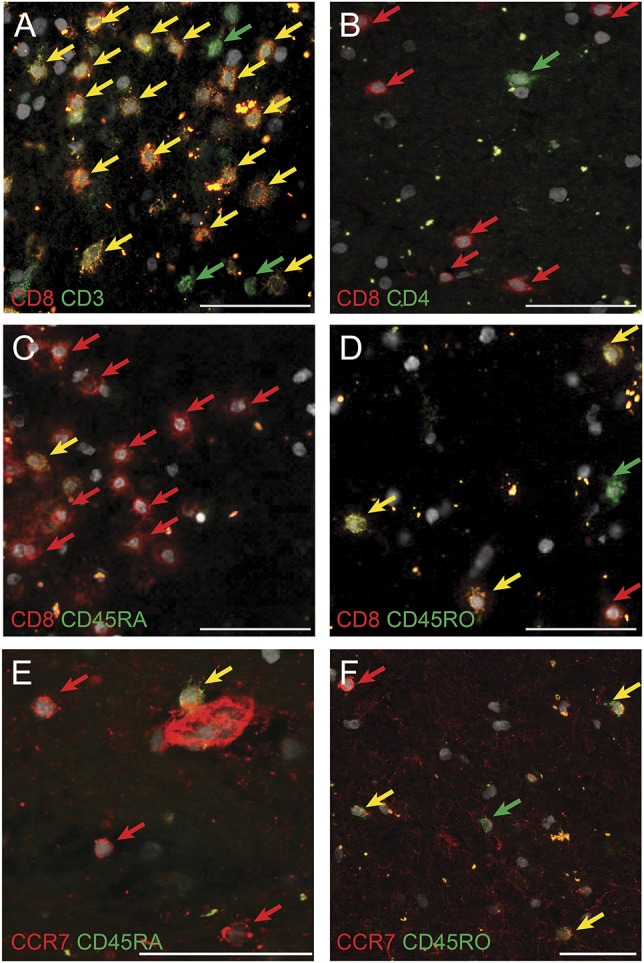
Immunofluorescence staining of brain-infiltrating immune cells. All nuclei are stained with 4′,6-diamidino-2-phenylindole (white). Green and red dyes were used. Double-positive cells are therefore shown in yellow. Scale bars 50 µm. (A) Double staining for CD3 (green) and CD8α (red). Most CD3+ T cells coexpress CD8α. (B) CD8+ (red) T cells outnumber CD4+ (green) T cells. (C) Low numbers of CD45RA+ (green) CD8+ (red) double-positive T cells in multiple sclerosis (MS) brain tissue. (D) Many CD8+ T cells (red) coexpress CD45RO (green). (E) Naive T cells double-positive for CCR7 (red) and CD45RA (green) are mostly found within blood vessels and are barely detectable in the parenchyma of MS CNS. (F) Effector memory (CD45RO+CCR7−) (green arrow) and central memory (CD45RO+CCR+) (yellow arrow) T cells in MS lesions.
MAIT cells are detected in parenchymal MS lesions.
To investigate phenotypic features of the MAIT-related cells, we stained brain sections for the expanded Vβ1+ TCRβ chain and MAIT cell–associated markers Vα7.2, CD161, and CD8α. Forty-two percent of the parenchymal Vα7.2+ cells coexpressed Vβ1; however, only 13% of Vβ1+ T cells expressed Vα7.2 (figure 3A). Seventy percent of the Vα7.2+ cells also expressed CD8α (figure 3B), and 69% of Vα7.2+ cells coexpressed CD161 (figure 3C), identifying them as MAIT cells.13 Of all CD161+Vα7.2+ MAIT cells, 74% were found in the brain parenchyma. Comparing the number of parenchymal CD161+Vα7.2+ cells to the total number of parenchymal CD8α+ cells in consecutive sections, we found that only about 1% of all CD8α+ cells belong to the CD161+Vα7.2+ MAIT cell subset. However, CD161 was found on 21% of CD8α+ T cells. Conversely, about 80% of the CD161-expressing cells were CD8α+ (figure 3D). Furthermore, about 80% of Vβ1+ T cells expressed CD161+ (data not shown). This underlines the fact that many of the CD161+ cells are not MAIT cells.23
Figure 3. MAIT cells can be detected within parenchymal lesions of MS brain.

