

Abstract
The main objective of this work is to demonstrate the feasibility of using bone marrow-derived stem cells in treating a neurodegenerative disorder such as Friedreich's ataxia. In this disease, the dorsal root ganglia of the spinal cord are the first to degenerate. Two groups of mice were injected intrathecally with mesenchymal stem cells isolated from either wild-type or Fxntm1Mkn/Tg(FXN)YG8Pook (YG8) mice. As a result, both groups presented improved motor skills compared to nontreated mice. Also, frataxin expression was increased in the dorsal root ganglia of the treated groups, along with lower expression of the apoptotic markers analyzed. Furthermore, the injected stem cells expressed the trophic factors NT3, NT4, and BDNF, which bind to sensory neurons of the dorsal root ganglia and increase their survival. The expression of antioxidant enzymes indicated that the stem cell-treated mice presented higher levels of catalase and GPX-1, which are downregulated in the YG8 mice. There were no significant differences in the use of stem cells isolated from wild-type and YG8 mice. In conclusion, bone marrow mesenchymal stem cell transplantation, both autologous and allogeneic, is a feasible therapeutic option to consider in delaying the neurodegeneration observed in the dorsal root ganglia of Friedreich's ataxia patients.
Introduction
Friedreich's ataxia (FA) is a neurodegenerative disorder characterized by the progressive loss of motor functions and coordination, due to low levels of frataxin expression.1 The individuals affected by this disease present a GAA-repeat expansion in intron 1 of the frataxin gene. Frataxin is involved in iron homeostasis in the mitochondria, and when its expression is affected, iron is accumulated in the mitochondria, ultimately causing cell death.2 The most sensitive cell types due to frataxin alteration are certain neurons (such as the large sensory neurons of the dorsal root ganglia (DRG) in the spinal cord and the neurons in the deep cerebellar nuclei) and cardiomyocytes.3 There is currently no cure for this disease, and the treatments are not capable of ameliorating the progressive neurodegeneration.
There is evidence that oxidative stress plays a major role in FA.4,5 This has caused many of the current treatments to be based on counteracting oxidative stress. For example, coenzyme Q10 along with vitamin E seemed to give positive results in the cardiac and skeletal muscle.6,7,8 Idebenone is a free-radical scavenger that has been shown in several clinical trials to protect the heart-related problems of the disease.9,10,11,12 However, none of the current treatments have demonstrated to have any effect in the neurological aspects of FA.
In this work, we developed an experimental approach to possibly stall the neuronal degeneration observed in the DRG. Previous works have demonstrated that mesenchymal stem cells (MSCs) are capable of protecting FA cells from premature death when submitted to oxidative stress,13,14 as they are especially susceptible to this condition.5 This has been proven in vitro both with human and mouse cells. The effect observed by the stem cells, both from bone marrow and adipose tissue, was due to the secretion of various neurotrophic factors, mainly NT3, NT4, and BDNF. Also, we demonstrated that stem cells isolated from wild-type and mice with FA presented similar properties and gave similar results in vitro. The objective of this current work is to confirm this same effect in vivo, specifically in an FA mouse model. To this end, bone marrow-derived MSCs were isolated from wild-type and FA mice and transplanted intrathecally into the subarachnoid space of the spinal cord of ataxic mice. These mice were analyzed through several behavior tests, and the stem cells tracked in vivo by magnetic resonance. Five months after the surgical intervention, the mice were sacrificed and the DRG extracted and analyzed for prosurvival/apoptosis markers, neuronal markers, and neurotrophic factors. The data presented here may indicate a possible autologous or allogeneic stem cell-based therapeutic applicability to treat the neuronal degeneration observed in FA.
Results
YG8 mice treated with MSCs improve in their behavior tests
Both wild-type and YG8 mice were analyzed monthly on the rotarod and treadmill (Figure 1a). As a result, significant differences were detected in the rotarod and to a lesser degree in the treadmill. For the MSC-transplanted studies, stem cells were isolated from green fluorescent protein (GFP) transgenic mice and YG8 mice. In this sense, the injection of YG8-derived MSCs would emulate an autologous intervention in humans, while GFP-derived stem cells would be similar to an allogeneic procedure. The results of the behavior tests are shown in Figure 1b. Both stem cell-treated groups improved in the behavior tests compared to the control group. Furthermore, there were no significant differences in the results of the behavior tests comparing both stem cell-injected groups, although there seemed to be a tendency of further improvement in the YG8-stem cell group compared to the GFP-stem cell group.
Figure 1.

Behavior analysis of Freidreich's ataxia mice. (a) Behavior tests (rotarod, treadmill) performed on a monthly basis on wild-type (WT) and Freidreich's ataxia (YG8) mice. (b) Behavior tests performed on a monthly basis on vehicle-injected controls (TX sham), mice treated with mesenchymal stem cells of GFP mice (TX MSC-GFP), and mice treated with mesenchymal stem cells of YG8 mice (TX MSC-AtF). Vertical black bar indicates the moment the surgical procedure was performed. All values in a and b are with respect to the initial value obtained in the first month (normalized to 1). n = 8 in each case. *P < 0.05; **P < 0.01.
Thus, the results in this case indicate that YG8 mice present significant differences in their behavior tests compared to their wild-type counterparts. Furthermore, the MSCs-treated YG8 mice presented higher scores compared to nontreated ataxic mice, with no differences observed with either GFP- or YG8-derived stem cells.
