

Abstract
Adoptive immunotherapy with antigen-specific T cells has shown promise for the treatment of malignancies. However, infused T cells are unable to redirect resident T cells, limiting potential benefit. While the infusion of bispecific T-cell engagers can redirect resident T cells to tumors, these molecules have a short half-life, and do not self amplify. To overcome these limitations, we generated T cells expressing a secretable T-cell engager specific for CD3 and EphA2, an antigen expressed on a broad range of human tumors (EphA2-ENG T cells). EphA2-ENG T cells were activated and recognized tumor cells in an antigen-dependent manner, redirected bystander T cells to tumor cells, and had potent antitumor activity in glioma and lung cancer severe combined immunodeficiency (SCID) xenograft models associated with a significant survival benefit. This new class of tumor-specific T cells, with the unique ability to redirect bystander T cells, may be a promising alternative to current immunotherapies for cancer.
Introduction
Immunotherapy with antigen-specific T cells has shown promise for the therapy of viral-associated diseases and malignancies.1,2 Genetic modification of T cells with chimeric antigen receptors (CARs) has enabled the rapid ex vivo generation of tumor-specific T cells, and clinical studies with CD19-specific CAR T cells have shown impressive responses for patients with CD19-positive malignancies.3,4,5,6 However, the efficacy of CAR T-cell therapy depends on significant in vivo expansion, which may not always be possible, for example, in the immuosuppressive environment of a tumor.7,8,9 In addition, adoptively transferred T cells, including CAR T cells, do not redirect the vast reservoir of resident T cells to tumors.
One successful strategy to redirect resident T cells to tumors is the infusion of recombinant proteins encoding T-cell engagers that are specific for CD3 expressed on T cells and an antigen expressed on the cell surface of tumor cells.10,11,12,13 Of these, BiTEs, consisting of two single chain variable fragments (scFVs) connected by a short linker, have been the most successful with promising antitumor activity against CD19-positive malignancies in clinical studies.14,15 While effective, BiTEs have a short half-life necessitating continuous, systemic infusion that may be associated with toxicities, lack active biodistribution, and similar to conventional monoclonal antibodies (MAbs) do not self amplify.12,13
Here we report the generation of T cells that themselves secrete a bispecific T-cell engager (ENG T cells) specific both for CD3 and the tumor-associated antigen erythropoietin-producing hepatocellular carcinoma A2 (EphA2), a member of the Eph family of receptor tyrosine kinases that is overexpressed in a broad range of malignancies including breast, lung, prostate, and glioblastoma.16,17 These EphA2-specific ENG T cells produced immunostimulatory cytokines and proliferated in an antigen-specific manner, killed EphA2-positive targets in vitro, redirected bystander T cells to tumor cells, secreted more engager molecules upon activation, and had potent antitumor activity in both loco-regional and systemic severe combined immunodeficiency (SCID) xenograft tumor models.
Results
Generation of engager T cells
A bispecific EphA2-specific T-cell engager consisting of EphA2- and CD3-specific scFVs connected by short linker was cloned into a retroviral vector upstream of an internal ribosomal entry site (IRES) and mOrange (Figure 1a). To generate T cells secreting EphA2-specific engagers (EphA2-ENG T cells), CD3/CD28-activated T cells were transduced with RD114-pseudotyped retroviral particles. Five to 7 days post-transduction mOrange expression was determined by fluorescence-activated cell sorting (FACS) analysis. 57.4 ± 12.2% (n = 23) of cells were positive for mOrange (Figure 1b), and CD4- as well as CD8-positive T cells were transduced (Supplementary Figure S1). Transduced T cells expressed engager molecule mRNA as judged by qRT-PCR (Figure 1c). To confirm expression by FACS analysis, we generated an engager molecule with a 6xHIS-myc tag (Supplementary Figure S2a,b). We demonstrated cell surface binding of engagers using a myc-specific MAb and secretion using HIS-Mag beads followed by detection of engager molecules with anti-myc western blot (Supplementary Figure S2c,d).
Figure 1.