Double fluorescence immunohistochemistry identifies brain-infiltrating mucosal-associated invariant T (MAIT) cells in multiple sclerosis (MS) lesions of patient A. Nuclei are stained with 4',6-diamidino-2-phenylindole (white). Green and red dyes were used. Double-positive cells are therefore shown in yellow. Scale bars 20 µm. (A) T cells expressing the T-cell receptor Vα7.2 (red) and Vβ1 chains (green). This combination (see figure 1B) was identified by single-cell PCR. (B) Most Vα7.2+ (green) T cells belong to the CD8+ (red) T-cell subset. (C) MAIT cells expressing Vα7.2 (red) and CD161 (green) in the parenchyma of MS brain. (D) The vast majority of CD161+ (green) cells in MS CNS coexpress CD8α (red).
Changes in the MAIT cell–related TCRα and TCRβ chain repertoire in blood between 1996 and 2014.
To follow the longitudinal development of the peripheral T-cell repertoire from 1996 to 2014 (figure e-2), we analyzed the 1996 brain sample as well as different subsets of peripheral T cells from 2013 to 2014 by pyrosequencing.
Tracking of TCR chains identified by single-cell PCR in the 1996 biopsy specimen revealed that the Vβ1-Jβ2.3 chain that was expanded in brain and blood samples from 1996, 2001, and 20035,6 was still detectable by pyrosequencing in peripheral T cells 18 years after clinical onset, but it was no longer expanded. Furthermore, the canonical Vα7.2-CAVRDSNYQLIW-Jα33 MAIT cell clone found in the 1996 brain sample by single-cell PCR was still present in the 2014 CD161+Vα7.2+ MAIT PBMC subset, as found by pyrosequencing.
Next, we compared the CDR3 repertoires of Vα7.2+ chains. We show the distribution of the N and J regions of the 1996 brain specimen in figure 4A. One of the 3 dominant clones is a canonical MAIT cell Vα7.2-Jα33 clone, although it differs from the clone found in the single cells. In peripheral blood from 2014, clonal expansions were found in only the CD161+Vα7.2+ subset, with the MAIT cell canonical and noncanonical clones dominating (blue in figure 4B). In contrast, the CD161−Vα7.2+ subset is polyclonal (figure 4C). We also detected clonal expansions including some typical sequences for MAIT cells in the entire pool of CD8+ T cells (figure 4D), whereas CD4+ T cells were polyclonal (figure 4E).
Figure 4. Vα7.2 TCR chain repertoire.

Analysis of the T-cell receptor (TCR) Vα7.2 repertoire of patient A by pyrosequencing shows oligoclonal T-cell expansions in different samples. Each pie chart represents the total number of nucleotide sequences found in the Vα7.2 repertoire of a certain compartment and each sector represents one distinct T-cell clone. We compared (A) samples from CNS tissue from the biopsy taken in 1996, (B) Vα7.2+CD161+ mucosal-associated invariant T (MAIT) cells, (C) Vα7.2+CD161− cells, (D) CD8+ cells, and (E) CD4+ T cells from peripheral blood taken in 2013 and 2014. For each population we list the designation of the Jα elements and their relative percentage. Clones carrying the MAIT canonical TCR α chain (CAXXDSNYQLIW) are marked in bright blue, and clones containing the noncanonical α chains characterized by Jα12 (CAXXDSSYKLIF) and Jα20 (CAVXXDYKLSF) are marked in dark blue and light blue, respectively. (F) Percentages of identical complementarity determining region 3α amino acid sequences within the TCR Vα7.2+ repertoire between the different samples defined above (A–E). The numbers are percentages indicating how often a particular sequence detected in one sample was also found in another sample. The greatest overlap was between Vα7.2+CD161+ and CD8+ T cells from peripheral blood, but there was also significant overlap between the CNS sample and the Vα7.2+CD161+ sample from 2013 to 2014, as highlighted by the red and yellow colors. PBMC = peripheral blood mononuclear cell.