Magnetic resonance imaging analysis demonstrate that the injected cells enter the DRG as early as 1–2 weeks posttransplantation
In several cases, the MSCs isolated from GFP mice were incubated for 16 hours with superparamagnetic iron oxide particles before transplanting into the ataxic mice. In this manner, it was possible to track in vivo the cells using magnetic resonance imaging (MRI) (Figure 2a). YG8 mice were analyzed at the moment of injection, then 7 and 14 days posttransplantation. As a result, the cells were detected in the area around the spinal cord (green arrow in Figure 2a) at the moment of injection, indicated by a negative contrast in the image. There was a similar negative stain in the pathway of the needle (red arrow), which gradually disappeared at day 7 and 14, while the stain in the spinal cord remained intact. Also, it was possible to detect the stem cells in the area where the DRG were present, indicating the possibility that the stem cells had entered this tissue. There was no indication of stem cells in other regions, including the muscle and spinal cord.
Figure 2.

Magnetic resonance imaging (MRI) of the YG8 mice. (a) MRI of vehicle-injected (sham) and YG8 mice treated with mesenchymal stem cells of GFP mice (TX MSC), analyzed at days 0, 7, and 14 postsurgery. In the TX MSC groups, the stem cells can be observed due to the presence of iron nanoparticles in the cells (see red and green arrows at day 0). The second row of images in the sham and TX MSC correspond to close-ups of the spinal cord observed in the first row of images. In the sham group, a dark staining to the right of the spinal cord can be observed at day 0, corresponding to hemorrhage due to the surgical intervention (red circle), which disappears at days 7 and 14. In the TX MSC group, dark staining is observed in the spinal cord (green arrow), and the path of the needle (red arrow and red circle). Both stains remain at 7 and 14 days, corresponding to the injected stem cells (red circle), although the marked cells in the muscle progressively lose signal intensity while the ones in the spinal cord remain. The images to the left (below the SHAM and TX MSC titles) are drawings identifying the various anatomical parts of the MRI images. (b) Immunohistochemistry of TX MSC-treated mice, performed at day 0 and 14 postsurgery. At day 0, the grafted cells were observed in the subarachnoid space, while at day 14, many cells were detected in the dorsal root ganglia. In the images, green is GFP (mesenchymal stem cells), red is Tuj1, and blue is DAPI staining. Similar results were observed in MRIs of four mice. AM, arachnoid mater; AS, arachnoid space; DM, dura mater; DRG, dorsal root ganglia; NR, nerve root; SC, spinal cord.
Several YG8 mice were sacrificed at day 0 and day 14 after the surgical intervention in order to confirm the MRI observations. Immunohistochemistry was performed for GFP staining in order to locate the MSCs (Figure 2b). At day 0, the grafted MSCs were detected in the cerebrospinal fluid around the spinal cord. However, at day 14, it was possible to detect MSCs inside or near the DRG. No stem cells were detected in other regions, and there were no signs of proliferation.
Thus, the immunohistochemistry analysis confirmed the results observed in the MRI, where the MSCs, when injected intrathecally into the subarachnoid space of the spinal cord, are capable of entering the DRG as early as 2 weeks after injection. This confirms the feasibility of this approach for stem cell injection in the DRG without causing damage to the nervous tissue.
MSCs persist 5 months after transplantation and exert a neuroprotective effect on the DRG
In the MRI, the ataxic mice were sacrificed and analyzed at 2 weeks after the surgical procedure. However, the mice submitted to behavior tests were sacrificed and analyzed 5 months after the intervention. Immunohistochemical analysis confirmed the presence of the grafted MSCs in the DRG (Figure 3a–f). These cells were detected not only near the neurons of the DRG (Figure 3a–c) but also in the surrounding blood vessels (Figure 3d), in the nerve root (Figure 3e), and even in the inner wall of the vertebrae, in the subarachnoid space (Figure 3f). This was observed in both groups of stem cell-treated mice, where GFP staining was used in the case of the MSC isolated from GFP transgenic mice, while CD44 and CD90 were used as MSC-specific markers in the case where stem cells from YG8 mice were transplanted. Furthermore, additional experiments were performed to confirm the presence of the MSCs in the dorsal root ganglia of the treated mice. Specifically, DRG were isolated from the treated mice to analyze GFP, CD90, and CD44 expression both by PCR and by western blot (Figure 3g–i). As a result, GFP was only detected in the mice treated with MSCs isolated from GFP mice. Also, CD90 and CD44, which were slightly expressed in the DRG,15 were more strongly detected in the stem cell-treated groups (both from GFP and YG8 mice). These results confirm that the MSCs that were initially injected into the cerebrospinal fluid of the spinal cord entered the DRG and remained there 5 months after transplantation.
Figure 3.

Analysis of stem cell markers in the dorsal root ganglia. (a–f) Immunohistochemical staining of the dorsal root ganglia (DRG) of YG8 mice treated with mesenchymal stem cells (labeled with an arrow). (a) DRG of an YG8 mouse where GFP-expressing mesenchymal stem cells were grafted (in green, blue is nuclear DAPI staining). (b,c) DRG of YG8 mice where mesenchymal stem cells isolated from ataxic mice were grafted, staining for specific markers CD90 and CD44, respectively. (d) Stem cells in a DRG surrounding a blood vessel. (e) Stem cells in the nerve root. (f) Stem cells from YG8 mice detected in the inner wall of the spinal cord (in the subarachnoid space). (g) GFP expression as analyzed by PCR in the TX MSC-GFP and control groups. Each lane corresponds to an individual mouse. Non-GFP and GFP mesenchymal stem cells were used as negative and positive controls (NEG, POS), respectively. (h) CD90 and CD44 expression analyzed by QPCR. RQ, relative quantity. (i) Western blot analysis of CD44, CD90, and GFP, using actin as control. Scale bar = 100 µm in a and e, 50 µm in b–d, and 250 µm in f. n = 4 in each case. *P < 0.05; **P < 0.001.