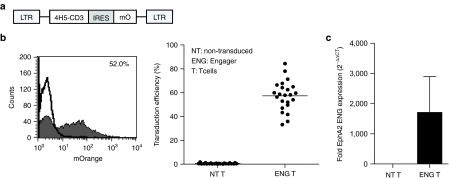
Generation of EphA2-ENG T cells. (a) Scheme of retroviral vector (IRES, internal ribosomal entry site; mO, mOrange). (b) Transduction efficiency was determined by FACS analysis for mO of transduced (filled) and nontransduced (NT; line) T cells. Representative FACS plot and summary of all data in shown in scatter plot format (n = 23). (c) qRT-PCR for EphA2-engager mRNA of transduced and NT T cells.
EphA2-ENGT cells recognize and kill EphA2-positive tumor cells
EphA2-ENG T cells were cocultured with EphA2-positive (U373, A549, K562-EphA2) or EphA2-negative (K562) tumor cells,18,19 and after 24 hours, we determined the concentration of IFN-γ and IL-2 in cell culture supernatants by ELISA. Nontransduced (NT) T cells and T cells expressing a CD19-specific T-cell engager served as controls (Supplementary Figure S3). EphA2-ENG T cells were only activated by EphA2-positive tumor cells as judged by IFN-γ (P < 0.001) and IL-2 (P < 0.05) production (Figure 2a), while CD19-ENG and NT T cells produced neither IFN-γ nor IL-2. When stimulated with EphA2-positive (U373, A549) tumor cells, only EphA2-ENG T cells expanded four to fivefold within 1 week after stimulation in contrast to CD19-ENG or NT-T cells (P < 0.001; Figure 2b). Importantly, there was no significant difference between CD19-ENG or NT-T cells, indicating that Engager-modified T cells do not proliferate autonomously. To validate this for EphA2-ENG T cells, EphA2-ENG T cells or NT T cells were labeled with carboxyfluorescein diacetate succinimidyl ester and cultured with or without irradiated K562 cells for 7 days. Within 7 days, only 25% of EphA2-ENG T cells were alive and of the viable cells only 23% had proliferated in the presence of K562 cells, indicating that EphA2-ENG T cells also do not significantly proliferate in the absence of antigen (Supplementary Figure S4). In standard cytotoxicity assays, EphA2-ENG T cells but not CD19-ENG or NT T cells killed EphA2-positive target cells (P < 0.001), confirming that the recognition and killing of tumor cells depends on the expression of EphA2 on tumor cells and on EphA2-Engager production by T cells (Figure 2c).
Figure 2.

EphA2-ENG T cells secrete cytokines, proliferate and kill target cells in an antigen-specific manner. (a) EphA2-ENG, CD19-ENG, and NT T cells were cocultured with EphA2-positive (U373, A549, K562-EphA2) or -negative (K562) tumor cells at a ratio of 10:1. After 24 hours, interferon (IFN)-γ and interleukin (IL)-2 production was determined by enzyme-linked immunosorbent assay (n = 4). (b) EphA2-ENG, CD19-ENG, and NT T cells were cocultured with EphA2-positive (U373, A549) tumor cells at a ratio of 10:1. After 7 days, number of viable T cells were enumerated by trypan blue staining (n = 4). (c) Cytotoxicity assays were performed using EphA2-ENG, CD19-ENG, and NT T cells as effectors and EphA2-positive (U373, A549, K562-EphA2) and -negative (K562) tumor cells as targets at a E:T ratio of 10:1 (mean ± SD; n = 4).
EphA2-ENG T cells redirect bystander T cells to EphA2-positive tumor cells
To demonstrate that EphA2-ENG T cells redirect bystander T cells to EphA2-positive target cells, we performed 2 sets of experiments. Media from EphA2-ENG or NT T cells was mixed with EphA2-positive tumor cells (U373) and NT T cells. After 24 hours, IFN-γ was measured in the cell culture supernatants. IFN-γ production in cocultures of NT T and U373 tumor cells was only increased (P < 0.001) when these NT T cells were redirected to tumor by media from EphA2-ENG T cells (Figure 3a). To assess long-term EphA2-ENG expression, transduced and NT T cells were expanded with IL-2 for 33 days post-transduction. Cells were replated in fresh media days 5, 13, and 33 post-transduction, and after 48 hours, culture supernatants were mixed with NT T and U373 tumor cells as described above. At all three time points tested, NT T cells were redirected to tumor cells by media only from EphA2-ENG T cells as judged by IFN-γ secretion (Supplementary Figure S5). We then used transwell assays containing inserts that do not allow T-cell migration (Figure 3b). When NT T cells and tumor cells were present in the bottom well, tumor cell killing was only seen when EphA2-ENG T cells were present in the insert well demonstrating the ability of a diffusible product from EphA2-ENG T cells to redirect NT T cells to EphA2-positive tumor cells (Figure 3c). No cell killing was observed if CD19-ENG T cells were plated in the insert well, indicating that killing is antigen specific.