We then set out to identify distinct clones shared between these different subsets (figure 4F). Within the TCR Vα7.2 repertoire, several clones that were present in the 1996 brain sample can still be found in blood T cells obtained 18 years after onset, especially in the CD161+Vα7.2+ MAIT cell subset. The greatest overlap was observed between the CNS sample and the PBMC MAIT cell sample from 2014, in which 2.4% of all sequences were identical at the amino acid level. It is notable that the canonical Vα7.2-CAVRDSNYQLIW-Jα33 sequence could also be detected in T-cell subsets other than the MAIT cells.
DISCUSSION
For this in-depth analysis of CD8+ T cells in MS, we had the unique opportunity to investigate a patient whose initial inflammatory lesion was resected under the misdiagnosis of glioma5 and whom we have been following for 18 years. By combining immunohistochemistry, laser microdissection, and single cell PCR analysis of brain-infiltrating Vβ1+Jβ2.3+ cells, we show that the expanded and persistent Vβ1-Jβ2.3+ T-cell population6 comprises several distinct Vα7.2+ T-cell clones, all of which bear striking similarities in their CDR3α regions. In addition to a canonical MAIT cell Vα7.2-Jα33 chain, we identified 3 MAIT cell–atypical Vα7.2 chains with different yet related CDR3α regions that form an oligoclonal spectrum of heterodimeric αβ TCRs.
Transcripts of canonical Vα7.2-Jα33 MAIT cells have been identified in MS brain autopsies,24 but it remained unclear whether the MAIT cells indeed infiltrated the parenchyma. Later, phenotypically defined (CD3+CD161+Vα7.2+ or CD8+CD161+Vα7.2+) bona fide MAIT cells were detected in brain and meningeal infiltrates, mostly of patients with long-standing progressive disease.25–28 Consistent with these studies, we found that CD161+Vα7.2+ MAIT cells represented only about 1% of CD8+ T cells present in the brain specimen of our patient. This is lower than the typical MAIT cell frequency of 3%–10% of all T cells in blood,13,29 suggesting that MAIT cells are not selectively recruited into the CNS. CD161 expression is not limited to MAIT cells,23 and we found CD161 expression on 21% of all CD8+ T cells in our patient's brain lesions. Furthermore, we found more CD161+Vβ1+ cells than Vα7.2+Vβ1+ T cells, indicating that most brain-infiltrating CD161+Vβ1+ cells do not belong to the CD161+Vα7.2+ MAIT cell subset. However, despite their low numbers, the brain-infiltrating Vβ1+Vα7.2+ cells are unlikely to represent a randomly recruited bystander population. First, their clonal features strongly point to an antigen-driven process: a single strikingly expanded TCR Vβ1 chain was found to be associated with 4 different Vα7.2 chains that all shared highly homologous CDR3 regions. Second, the expanded Vβ1+ T cells formed conspicuous local clusters, consistent with local recognition of shared antigen(s). Third, the brain specimen was obtained at clinical disease onset and contains early active lesions, pointing to an early role of these inflammatory cells. Fourth, Vβ1-Jβ2.3+ clones were still expanded in CSF and/or blood 7 years after disease onset.6
Because the MAIT TCR repertoire depends on the commensal gut microbiota17 and may be shaped by microbes,19 the presence of MAIT cells in MS brain would be consistent with the hypothesis of a “gut–brain axis.”30 Recent evidence indicates that some MAIT cells recognize bacterial vitamin B metabolites presented by MR1,15,16,31,32 but these clones express canonical N-Ja elements and β chains that are very different from our Vβ1 chain. However, our brain-derived T-cell clones do not react with these metabolites nor do they recognize Escherichia coli–infected HeLa cells in a standard MAIT assay (unpublished observations). Another possibility is that these cells recognize conventional peptide antigens, but our efforts to identify such peptides have not been successful. A third possibility is that they might recognize other, still-unknown low-molecular-weight ligands. A recent study19 showed that exposure to different microbes evoked expression of atypical Jα and Vβ elements of MAIT cells. This would be consistent with our observation that except for one clone with a canonical Jα33+ α-chain, our clones express α chains with varying CDR3α lengths and Jβ elements that are atypical for MAIT cells. Furthermore, the Jα16, Jα24.1, and Jα58 chains lack the conserved Tyr 95α required for MAIT cell antigen recognition,15 and all 4 of our brain-derived T clones express Vβ1, which is very atypical for MAIT cells.