Semi-thin sections for electron microscopy were used to analyze the small cell to large cell ratio (Figure 4). The DRG from wild-type mice presented a ratio of ~1:1, small cells being mainly satellite cells (arrowheads and red colored cells in Figure 4a–c,e) and large cells being neurons (yellow colored cells). However, YG8 mice presented a much higher ratio, indicating that there were more satellite cells than neurons. The increase in small cell to large cell ration has also been detected in the DRG of human patients.16 However, this work is the first to report this observation in the YG8 model. As for the ataxic mice treated with MSCs, the ratio was similar to that detected in the wild-type mice. The ratio was not significantly different in the nontreated and treated ataxic mice, but there was a tendency for a reduced ratio, indicating a posible neuroprotective effect in the treated ataxic mice.
Figure 4.

Small cell to large cell ratio of the dorsal root ganglia. (a–c) Semi-thin images of the dorsal root ganglia of (a) wild-type, (b) YG8, and (c) stem cell-grafted YG8 mice, analyzed 5 months after injection. Arrowheads indicate satellite cells (in red, yellow cells indicate neurons). (d) Histogram depicting the ratio of small cells to large cells in each group of mice (ATX, YG8 mice; TX MSC, stem cell-treated YG8 mice; WT, wild-type mice). n = 3 in each case. *P < 0.05. (e) Dorsal root ganglia indicating by colors the small cells (in red) and large cells (in yellow) that were counted. The small cells are primarily satellite cells, while the large cells are neurons.
MSCs from GFP and YG8 mice express neurotrophic factors NT3, NT4, and BDNF in the DRG
Previous studies by our laboratory indicated that MSCs were capable of expressing and releasing NT3, NT4, and BDNF.14 These factors are capable of binding to the Trk receptors of the sensory neurons in the DRG and are known to play several roles, including activation of prosurvival mechanisms. In this study, we analyzed the expression of these trophic factors in the MSC-grafted DRG of ataxic mice 5 months after transplantation (Figure 5a), confirming the results previously observed in vitro. Furthermore, the expression of these trophic factors were analyzed both at the genetic and protein levels (Figure 5b,c). In the protein, several bands can be detected in each of the three trophic factors, generally corresponding to a smaller band (the mature form in monomer form, which is generally located in the interior of the cells), a larger band (the precursor or proform, also located inside the cells), and an intermediate band, which corresponds to the homodimer.17,18,19 The latter is the form in which the trophic factors are generally secreted. Higher levels of BDNF and NT3 expression was detected in the DRG of the MSC-treated mice, compared to the sham group. However, NT4 expression seemed similar in all three mice groups (Sham, MSC-GFP treatment, and MSC-YG8 treatment).
Figure 5.

Neurotrophic analysis in the dorsal root ganglia. (a) Immunohistochemistry of expression of BDNF, NT3, and NT4 in the dorsal root ganglia of a mouse treated with mesenchymal stem cells isolated from GFP mice, analyzed 5 months after injection. The stem cells are stained in green (GFP), neurotrophic factors in red, and DAPI staining in blue. Scale bar = 100 µm. DRG, dorsal root ganglia; NR, nerve root. (b) Neurotrophic factor expression measured in the dorsal root ganglia by western blot analysis of vehicle-injected mice (sham), treatment with mesenchymal stem cells of GFP mice (TX MSC-GFP) and of Friedreich's ataxia mice (TX MSC-AtF). In all cases, several bands appear, corresponding either to the proform, homodimer (secreted form), or the mature form (cytosolic form). (c) QPCR analysis of the trophic factors in the sham, TX MSC-GFP, and TX MSC-AtF groups. RQ, relative quantity. n = 4 in each case, *P < 0.05.
The western blot results were compared to the quantitative polymerase chain reaction (QPCR) results for the expression of the same trophic factors. In this case, the DRG of the mice treated with MSCs from both sources (GFP and YG8) presented higher levels of expression of the three trophic factors with respect to the sham control. No differences were detected in the trophic factor expression, both by western blot and by QPCR, of control nontreated mice and sham controls of the same age (data not shown).
These results indicate that MSCs express and release the trophic factors NT3, NT4, and BDNF in the DRG, inducing an increased expression of these trophic factors in this region. As our previous in vitro work has demonstrated, the expression of these trophic factors is known to protect DRG neurons from cell death14 and is the most probable cause for the effect observed by the stem cell transplantation.
DRG of treated mice present less apoptosis, more neuronal and glial markers, and increased frataxin expression
The previous results indicated that the grafted cells expressed the neurotrophic factors analyzed, which are known to be implicated in cell survival mechanisms. Thus, to prove that this was the case, the expression of proapoptotic (caspase-3 and Bax), prosurvival (Bcl2), neuronal (Tuj1 and MAP2), and glial markers (GFAP) were studied (Figure 6a). In the MSC-GFP group, there was more expression of all the glial and neuronal markers analyzed, while in the MSC-ATF group, only MAP2 expression was increased. Also, the DRGs of these groups presented low levels of proapoptotic markers caspase-3 and Bax, while expressing very high levels of prosurvival marker Bcl2. The sham group presented similar levels of expression of all the neuronal, glia, and apoptosis/survival markers analyzed compared to the control group, except for caspase-3 and Bax, which were higher.
Figure 6.
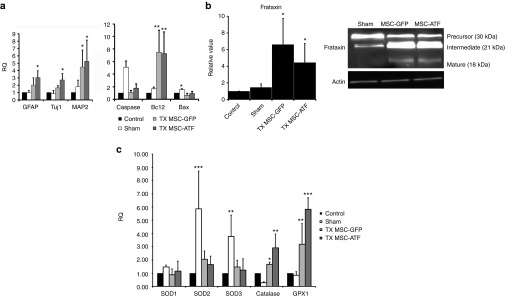
Dorsal root ganglia analysis. (a) QPCR analysis of the dorsal root ganglia in nontreated Friedreich's ataxia (control), vehicle-injected (sham), treatment with mesenchymal stem cells of GFP mice (TX MSC-GFP) and of Friedreich's ataxia mice (TX MSC-AtF), 5 months after injection. The genes analyzed are shown in the histograms. RQ, relative quantity. (b) Frataxin expression measured by QPCR (histogram) and western blot (right image). (c) QPCR analysis of the oxidative stress markers SOD1, SOD2, SOD3, catalase, and GPX-1 in the control, sham TX MSC-GFP, and TX MSC-AtF mice. n = 4 in each case. *P < 0.05; **P < 0.01; ***P < 0.001. SOD, superoxide dismutase.