Figure 3.

EphA2-ENG T cells redirect bystander T cells to tumor cells. (a) A549 cells were cocultured with or without NT T cells and media of NT T cells or EphA2-ENG T cells. After 24 hours, IFN-γ production was determined by enzyme-linked immunosorbent assay (mean ± SD; n = 4). (b) Scheme of coculture transwell assays. (c) 1 × 106 NT T cells and 2.5 × 105 U373 cells were plated in the bottom well, and EphA2-ENG T cells in the insert well. The number of plated EphA2-ENG T cells ranged from 103 to 106. CD19-ENG T cells in the insert well and bottom wells without NT T cells served as controls. After 48 hours, live U373 cells were visualized by crystal violet staining. The experiment was performed three times and results of one representative experiment are shown.
To demonstrate that T-cell activation results in increased secretion of engager molecules, EphA2-ENG T cells were cultured on EphA2 or HER2 recombinant protein-coated plates. Phosphate-buffered saline (PBS)-treated plates served as controls. EphA2-ENG T cells were activated by EphA2 but not HER2 protein as judged by interferon (IFN)-γ secretion (P < 0.05; Figure 4a). Following exposure to EphA2, EphA2-ENG T cells expressed higher levels (P < 0.05) of engager molecule mRNA as judged by qRT-PCR (P < 0.05; Figure 4b). The effects of activation on the ability of EphA2-ENG T cells to redirect bystander T cells to eGFP-fire fly luciferase-expressing U373 (U373.eGFP.ffLuc cells) were also evaluated in transwell coculture assays. ffLuc-expressing EphA2-negative BV173 (BV173.ffLuc) cells served as controls. Antigen activation with recombinant EphA2 protein of EphA2-ENG T cells induced a ~10-fold increase in their ability to redirect bystander T cells to U373 cells (P < 0.00001). In summary, our in vitro studies indicate that ENG molecules are secreted and bind to transduced, and bystander T cells. The monovalent binding does not induce T-cell activation unless target antigen binding occurs. Once activated, T cells increase engager molecules mRNA production (Figure 5).
Figure 4.

Antigen-specific activation of EphA2-ENG T cells enhances their ability to redirect bystander T cells to tumor cells. (a,b) NT T cells and EphA2-ENG T cells were cultured on EphA2 (activated) or HER2 (nonactivated) protein-coated plates. Phosphate-buffered saline–treated plates served as controls. (a) IFN-γ production was determined by enzyme-linked immunosorbent assay after 24 hours (mean + SD; n = 3). (b) After 72 hours, expression of EphA2-ENG mRNA was determined by qRT-PCR (mean + SD; n = 3). (c) U373.eGFP.ffLuc or BV173.ffLuc cells were cocultured with NT T cells and increasing numbers of transwell-separated activated or non-activated EphA2-ENG T cells. After 48 hours, live tumor cells were determined by luciferase assay (n = 3; duplicate assay for each donor; activated versus nonactivated T cells: for U373: P < 0.00001; for BV173: P = 0.12).
Figure 5.

Model of ENG T-cell mode of action. (a) ENG T cells (light green) express and secrete engager molecules that bind to ENG and bystander (light blue) T cells. T cells are not activated since the engager molecule is monovalent. (b) Once engager bind antigen, ENG and bystander T cells are activated (dark green and dark blue respectively). Once activated, ENG T cells increase engager molecule mRNA production.
EphA2-engagers induce the expansion of transduced and bystander T cells in vivo
We used the human A549 lung cancer SCID xenograft model20 to demonstrate that EphA2-ENG T cells expand in vivo. Tumor-bearing (n = 5) or control (n = 5) mice were injected intravenously (i.v.) with an admixture of 5 × 106 eGFP.ffluc-expressing EphA2-ENG T cells (Supplementary Figure S6) and 5 × 106 unmodified T cells, and received one intraperitoneal (i.p.) dose of IL2. While EphA2-ENG T cells expanded in tumor-bearing mice, no expansion was observed in the absence of tumors (Figure 6a). To demonstrate that EphA2-ENG T cells induce the expansion of bystander T cells in vivo, we injected tumor-bearing mice i.v. with an admixture of 5 × 106 EphA2-ENG T cells and 5 × 106 eGFP.ffLuc expressing T cells (n = 5) or 5 × 106 CD19-ENG T cells and 5 × 106 eGFP.ffLuc expressing T cells (n = 5). eGFP.ffLuc-expressing T cells only expanded when coinjected with EphA2-ENG T cells as judged by bioluminescence imaging (Figure 6b). These results indicate that EphA2-engagers induced the expansion of transduced and bystander T cells in an antigen-dependent manner in vivo.