Our results reveal several novel features of CD8+ T cells in MS. First, the complexity of the αβ TCR repertoire is far greater than could be assumed from the TCRβ chain repertoire alone. Second, the αβ TCR dimers we obtained from individual brain-invading CD8+ T cells show MAIT-related features. Third, the brain-infiltrating MAIT-related cells themselves are heterogeneous; they express either a public “canonical” Vα7.2 chain or private MAIT-atypical Vα7.2 chains. Fourth, although histochemical stainings indicate that the MAIT-related T cells constitute only a small fraction of the infiltrate, the striking homology of the TCR CDR3 regions of the identified panel of Vα7.2-Vβ1+ T-cell clones points to their antigen-driven recruitment into early MS lesions. Fifth, follow-up of the peripheral TCR repertoire of the index patient by next-generation sequencing reveals that some MAIT-related TCR sequences are still detectable in blood 18 years after clinical onset. Sixth, the observation that a small yet clonally conspicuous fraction of CD8+ T cells in early MS lesions consist of MAIT or MAIT-related cells provides a hypothetical link between the brain-infiltrating T cells and the gut microbiota. Currently, the precise functional role of the brain-infiltrating MAIT cells is unknown: they could be harmful, protective, or inert. Future studies will attempt to tackle the complex interplay between the different CNS-invading T cells and their target tissue.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Hans Lassmann for discussion, Joachim Malotka and Ingrid Eiglmeier for expert technical assistance, Sabine Seitz for help, and Naoto Kawakami and Edgar Meinl for comments on the manuscript. The authors thank Lars Kjier-Nielsen, Jamie Rossjohn, and James McCluskey for providing the candidate MAIT cell antigens RL-6M and rRL-6HM.
GLOSSARY
- CDR
complementarity determining region
- FITC
fluorescein isothiocyanate
- MAIT
mucosal-associated invariant T
- MR1
MHC-related molecule 1
- MS
multiple sclerosis
- PBMC
peripheral blood mononuclear cell
- PBS
phosphate-buffered saline
- TCR
T-cell receptor
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
K. Held contributed to data collection, analysis and interpretation of the data, design of the study, and drafting and writing the manuscript. L. Bhonsle-Deeng contributed to data collection, analysis and interpretation of the data, and the design of the study. K. Siewert contributed to data collection, analysis and interpretation of the data, and the design of the study. W. Sato contributed to data collection and analysis and interpretation of the data. E. Beltrán contributed to data collection, analysis and interpretation of the data, and the design of the study. S. Schmidt contributed to patient recruitment, data collection, interpretation of the data, and writing the manuscript. G. Rühl contributed to data collection and analysis and interpretation of the data. J.K.M. Ng contributed to data collection and analysis and interpretation of the data. P. Engerer contributed to confocal microscopy data collection and analysis. M. Moser contributed to data collection and analysis of the data. W.E.F. Klinkert contributed to data collection and analysis of the data. H. Babbe contributed to data collection and interpretation of the data. T. Misgeld contributed to confocal microscopy data collection and analysis. H. Wekerle contributed to study design and writing and revising the manuscript. D.A. Laplaud contributed to data collection, analysis and interpretation of the data, and design of the study. R. Hohlfeld contributed to conceiving the study and drafting and revising of the manuscript. K. Dornmair contributed to conceiving and designing the study, interpretation of the data, and drafting and revising the manuscript.