Also, frataxin expression was analyzed both at the gene expression and protein levels (Figure 6b). Three forms were detected: the precursor, intermediate, and mature form.20 The DRGs of the MSC-treated groups expressed higher levels of frataxin than control and sham groups. Furthermore, there was more expression of the mature form of the protein, while almost no expression was detected in the sham group. Thus, MSCs injection increased frataxin expression, as previously observed in the in vitro studies.14
Since oxidative stress plays a major role in FA,5 several of the most important markers was analyzed (Figure 6c). Previous results in vitro demonstrated that catalase and GPX-1 expression levels were significantly lower in the DRGs of YG8 mice compared to wild-type mice of the same age, while superoxide dismutase (SOD) levels remained mainly unchanged.14 In this work, the MSC-treated ataxic mice expressed similar levels of SODs as the controls, while catalase and GPX-1 levels were increased, as a possible compensatory mechanism to reach levels similar to wild-type mice.
Interestingly, in the sham controls, SODs expression was very high. This could be a compensation mechanism due to the high apoptotic levels detected in this experimental group (Figure 6a). The sham group presented higher levels of apoptosis, indicated by the increased expression of active caspase-3 and Bax. Increased cell death gives rise to increased oxidative stress, as free radicals are released from the degenerating neurons. Thus, the aberrant levels of SOD2 and SOD3 may be a compensatory mechanism to remove the free radicals. The fact that the expression levels of these antioxidants were higher than that in nontreated controls may be due to the surgical intervention. The intervention can initially cause tissue damage, as well as an inflammatory process which may cause, at the long run, further damage. The stem cell injection may be avoiding this due to their immunomodulatory properties. On the other hand, catalase and GPX-1 levels remained unchanged or were even lower than controls. These observations have one additional very important connotation: an uncontrolled intervention (such as, injecting an inadequate source of cells or an incorrectly performed surgical intervention) may cause significant damage in the nervous tissue, which can continue much after the intervention (5 months in this case).
MSCs grafted in the subarachnoid space of the spinal cord of wild-type mice do not penetrate into the DRG, nor increase neurotrophic factor expression
The same surgical procedure with MSCs was performed in wild-type mice, in order to confirm if the trophic effect observed in the YG8 mice were due to the stem cells being injected into a neurodegenerative niche or if it was a default effect of the grafted cells. As a result, in vivo tracking by MRI analysis confirmed that the grafted cells remained in the area injected of the spinal cords of wild-type mice (Figure 7a). However, immunohistochemical analysis 2 weeks after surgery indicated that the stem cells did not enter the DRG (Figure 7b), but rather remained in the cerebrospinal fluid of the spinal cord. Furthermore, real-time PCR indicated that the MSCs did not increase the expression of neurotrophic factors NT3 and NT4, with a slight increase in BDNF (Figure 7c). This differs from the data obtained when grafting MSCs in YG8 mice, where the DRG presented increased expression levels of the three trophic factors analyzed.
Figure 7.

Stem cell injection in the spinal cord of wild-type mice. (a) MRI images of the superparamagnetic iron oxide-incubated stem cells in the spinal cord of wild-type mice, at days 0, 7, and 14. The stem cells can be observed as dark spots in the spinal cord region. (b) Immunohistochemistry for GFP, staining the grafted stem cells, 2 weeks after injection. No grafted cells were detected in the DRG. Images taken at ×100. DRG, dorsal root ganglia; MSC, mesenchymal stem cells; NR, nerve root; SC, spinal cord. (c) QPCR analysis of neurotrophic factor expression in the dorsal root ganglia of control mice (CT) versus those treated with mesenchymal stem cells (TX MSC) in wild-type (left histogram) and YG8 (right histogram hosts). n = 4 in all cases. *P < 0.05; **P < 0.01.
These results indicate that the bone marrow-derived MSCs, when in the presence of a neurodegenerative niche such as the DRG of YG8 mice, are capable of integrating into the DRG and increase the expression and secretion of neurotrophic factors known to be implicated in neuronal survival.
Discussion
Our results indicate that MSCs injected intrathecally into the lumbar region of the spinal cord migrate toward the DRG, where they integrate and release various neurotrophic factors, increasing the survival rate of the sensory neurons in a FA mouse model. Using this treatment, the mice presented improved motor skills compared to their untreated counterparts, as well as increased response to oxidative stress. These results corroborate with those we have previously demonstrated in vitro using DRG primary cultures.14 Also, stem cells from both wild-type and FA mice gave similar results.