Figure 6.

Antigen-dependent expansion of EphA2-ENG T cells and bystander T cells in vivo. (a) A549 tumor- or nontumor-bearing mice received an intravenous (i.v.) injection of an admixture of 5 × 106 eGFP.ffLuc-expressing EphA2-ENG and 5 × 106 NT T cells, and one intraperitoneal (i.p.) dose of IL-2 (1,500 units). Serial bioluminescence imaging was performed to track T cells (means ± SD are shown; n = 5 per group; *P < 0.05, **P < 0.01, ***P < 0.005). (b) A549 tumor-bearing mice received an i.v. injection of an admixture of 5 × 106 EphA2-ENG and eGFP.ffLuc-expressing 5 × 106 NT cells or 5 × 106 CD19-ENG and eGFP.ffLuc-expressing 5 × 106 NT cells, and one i.p. dose of IL-2 (1,500 units). Serial bioluminescence imaging was performed to track T cells (means ± SD are shown; n = 5 per group; *P < 0.05, ***P < 0.005).
EphA2-ENG T cells have potent antitumor activity in vivo
We evaluated the antitumor activity of EphA2-ENG T cells in human glioma and lung cancer SCID xenograft models.18,20 Seven days after intracranial injection of 1 × 105 U373.eGFP.ffLuc cells, mice were stereotactically injected at the tumor site with 2 × 106 EphA2-ENG T cells (n = 8), or CD19-ENG T cells (n = 5). Untreated animals served as controls (n = 5). Serial bioluminescence imaging was used to track tumor growth (Figure 7a,b). Mice treated with EphA2-ENG T cells had a 2 log or greater reduction in their tumor signal with an increase in weight over time (Supplementary Figure S7a), resulting in a long-term tumor free survival of five out of eight mice (P < 0.0005) (Figure 7c).
Figure 7.

EphA2-ENG T cells have potent antitumor activity in vivo. (a–c) Antitumor activity of EphA2-ENG T cells in U373 glioma SCID xenograft model. Seven days after intracranial injection of 1 × 105 U373.eGFP.ffLuc cells, 2 × 106 EphA2-ENG (n = 8) or CD19-ENG (n = 5) T cells were injected intracranially in the same site. Untreated animals served as controls (n = 5). Tumor growth was followed by bioluminescence imaging. (a) Images of representative animals; (b) Quantitative bioluminescence imaging results for each mice (radiance = photons/sec/cm2/sr) over time. There was no tumor signal outside the brain for all groups of mice during the entire length of the experiment. The region of interest (ROI) for bioluminescence quantification was therefore restricted to the head of mice; (c) Kaplan–Meier survival curve. (d–f) Antitumor activity of EphA2-ENG T cells in A549 lung tumor SCID xenograft model. Seven, 14, and 21 days after intravenous (i.v.) injection of 2.5 × 106 A549.eGFP.ffLuc cells mice received an i.v. dose of 1 × 107 EphA2-ENG (n = 5) or CD19-ENG T cells (n = 4) and an intraperitoneal (i.p.) dose of IL-2 (1,500 units). Untreated animals served as controls (n = 5). Tumor growth was followed by bioluminescence imaging. (d) Images of representative animals; (e) Quantitative, systemic bioluminescence imaging results for each mice; (f) Kaplan–Meier survival curve.
Finally, we evaluated the antitumor efficacy of EphA2-ENG T cells in the A549.eGFP.ffLuc metastatic lung cancer model. 2.5 × 106 A549.eGFP.ffLuc cells were injected i.v. on day 0, and on day 7, 14, 21 mice received 1 × 107 EphA2-ENG T cells (n = 5) or CD19-ENG T cells (n = 4) i.v. with one i.p. dose of IL2. Untreated animals served as controls (n = 5). Mice treated with EphA2-ENG T cells had a significant reduction (P < 0.005) in their tumor signal as early as 5 days post the first T-cell dose (Figure 7d,e), while in the other groups of mice tumors rapidly progressed with accompanying weight loss (Supplementary Figure S7b). This resulted in a survival advantage of EphA2-ENG T-cell treated mice in comparison to untreated mice and mice treated with CD19-ENDGT cells (P < 0.005) (Figure 7f).