STUDY FUNDING
This work was supported by the Deutsche Forschungsgemeinschaft (Munich Cluster for Systems Neurology [EXC 1010 SyNergy], CRC-TR-128-A5, -128-A1, and -128-B8), the Deutsche Multiple Sklerose Gesellschaft (DMSG), the Fondation pour l'Aide à la Recherche sur la Sclérose en Plaques (ARSEP), the Gemeinnützige Hertie Stiftung, and the Bundesministerium für Bildung und Forschung (Kompetenznetz MS, TP B8.1). D.A.L. has been also funded by Lilly Neurosciences and the Ligue Française contre la Sclérose en Plaques (LFSEP).
DISCLOSURE
K. Held, L. Bhonsle-Deeng, and K. Siewert report no disclosures. W. Sato received research support from Novartis. E. Beltrán reports no disclosures. S. Schmidt is on the scientific advisory board for Novartis, Merck Serono, Bayer Vital, Biogen Idec, Genzyme, and Teva; received travel funding and/or speaker honoraria from Novartis, Merck Serono, Bayer Vital, Biogen Idec, Genzyme, and Teva; and received research support from Bayer Vital. G. Rühl, J.K.M. Ng, P. Engerer, M. Moser, and W.E.F. Klinkert report no disclosures. H. Babbe is employed by Janssen Research & Development LLC and his spouse is employed by Coriell Institute for Medical Research. T. Misgeld received research support from RTG, DFG, CRC, and ERC. H. Wekerle is on the scientific advisory board for Myelin Repair Foundation, is on the editorial board for Annals of Neurology, and received research support from KKNMS, German Ministry of Science. D.A. Laplaud is on the scientific advisory board for Biogen Idec, Teva Pharma, Novartis, and Genzyme; received travel funding and/or speaker honoraria from Biogen Idec, Novartis, and Genzyme; is an editor for La Presse Medicale and coeditor for SEP & Neuroscience; and received research support from Biogen Idec, Novartis, LFSEP, and Fondation ARSEP. R. Hohlfeld served on the scientific advisory board for Novartis, Biogen Idec, Bayer-Schering, Merck Serono, Sanofi-Aventis, and Teva; served on the data safety monitoring board for Novartis, Merck Serono, and CSL Behring; received travel funding from Novartis, Biogen Idec, Bayer-Schering, Merck Serono, Sanofi-Aventis, Teva, and Genzyme; is an editorial board member for Neurology, Brain, Clinical and Experimental Immunology, Deutsche Medizinische Wochenschrift, Expert Opinion on Biological Therapy, Journal of Neuroimmunology, Multiple Sclerosis Journal, Nervenarzt, Practical Neurology, Seminars in Immunopathology, and Therapeutic Advances in Neurological Disorders; received consulting fees from Novartis, Biogen Idec, Bayer-Schering, Merck Serono, Sanofi-Aventis, Teva, and Genzyme; and received research support from Novartis, Biogen Idec, Bayer-Schering, and Teva. K. Dornmair received research support from Deutsche Forschungsgemeinschraft, Deutsche Multiple Sklerose Gesellschaft, and Fondation pour l'Aide a la Recherche sur la Sclerose en Plaques (ARSEP). Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol 2009;9:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol 2005;23:683–747. [DOI] [PubMed] [Google Scholar]
- 3.Hohlfeld R, Wekerle H. Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: from pipe dreams to (therapeutic) pipelines. Proc Natl Acad Sci USA 2004;101(suppl 2):14599–14606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol 1986;19:578–587. [DOI] [PubMed] [Google Scholar]
- 5.Babbe H, Roers A, Waisman A, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 2000;192:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skulina C, Schmidt S, Dornmair K, et al. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci USA 2004;101:2428–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Junker A, Ivanidze J, Malotka J, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain 2007;130:2789–2799. [DOI] [PubMed] [Google Scholar]
- 8.Friese MA, Fugger L. Pathogenic CD8(+) T cells in multiple sclerosis. Ann Neurol 2009;66:132–141. [DOI] [PubMed] [Google Scholar]
- 9.Saxena A, Martin-Blondel G, Mars LT, Liblau RS. Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett 2011;585:3758–3763. [DOI] [PubMed] [Google Scholar]