The injected stem cells secreted neurotrophic factors BDNF, NT3, and NT4, which in turn are known to increase the survival of the sensory neurons in the DRG.21 The surgical intervention resulted in increased cell survival markers and subsequently decreased apoptotic markers. Also, upregulation of certain neuronal and satellite/Schwann cell markers were detected in the DRG of the treated mice. This is important since even though the proprioceptive neurons are the first to degenerate in FA, the rest of the neurons as well as the Schwann cells are affected.22 This is partly due to oxidative stress. In FA, the antioxidant mechanisms are affected, which is exacerbated by the lack of frataxin, causing iron overload and increased free-radical production.23 The mouse model used in this work also present the deficient antioxidant mechanisms.24 This is the reason several oxidative stress markers were analyzed in the DRG of the treated mice. Specifically, catalase and GPX-1 were upregulated in the treated mice. Catalase is an important antioxidant enzyme involved in removing hydrogen peroxide, converting it to water and oxygen.25 GPX-1, on the other hand, plays a major role in removing peroxides in the cell.26 In our in vitro work,14 we demonstrated that these genes were downregulated in YG8 mice compared to wild-type mice of the same age. This was also corroborated in a recent work,27 where a direct correlation between frataxin and antioxidants levels was observed (e.g., lower frataxin levels correlated with lower catalase levels). Consequently, higher frataxin levels may help to increase antioxidant levels, which may be the cause of the increased catalase and GPX-1 levels detected. The expression of SODs, however, was not increased. This may be due to the fact that in our in vitro study, the transcriptions of SODs were unaffected in the YG8 mice compared to wild-type mice. On the other hand, in the sham control, they were significantly increased. As previously commented in the Results section, this may be due to increased oxidative stress levels in the DRG of the sham controls, which coincided with increased levels of apoptosis. In this group, catalase and GPX-1 levels were unaffected.
In conclusion, our study indicates that bone marrow MSCs, either from wild-type or FA mice, are capable of inducing a neuroprotective effect on the sensory neurons of the DRG in ataxic mice. This results in improved behavior tests, increased frataxin levels, improved cell survival, and increased levels of certain antioxidants. The results shown here may be considered as a step further into a possible therapeutic approach using autologous stem cells to decrease the progression of the neurological degeneration observed in FA. Furthermore, as oxidative stress is important in other neurodegenerative disease, including amyotrophic lateral sclerosis, Parkinson's disease, Alzheimer's disease, and Huntington's disease,28 this therapeutic approach may also be viable for these disorders.
Materials and Methods
Animals. All the experiments with animals have been performed in compliance with the Spanish and European Union laws on animal care in experimentation (Council Directive 86/609/EEC) and have been analyzed and approved by the Animal Experimentation Committee of the University Miguel Hernandez and Neuroscience Institute, Alicante, Spain (Reference IN-JJ-001-13). All efforts were made to minimize suffering. Mice were bred and maintained in our animal facilities. Eight-month-old Fxntm1Mkn/Tg(FXN)YG8Pook (YG8, originally purchased from Jackson Laboratory, Bar Harbor, ME) transgenic mice were used. These mice present a knock-out mutation of the Frataxin gene with a human knock-in Frataxin gene to rescue the phenotype.29 The inserted gene presents the same mutation as in humans, that is, the extended GAA triplet repeats. As a result, the mice present reduced levels of frataxin, causing degeneration in the heart, DRG, and, in some cases, the pancreas. For the MSCs, the bone marrows of 2–3-month-old GFP transgenic mice and YG8 mice were used.
Bone marrow mesenchymal stem cells isolation and culture. The protocol used was similar to our previously published work.14 Briefly, femurs were dissected from 2–3-month-old mice, sacrificed by cervical dislocation. Bone marrow was extracted, and single-cell suspensions were obtained by mechanical dissociation. Then, the suspension was washed and centrifuged, and the pellet resuspended in D-MEM (Invitrogen, Life Technologies, Baseley, UK) supplemented with 15% fetal bovine serum (Biochrom AG, Berlin, Germany) and 100 U/ml penicillin/streptomycin (Sigma-Aldrich, St Louis, MO). These cells were placed in culture flasks, and the plastic-adherent population was isolated and allowed to proliferate for 3–4 weeks, changing the media every 2–3 days and replating when needed.
Surgical intervention. Depending on the mouse group, either culture medium or bone marrow MSCs were injected with a Hamilton syringe into the mutant mice (Hamilton, Bonaduz, Switzerland). A total of six injections (2 µl/each, 3 × 105 cells per injection) were performed through the interlaminar (yellow) ligaments of two consecutive lumbar vertebrae. The cells were injected into the cerebrospinal liquid by subarachnoid punction, with the mouse inclined 30° head-down. In this manner, the cells diffuse to dorsal levels of the spinal cord subarachnoidal space, attach to the pial surface of dorsal roots, and penetrate into the DRG, the first area affected in these mice. The injections were performed in a slow, mechanically controlled speed so as to avoid swelling and thus damaging the spinal cord.
Magnetic resonance imaging. Twenty-four hours before the surgical intervention, the MSCs were previously cultured in 7 µg/ml of Feraspin XL (Viscover Imaging, Miltenyi Biotec, Cologne, Germany). This product consists in nanoparticles containing iron and enveloped in dextran, allowing its diffusion to the cells. For the MRI experiments, mice were anesthetized in an induction chamber with 3–4% isoflurane (Esteve Veterinary, Milan, Italy) in medical air and maintained with 1–2% isoflurane during the process. Anesthetized animals were placed in a custom-made animal holder with movable bite and ear bars and positioned fixed on the magnet chair. This allowed precise positioning of the animal with respect to the coil and the magnet and avoided movement artifacts. The body temperature was kept at ~37 °C using a water blanket, and the animals were monitored using a MRI compatible temperature control unit (MultiSens Signal conditioner, OpSens, Quebec, Canada). Experiments were carried out in a horizontal 7 Tesla scanner with a 30 cm diameter bore (Biospec 70/30v; Bruker Medical, Ettlingen, Germany). The system had a 675 mT/m actively shielded gradient coil (Bruker Medical; BGA 12-S) of 11.4 cm inner diameter. A 1H rat brain receive-only phase array coil with integrated combiner and preamplifier, no tune/no match, in combination with the actively detuned transmit-only resonator (Bruker BioSpin MRI) was employed. Data were acquired with a Hewlett-Packard console running Paravision software (Bruker Medical) operating on a Linux platform.