Discussion
Here we describe the development and characterization of a new class of T cells that secrete bispecific T-cell engagers, and show that T cells secreting EphA2-engagers effectively target EphA2-positive cells. These EphA2-ENG T cells produce immunostimulatory cytokines and proliferate in response to tumor antigen by virtue of CD3 binding of the secreted bi-specific engager molecule, induce tumor cytolysis when cocultured with EphA2-positive targets, redirect bystander T cells to EphA2-positive tumor cells, and have potent antitumor activity in vivo.
Genetic modification of T cells with CARs or T cell receptors (TCRs) is an attractive strategy to rapidly generate antigen-specific T cells.1,21 While neither CAR nor TCR T cells are able to redirect bystander T cells to cancer cells directly, T-cell mediated tumor destruction can result in the activation of tumor antigen-specific T cells through antigen spreading,22,23 Several groups of investigators have developed bispecific antibodies, including BiTEs, DARTs, and diabodies, to redirect resident T cells to tumor cells.12,13 Among these, the CD19-specific BiTE, blinatumomab, has shown encouraging results in Phase I and II clinical studies for patients with hematological malignancies.24,25 However, BiTEs have to be given as a continuous infusion, which can be associated with systemic toxicities.25 In addition, like regular MAbs, BiTEs lack active biodistribution or self amplify once infused. In addition, they do not penetrate tissue planes, which might explain the so far limited activity of BiTEs in humans with solid tumors. While in our systemic model ENG T cells trafficked to tumor sites (Supplementary Figure S8), the homing of T cells to tumor sites depends in part on the pattern of chemokine expression by tumors. Several studies have shown that trafficking of T cells to tumor cells can be increased by transgenic expression of chemokine receptors.26,27,28
T cells secreting engager molecules can overcome many limitations of bispecific MAbs since they are able to persist and expand post infusion, actively traffic to tumor sites,29,30 and increase transgene expression upon activation. Indeed, EphA2-ENG T cells expanded in vivo, obviating the need for continuous infusion of engager molecules. Once activated, EphA2-ENG T cells increased the production of the engager molecules resulting in an enhanced ability to redirect bystander T cells to tumor cells. These favorable characteristic of T cells should result in high concentrations of engager molecules at tumor sites while minimizing systemic exposure, which can be toxic. Indeed, we were not able to detect human cytokines in the peripheral blood of A549 tumor bearing mice 4 and 10 days post-EphA2-ENG T-cell injection (Supplementary Figure S9). In addition, we were unable to detect sufficient levels of engager molecules in the peripheral blood of mice using a bioassay in which human T cells are mixed with EphA2-positive tumor cells and serum of mice (Supplementary Figure S10). While these results are encouraging in regards to the potential safety of ENG T cells, recent clinical studies have highlighted the limited value of murine models to predict the safety profile of cell therapies or MAbs.31,32 However, the inability to accurately evaluate the safety of biotherapeutics in preclinical models should not, and has not, prevented their safe and successful introduction into clinical practice.33
In vivo, EphA2-ENG T cells had potent antitumor activity in two animal models. While in the local glioma model, five of eight mice remained tumor free, in the systemic model, tumors eventually recurred. Tumor recurrence is most likely explained by limited EphA2-ENG T-cell expansion and persistence in vivo. We have previously observed tumor progression in this aggressive xenograft model with EphA2-CAR T cells without evidence of selecting EphA2-negative A549 escape variants.19 While the murine environment is not conducive for long-term persistence of human T cells, the secreted EphA2-specific engager molecules similar to bispecific MAbs do not provide costimulation. Analogous to first-generation CAR T cells, providing costimulation34,35,36 or additional genetic modifications of ENG T cells such as the transgenic expression of IL12 or IL15,37,38,39,40 has the potential to enhance T-cell expansion and persistence in vivo. In addition, providing costimulation and/or cytokines should prevent the activation of inhibitory T cells and reverse the immunosuppressive tumor environment. While our results highlight some of the potential advantages of ENG T cells in comparison to the administration of recombinant engager protein, we did not perform a formal comparison due to the used xenograft models, which would have required the coinfusion of human peripheral blood mononuclear cells (PBMCs) and recombinant protein.