- 10.Montes M, Zhang X, Berthelot L, et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin Immunol 2009;130:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seitz S, Schneider CK, Malotka J, et al. Reconstitution of paired T cell receptor alpha- and beta-chains from microdissected single cells of human inflammatory tissues. Proc Natl Acad Sci USA 2006;103:12057–12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tilloy F, Treiner E, Park SH, et al. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med 1999;189:1907–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin E, Treiner E, Duban L, et al. Stepwise development of MAIT cells in mouse and human. PLoS Biol 2009;7:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gold MC, Cerri S, Smyk-Pearson S, et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol 2010;8:e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reantragoon R, Corbett AJ, Sakala IG, et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J Exp Med 2013;210:2305–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lepore M, Kalinichenko A, Colone A, et al. Parallel T-cell cloning and deep sequencing of human MAIT cells reveal stable oligoclonal TCRβ repertoire. Nat Commun 2014;5:3866. [DOI] [PubMed] [Google Scholar]
- 17.Treiner E, Duban L, Bahram S, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003;422:164–169. [DOI] [PubMed] [Google Scholar]
- 18.Le Bourhis L, Martin E, Peguillet I, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol 2010;11:701–708. [DOI] [PubMed] [Google Scholar]
- 19.Gold MC, McLaren JE, Reistetter JA, et al. MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J Exp Med 2014;211:1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arden B, Clark SP, Kabelitz D, Mak TW. Human T-cell receptor variable gene segment families. Immunogenetics 1995;42:455–500. [DOI] [PubMed] [Google Scholar]
- 21.Kim SM, Bhonsle L, Besgen P, et al. Analysis of the paired TCR α- and β-chains of single human T cells. PloS One 2012;7:e37338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alamyar E, Giudicelli V, Li S, Duroux P, Lefranc MP. IMGT/HighV-QUEST: the IMGT(R) web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res 2012;8:26. [Google Scholar]
- 23.Fergusson JR, Fleming VM, Klenerman P. CD161-expressing human T cells. Front Immunol 2011;2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Illés Z, Shimamura M, Newcombe J, Oka N, Yamamura T. Accumulation of Valpha7.2-Jalpha33 invariant T cells in human autoimmune inflammatory lesions in the nervous system. Int Immunol 2004;16:223–230. [DOI] [PubMed] [Google Scholar]
- 25.Annibali V, Ristori G, Angelini DF, et al. CD161(high)CD8+T cells bear pathogenetic potential in multiple sclerosis. Brain 2011;134:542–554. [DOI] [PubMed] [Google Scholar]
- 26.Miyazaki Y, Miyake S, Chiba A, Lantz O, Yamamura T. Mucosal-associated invariant T cells regulate Th1 response in multiple sclerosis. Int Immunol 2011;23:529–535. [DOI] [PubMed] [Google Scholar]
- 27.Abrahamsson SV, Angelini DF, Dubinsky AN, et al. Non-myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes IL-17 producing mucosal-associated invariant T cells in multiple sclerosis. Brain 2013;136:2888–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willing A, Leach OA, Ufer F, et al. CD8(+) MAIT cells infiltrate into the CNS and alterations in their blood frequencies correlate with IL-18 serum levels in multiple sclerosis. Eur J Immunol 2014;44:3119–3128. [DOI] [PubMed] [Google Scholar]
- 29.Dusseaux M, Martin E, Serriari N, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 2011;117:1250–1259. [DOI] [PubMed] [Google Scholar]
- 30.Wekerle H, Berer K, Krishnamoorthy G. Remote control-triggering of brain autoimmune disease in the gut. Curr Opin Immunol 2013;25:683–689. [DOI] [PubMed] [Google Scholar]
- 31.Kjer-Nielsen L, Patel O, Corbett AJ, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012;491:717–723. [DOI] [PubMed] [Google Scholar]
- 32.Patel O, Kjer-Nielsen L, Le Nours J, et al. Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat Commun 2013;4:2142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.