T2-weighted anatomical images to position the animal were collected in the three orthogonal orientations using a rapid acquisition relaxation enhanced sequence, applying the following parameters: field of view: 40 × 40 mm, 15 slices, slice thickness: 1 mm, matrix: 256 × 256, effective echo time (TEeff) 56 milliseconds, repetition time: 2 seconds, rapid acquisition relaxation enhanced sequence factor of 8, 1 average and a total acquisition time of 1 minute 4 seconds. To detect superparamagnetic iron oxide labeled cells, a T2* multigradient echo images were acquired in the three orthogonal orientations with the following parameters: repetition time: 1,500 milliseconds, echo time: 3 milliseconds, flip angle: 30°, field of view: 20 × 20 mm, 20 slices, slice thickness: 0.5 mm, matrix: 256 × 256, two averages and a total acquisition time of 12 minutes 48 seconds.30
Behavior assays. Rotarod and treadmill tests were performed similar to our previous work.31 The tests were performed on a monthly basis for up to five trials before the surgical intervention. Then, and after 1 month of surgical recovery, the mice continued the monthly tests every month for another five trials.
In the case of the rotarod (model 8500, Letica Scientific Instruments, Barcelona, Spain), it was set to accelerate from 4 to 40 rpm in a 5-minute time span. Each mouse was placed five times (with adequate rests between trials), and both the maximum speed and time on the rod were noted. This was performed for four consecutive days, once a month, and the values obtained in the fourth day was used.
For the treadmill test, an LE 8700 model was used (Letica Scientific Instrument). The treadmill test consisted of placing the mouse in a lane that pushed the animal to a shock grid (0.4 mA). In this manner, the animal must run to avoid the shock. Each mouse was placed on the treadmill five times, with adequate rests between trials, in order to obtain the average maximum speed.
The results were normalized with respect to the initial value obtained in the first month, which presented a value of 1, using the same approach as in our previous report.14 In this manner, a value above or below 1 indicated an improvement or worsening, respectively, with respect to the initial value.
Immunohistochemistry. The mice were anesthetized with isoflurane (Esteve Veterinary) and spinal cords fixed with 4% paraformaldehyde (Sigma-Aldrich) in phosphate buffer (pH 7.4) overnight. After fixation, the spinal cords were placed in Osteosoft (Merck Millipore, Billerica, MA) solution for 5 days, in order to decalcify the vertebrae. Then, the tissue was placed in paraffin, and transverse sections of 16 µm were obtained and mounted on slides.
The sections were first incubated at room temperature in 10% goat serum (Sigma-Aldrich), 5% bovine albumin (Sigma-Aldrich), 0.25% triton (Sigma-Aldrich), and phosphate-buffered saline to permeate the tissue and block nonspecific binding. Afterward, the sections were incubated overnight at room temperature with the primary antibody, diluted in blocking solution (10% goat serum, 5% bovine albumin, and phosphate-buffered saline). The following primary antibodies were used: mouse or rabbit anti-GFP (1:200; Molecular Probes, Eugene, OR), rat anti-CD90 (1:250; BD Biosciences, San Diego, CA), rat anti-CD44 (1:250; BD Biosciences), rabbit anti-parvalbumin (1:2,000; Swant, Marly, Switzerland), mouse anti-Tuj1 (1:1,000; Covance, Berkeley, CA), rabbit anti-BDNF (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), neurotrophin-4/5 (NT-4/5, 1:100; Chemicon/Millipore), and sheep anti-neurotrophin-3 (NT-3, 1:100; Chemicon/Millipore).
The following day, the sections were incubated with the secondary antibody. For GFP staining, anti-mouse Alexa Fluor 488 (1:500; Molecular Probes) was used, and for the other antibodies, biotinylated secondary antibodies were used (1:200, Vector Laboratories, Burmingham, CA) followed by an incubation with streptavidin conjugated with Cy3 (1:500). 4',6-diamidino-2-phenylindole (DAPI) (Molecular Probes) was used to stain nuclei. Histological samples were observed under a fluorescence microscope (Leica DMR, Leica Microsystems, Wetzlar, Germany).