In conclusion, ENG T cells present a new class of antigen-specific T cells with the unique ability to redirect bystander T cells to tumor cells in an antigen-dependent manner. ENG T cells induced the regression of established tumors in locoregional and systemic xenografts models, and thus have the potential to improve current immunotherapy for cancer.
Materials and Methods
Tumor cell lines. The lung cancer cell line A549, leukemia cell line K562, and glioblastoma line U373 were purchased from the American Type Culture Collection (ATCC, Manassas, VA). The leukemia cell line BV173 was purchased from the Leibniz Institute DSMZ—German Collection of Microoganisms and Cell Cultures (Braunschweig, Germany). The generation of K562 cells expressing human EphA2 (K562-EphA2) and ffLuc-expressing U373, A549 and BV173 cells were described previously.18,20,41
Construction of retroviral vectors encoding EphA2-specific and CD19-specific ENGs. The construction of the EphA2-specific engager containing the immunoglobulin heavy-chain leader peptide, the EphA2-specific scFv 4H5,42 a short serine-glycine linker and a CD3-specific scFV derived from OKT3 (ref. 43) is described elsewhere.44 Specifically, the EphA2-specific engager consists of the immunoglobulin heavy-chain leader peptide, the 4H5 heavy-chain, a glycine (G) serine (S) linker [(G4S)3], the 4H5 light-chain,42 a short G4S linker, the OKT3 heavy-chain, a (G4S)3 linker, and the OKT3 light-chain. EphA2-specific Engager was subcloned into pSFG-IRES-mO (provided by Dr Vera, Baylor College of Medicine). We used mO as a marker gene for FACS analysis since we used an eGFP.ffLuc construct. The CD19-specific engager, containing the immunoglobulin heavy-chain leader peptide, the CD19-specific scFv (FMC63),45 a short serine-glycine linker, and a CD3-specific scFV derived from OKT3 was synthesized by Invitrogen (Carlsbad, CA) and subcloned into pSFG-IRES-mOrange. As previously described, a retroviral vector encoding eGFP.ffLuc was used to transduce T cells for detection by bioluminescence imaging.19 RD114-pseudotyped retroviral particles were generated as previously described.18
Generation of ENG T cells. ENG- or CAR-expressing T cells were generated as previously described.18 Prior to blood collection, informed consent was obtained from healthy donors in accordance to protocols approved by the Institutional Review Board of Baylor College of Medicine. PBMCs were stimulated as previously described.18 Nontissue culture–treated 24-well plates were coated overnight with 1 µg/ml OKT3 (purified from hybridoma CRL-8001, ATCC) and 1 µg/ml CD28 (Becton Dickinson, Mountain View, CA) antibodies overnight. The next day, the antibody solution was removed, and the wells were washed once with PBS before plating PBMCs in Roswell Park Memorial Institute (Thermo Scientific HyClone, Waltham, MA) with 10% fetal calf serum (HyClone, Logan, UT) and 2 mmol/l GlutaMAX-I (Invitrogen). 100 U/ml human IL2 (Biological Resources Branch, National Cancer Institute, Frederick, MD) was added to cultures on day 2, and on day 3, T cells were transduced with retroviral particles on RetroNectin (Clontech, Mountainview, CA) coated plates in the presence 100 U/ml IL-2. T cells were subsequently expanded with IL-2. NT T cells were activated with OKT3/CD28 and expanded in parallel with 100 U/ml IL-2.
Flow cytometry. The expression of mOrange was detected by FACS analysis. For immunophenotyping, cells were stained with CD3-PerCP, CD4-FITC, and CD8-FITC monoclonal antibodies (BD Biosciences, San Jose, CA). Isotype controls were immunoglobulin G1-fluorescein isothiocyanate and IgG1-peridinin chlorophyll protein (both BD Biosciences). For each sample, 20,000 cells were analyzed by a FACSCalibur instrument (BD Biosciences) using Cell Quest Software (BD Biosciences).
Ex vivo functional analysis of T cells. EphA2-ENG, CD19-ENG, and NT T cells were plated at 10:1 ratio with tumor cells. IFN-γ and IL-2 production after 24 hours of coculture was measured using ELISA as per the manufacturer's instructions (R&D Systems, Minneapolis, MN). Standard chromium (51Cr) release assays with 6 hours incubation for adherent cells (A549 and U373) and 4 hours for nonadherent cells (K562 and BV173) were performed as previously described.46
Transwell assay. U373, U373.eGFP.ffLuc, or BV173.ffLuc cells were plated on bottom wells of 24-well plate. After 24 hours, NT T cells were added to bottom wells and EphA2-ENG or CD19-ENG T cells were added to transwell insert wells (6.5 mm in diameter, 0.4 µm pore, polycarbonate, Corning, Corning, NY). After 48 hours, viable tumor cells were detected by crystal violet staining for U373 or by luciferase assay for U373.eGFP.ffLuc or BV173.ffLuc cells.
MTS assay. U373 cells were plated in 96-well plates at a density of 1 × 104 cells per well. After 24 hours, T cells were added to plates. After 48 hours coculture, nonadherent cells were removed and viable cells were detected by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (CellTiter 96 aqueous one solution cell proliferation assay; Promega, Madison, WI).
Quantitative real-time PCR. RNA was extracted from T cells using the RNeasy Mini Kit (Qiagen, Valencia, CA). Relative quantification of EphA2-ENG mRNA expression was done using SYBR Green Reagents (Qiagen).
Animal models. All animal experiments followed a protocol approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Experiments were performed as described previously with minor modifications.18,19
Intracranial model. Male 8- to 12-week-old ICR-SCID mice were purchased from Taconic (IcrTac:ICR-Prkdcscid; Fox Chase C.B-17 SCID ICR; Taconic, Hudson, NY). Briefly, U373.eGFP.ffLuc cells (1 × 105 in 2.0 µl) were injected 3 mm deep to the bregma, corresponding to the center of the right caudate nucleus over 5 minutes. Seven days after tumor cell injection, animals were treated with 2 × 106 NT or ENG T cells from the same donor in 2 µl to the same tumor coordinates. No IL-2 was given to animals based on our past experience with T-cell therapy in this model.18 Photons emitted from the luciferase-expressing tumor cells were quantified using Living Image software (PerkinElmer, Waltham, MA). A constant region-of-interest was drawn over the tumor region and the intensity of the signal measured as total photon/second/cm2/steradian (p/s/cm2/sr). Animals were initially imaged every 2 days, and once a week thereafter. Mice were euthanized when the tumor radiance was >1 × 109 on two occasions or when they met euthanasia criteria (neurological deficits, weight loss, signs of distress) in accordance with the Center for Comparative Medicine at Baylor College of Medicine.
Systemic A549 tumor model (antitumor activity). Male 8- to 12-week-old SCID Beige mice were purchased from Charles River (CB17.Cg-PrkdcscidLystbg/Crl; Fox Chase SCIDR Beige mouse; Charles River Laboratories International, Wilmington, MA). 2.5 × 106 A549.eGFP.ffLuc cells in PBS were injected i.v. on day 0. Seven, 14, and 21 days after tumor cell injection, mice were treated i.v. with 1 × 107 CD19-ENG T cells or EphA2-ENG T cells. All animals received one i.p. dose of IL-2 (1,500 U) on the day of the T-cell injections as previously reported in this model,19 Untreated animals served as controls. Animals were imaged as described above. Mice were euthanized when they met euthanasia criteria in accordance with the Center for Comparative Medicine at Baylor College of Medicine.
Systemic A549 tumor model (T-cell expansion and persistence). To determine the expansion and persistence of EphA2-ENG T cells, male 8–12-week-old SCID Beige mice were injected i.v. with 2.5 × 106 A549 cells in PBS on day 0. On day 7, post-tumor challenge, five tumor-bearing mice and five controls were injected i.v. with an admixture of 5 × 106 eGFP.ffLuc-expressing EphA2 ENG T cells and 5 × 106 NT T cells. All mice received one i.p. dose of IL-2 (1,500 U), and expansion and persistence of T cells was monitored by serial bioluminescence imaging. To determine the expansion and persistence of bystander T cells, male 8–12-week-old SCID Beige mice were injected i.v. with 2.5 × 106 A549 cells on day 0. On day 7 post-tumor challenge, tumor-bearing mice were injected i.v. with an admixture of 5 × 106 EphA2 ENG T cells and 5 × 106 eGFP.ffLuc-expressing T cells or an admixture of 5 × 106 CD19 ENG T cells and 5 × 106 eGFP.ffLuc-expressing T cells (five mice per group). All mice received one i.p. dose of IL2 (1,500 U), and expansion and persistence of T cells was monitored by serial bioluminescence imaging.