Standard and real-time PCR. Total mRNA of the cells was isolated using the Trizol protocol (Invitrogen). In the case of standard PCR analysis, the mRNA was reverse transcribed using the Quantitect Reverse Transcription kit (QIAGEN, Silicon Valley, CA), processed with the QIAGEN Multiplex PCR kit, and run on the QIAxcel apparatus. In the case of real-time PCR, 5 µg of mRNA was reverse transcribed, and ~100 ng of cDNA was amplified using Power SYBR Green Master mix (Applied Biosystems, Foster City, CA). All the samples were run in triplicate using the StepOne Plus Real-Time PCR system (Applied Biosystems; 40 cycles) and analyzed with the StepOne Software (Applied Biosystems/Life Technologies, Madrid, Spain). Analyses were carried out using the ΔCt method and calculated relative to GAPDH (forward: AGGTCGGTGTGAACGGATTTG, reverse: GGGGTCGTTGATGGCAACA). The results were normalized with respect to the control condition, which presented a value of 1, using the same approach as in our previous report.14 The following primers were used, taken from the PrimerBank webpage (http://pga.mgh.harvard.edu/primerbank/): NT4 (forward: TGAGCTGGCAGTATGCGAC, reverse: CAGCGCGTCTCGAAGAAGT), BDNF (forward: TCATACTTCGGTTGCATGAAGG, reverse: GTCCGTGGACGTTTACTTCTTT), NT3 (forward: AGTTTGCCGGAAGACTCTCTC, reverse: GGGTGCTCTGGTAATTTTCCTTA), Frataxin (forward: CCACGCCCATTTGAACCTC, reverse: TCTTTCATACGCTGTCTCGTCT), GFAP (forward: CGGAGACGCATGACCTCTG, reverse: AGGGAGTGGAGGAGTCATTCG, GFP (forward: CTGCTGCCCGACAACCA, reverse: GAACTCCAGCAGGACGACGACCATGTG), Tuj1 (forward: TCAGCGATGAGCACGGCATA, reverse: CACTCTTTCCGCACGACATC), MAP2 (forward: AGCCGCAACGCCAATGGATT, reverse: TTTGTTCCGAGGCTGGCGAT), SOD1 (forward: ATGGCGATGAAAGCGGTGT, reverse: CCTTGTGTATTGTCCCCATACTG), SOD2 (forward: CAGACCTGCCTTACGACTATGG, reverse: CTCGGTGGCGTTGAGATTGTT), SOD3 (forward: CCTTCTTGTTCTACGGCTTGC, reverse: ACGTGTCGCCTATCTTCTCAA), Caspase-3 (forward: TGGTGATGAAGGGGTCATTTATG, reverse: TTCGGCTTTCCAGTCAGACTC), Catalase (forward: AGCGACCAGATGAAGCAGTG, reverse: TCCGCTCTCTGTCAAAGTGTG), GPX-1 (forward: CCACCGTGTATGCCTTCTCC, reverse: AGAGAGACGCGACATTCTCAAT), Bcl-2 (forward: ATGCCTTTGTGGAACTATATGGC, reverse: GGTATGCACCCAGAGTGATGC), Bax (forward: AGACAGGGGCCTTTTTGCTAC, reverse: AATTCGCCGGAGACACTCG), CD90 (forward: TGCTCTCAGTCTTGCAGGTG, reverse: TGGATGGAGTTATCCTTGGTGTT), and CD44 (forward: TCGATTTGAATGTAACCTGCCG, reverse: CAGTCCGGGAGATACTGTAGC).
Western blot analysis. The DRG of the treated mice were lysed, and the proteins separated on 15% sodium dodecyl sulfate–polyacrylamide gels and probed for rabbit anti-Frataxin (1:750; Santa Cruz Biotechnology), rat anti-CD44 (1:500; BD Pharmingen, San Diego, CA), rat anti-CD90(1:500; BD Pharmingen), mouse anti-GFP(1:1,000; Molecular Probes, Eugene, OR), rabbit anti-BDNF (1:500; Santa Cruz Biotechnology), anti-NT3 (1:250; Abcam, Cambridge, England), and anti-NT4 (1:250, Santa Cruz Biotechnology). Secondary antibodies were visualized by chemiluminescence (ECL, Amersham, England). For protein quantification, we used Quantity One software (BioRad, Hercules, CA). β-Actin was used as loading control.
Tissue processing for semi-thin sections. Unless otherwise stated, all reactants were purchased from Sigma-Aldrich. Animals were perfused intracardially with 0.9% saline followed by 4% paraformaldehyde and 0.5% glutaraldehyde. DRG were dissected and postfixed overnight in the same fixative solution. Then, whole ganglia were postfixed in 2% osmium tetroxide (OsO4) for 2 hours, rinsed, dehydrated, and embedded in Durcupan. To study the organization of the dorsal ganglia, semi-thin sections (1.5 µm) were cut with a diamond knife and stained with 1% toluidine blue. Subsequently, the area of interest was trimmed, and ultrathin sections (60–70 nm) were obtained with a diamond knife, stained with lead citrate and examined under a transmission electron microscope (Tecnai Spirit G2; FEI, Eindhoven, The Netherlands). In semi-thin sections, the small cell ratio was estimated dividing the number of small cells by the number of neurons. Three ganglia were analyzed per animal, and n = 3 per group (WT, ATX, and TX MSC).
Statistical analysis. Statistical significance between control and experimental groups were calculated with Sigmaplot v11.0 software (Systat Software, San Jose, CA), using the paired t-test and one-way ANOVA test where applicable, establishing the level of significance at P < 0.05. Values are measured as mean ± SD.
Acknowledgments
We thank M. Rodenas, P. Almagro, O. Bahamonde, and J. Martinez for their technical assistance. This work was supported by FARA/FARA Ireland (Friedreich's Ataxia Research Alliance), ASOGAF (Friedreich's ataxia association of Granada), Science and Innovation Ministry (MICINN BFU2010-27326), GVA Prometeo grant 2009/028, PROMETEOII GRANT 2014/014, Tercel (RD06/0010/0023 & RD06/0010/24), MEC-CONSOLIDER CSD2007-00023, Cinco P menos Foundation, EUCOMM, Fundacion Diogenes-Elche city government, Walk on Project, and MAPFRE Foundation.
References
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- Pandolfo M. Friedreich ataxia: the clinical picture. J Neurol. 2009;256 suppl. 1:3–8. doi: 10.1007/s00415-009-1002-3. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Boesch S, Bürk K, Dürr A, Giunti P, Mariotti C, et al. Diagnosis and treatment of Friedreich ataxia: a European perspective. Nat Rev Neurol. 2009;5:222–234. doi: 10.1038/nrneurol.2009.26. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Lodi R, Tonon C, D'Agata V, Sapienza M, Scapagnini G, et al. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia. J Neurol Sci. 2005;233:145–162. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Lodi R, Hart PE, Rajagopalan B, Taylor DJ, Crilley JG, Bradley JL, et al. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich's ataxia. Ann Neurol. 2001;49:590–596. [PubMed] [Google Scholar]
- Cooper JM, Schapira AH. Friedreich's ataxia: disease mechanisms, antioxidant and Coenzyme Q10 therapy. Biofactors. 2003;18:163–171. doi: 10.1002/biof.5520180219. [DOI] [PubMed] [Google Scholar]
- Hart PE, Lodi R, Rajagopalan B, Bradley JL, Crilley JG, Turner C, et al. Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol. 2005;62:621–626. doi: 10.1001/archneur.62.4.621. [DOI] [PubMed] [Google Scholar]
- Rustin P, von Kleist-Retzow JC, Chantrel-Groussard K, Sidi D, Munnich A, Rötig A. Effect of idebenone on cardiomyopathy in Friedreich's ataxia: a preliminary study. Lancet. 1999;354:477–479. doi: 10.1016/S0140-6736(99)01341-0. [DOI] [PubMed] [Google Scholar]
- Hausse AO, Aggoun Y, Bonnet D, Sidi D, Munnich A, Rötig A, et al. Idebenone and reduced cardiac hypertrophy in Friedreich's ataxia. Heart. 2002;87:346–349. doi: 10.1136/heart.87.4.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti C, Solari A, Torta D, Marano L, Fiorentini C, Di Donato S. Idebenone treatment in Friedreich patients: one-year-long randomized placebo-controlled trial. Neurology. 2003;60:1676–1679. doi: 10.1212/01.wnl.0000055872.50364.fc. [DOI] [PubMed] [Google Scholar]
- Buyse G, Mertens L, Di Salvo G, Matthijs I, Weidemann F, Eyskens B, et al. Idebenone treatment in Friedreich's ataxia: neurological, cardiac, and biochemical monitoring. Neurology. 2003;60:1679–1681. doi: 10.1212/01.wnl.0000068549.52812.0f. [DOI] [PubMed] [Google Scholar]
- Jones J, Estirado A, Redondo C, Bueno C, Martínez S. Human adipose stem cell-conditioned medium increases survival of Friedreich's ataxia cells submitted to oxidative stress. Stem Cells Dev. 2012;21:2817–2826. doi: 10.1089/scd.2012.0029. [DOI] [PubMed] [Google Scholar]
- Jones J, Estirado A, Redondo C, Martinez S. Stem cells from wildtype and Friedreich's ataxia mice present similar neuroprotective properties in dorsal root ganglia cells. PLoS One. 2013;8:e62807. doi: 10.1371/journal.pone.0062807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Wang SM, Yang SH, Jeng CJ. Role of Thy-1 in in vivo and in vitro neural development and regeneration of dorsal root ganglionic neurons. J Cell Biochem. 2005;94:684–694. doi: 10.1002/jcb.20341. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Morral JA, Davis AN, Qian J, Petrocine SV, Knutson MD, et al. The dorsal root ganglion in Friedreich's ataxia. Acta Neuropathol. 2009;118:763–776. doi: 10.1007/s00401-009-0589-x. [DOI] [PubMed] [Google Scholar]
- Mowla SJ, Farhadi HF, Pareek S, Atwal JK, Morris SJ, Seidah NG, et al. Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J Biol Chem. 2001;276:12660–12666. doi: 10.1074/jbc.M008104200. [DOI] [PubMed] [Google Scholar]
- Philo J, Talvenheimo J, Wen J, Rosenfeld R, Welcher A, Arakawa T. Interactions of neurotrophin-3 (NT-3), brain-derived neurotrophic factor (BDNF), and the NT-3.BDNF heterodimer with the extracellular domains of the TrkB and TrkC receptors. J Biol Chem. 1994;269:27840–27846. [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- Gakh O, Bedekovics T, Duncan SF, Smith DY, 4th, Berkholz DS, Isaya G. Normal and Friedreich ataxia cells express different isoforms of frataxin with complementary roles in iron-sulfur cluster assembly. J Biol Chem. 2010;285:38486–38501. doi: 10.1074/jbc.M110.145144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenghi G. Peripheral nerve regeneration and neurotrophic factors. J Anat. 1999;194 Pt 1:1–14. doi: 10.1046/j.1469-7580.1999.19410001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen AH. Friedreich's ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci. 2011;303:1–12. doi: 10.1016/j.jns.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparaco M, Gaeta LM, Santorelli FM, Passarelli C, Tozzi G, Bertini E, et al. Friedreich's ataxia: oxidative stress and cytoskeletal abnormalities. J Neurol Sci. 2009;287:111–118. doi: 10.1016/j.jns.2009.08.052. [DOI] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S, et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelikani P, Fita I, Loewen PC. Diversity of structures and properties among catalases. Cell Mol Life Sci. 2004;61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raes M, Michiels C, Remacle J. Comparative study of the enzymatic defense systems against oxygen-derived free radicals: the key role of glutathione peroxidase. Free Radic Biol Med. 1987;3:3–7. doi: 10.1016/0891-5849(87)90032-3. [DOI] [PubMed] [Google Scholar]
- Shan Y, Schoenfeld RA, Hayashi G, Napoli E, Akiyama T, Iodi Carstens M, et al. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich's ataxia YG8R mouse model. Antioxid Redox Signal. 2013;19:1481–1493. doi: 10.1089/ars.2012.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21:172–188. doi: 10.1021/tx700210j. [DOI] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Ruddle P, Carroll C, Webster Z, Pook M. GAA repeat instability in Friedreich ataxia YAC transgenic mice. Genomics. 2004;84:301–310. doi: 10.1016/j.ygeno.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Willenbrock S, Knippenberg S, Meier M, Hass R, Wefstaedt P, Nolte I, et al. In vivo MRI of intraspinally injected SPIO-labelled human CD34+ cells in a transgenic mouse model of ALS. In Vivo. 2012;26:31–38. [PubMed] [Google Scholar]
- Pastor D, Viso-León MC, Botella-López A, Jaramillo-Merchan J, Moraleda JM, Jones J, et al. Bone marrow transplantation in hindlimb muscles of motoneuron degenerative mice reduces neuronal death and improves motor function. Stem Cells Dev. 2013;22:1633–1644. doi: 10.1089/scd.2012.0487. [DOI] [PMC free article] [PubMed] [Google Scholar]