Statistical analysis. GraphPad Prism 5 software (GraphPad software, San Diego, CA) was used for statistical analysis. Measurement data were presented as mean ± standard deviation. For comparison between two groups, two-tailed t-test was used. For comparisons of three or more groups, the values were analyzed by one-way analysis of variance with Bonferroni's post-test. Linear regression analysis was performed to compare the antitumor activity of ENG and CAR T cells, and activated and nonactivated ENG T cells. The significance level used was p < 0.05. For the mouse experiments, five mice were planned to detect a large effect size of 2, which provided at least 80% power with 5% type-I error. Although no formal randomization was carried out, a cage of five mice was chosen randomly. Survival, determined from the time of tumor cell injection, was analyzed by the Kaplan–Meier method and by the log-rank test.
SUPPLEMENTARY MATERIAL Figure S1. Transduction of CD4- and CD8-positive T cells. Figure S2. Generation of T cells expressing EphA2-ENG with a 6xHis-Myc tag. Figure S3. Generation of CD19-ENG T cells. Figure S4. Minimal proliferation of EphA2-ENG T cells in the absence of antigen. Figure S5. Functional stability of EphA2-ENG T cells after transduction. Figure S6. FACS analysis of GFP.ffLuc and Engager expressing T cells. Figure S7. Weight change of mice treated with ENG T cells. Figure S8. T cells localize to lung tumors post injection. Figure S9. Peripheral blood cytokine levels in xenograft model. Figure S10. Bioassay of media or serum.
Acknowledgments
We thank Cliona Rooney, Malcolm Brenner, and Hao Liu for helpful discussions and advice. This work was supported by NIH grant P50 CA126752 and 1R01CA173750-01, Leukemia and Lymphoma Society SCOR in Lymphoma, Alex Lemonade Stand Foundation, St. Baldrick's Foundation, and the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine and the Adoptive T-cell Therapy for Lung Cancer Program. The Center for Cell and Gene Therapy has a research collaboration with Celgene and Bluebird Bio. KI, SK, MPV, FY, ZY, and SG have patent applications in the field of T-cell and gene-modified T-cell therapy for cancer.
Supplementary Material
References
- Morgan RA, Dudley ME, Rosenberg SA. Adoptive cell therapy: genetic modification to redirect effector cell specificity. Cancer J. 2010;16:336–341. doi: 10.1097/PPO.0b013e3181eb3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June C, Rosenberg SA, Sadelain M, Weber JS. T-cell therapy at the threshold. Nat Biotechnol. 2012;30:611–614. doi: 10.1038/nbt.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–145. doi: 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmüller G. Symmetry breaking: bispecific antibodies, the beginnings, and 50 years on. Cancer Immun. 2012;12:12. [PMC free article] [PubMed] [Google Scholar]
- Weidle UH, Tiefenthaler G, Weiss EH, Georges G, Brinkmann U. The intriguing options of multispecific antibody formats for treatment of cancer. Cancer Genomics Proteomics. 2013;10:1–18. [PubMed] [Google Scholar]
- Frankel SR, Baeuerle PA. Targeting T cells to tumor cells using bispecific antibodies. Curr Opin Chem Biol. 2013;17:385–392. doi: 10.1016/j.cbpa.2013.03.029. [DOI] [PubMed] [Google Scholar]
- Fournier P, Schirrmacher V. Bispecific antibodies and trispecific immunocytokines for targeting the immune system against cancer: preparing for the future. BioDrugs. 2013;27:35–53. doi: 10.1007/s40259-012-0008-z. [DOI] [PubMed] [Google Scholar]
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69:4941–4944. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- Biao-xue R, Xi-guang C, Shuan-ying Y, Wei L, Zong-juan M. EphA2-dependent molecular targeting therapy for malignant tumors. Curr Cancer Drug Targets. 2011;11:1082–1097. doi: 10.2174/156800911798073050. [DOI] [PubMed] [Google Scholar]
- Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6:1795–1806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21:629–637. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21:1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21:1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber A, Zhang T, Sentman CL. Immunotherapy with chimeric NKG2D receptors leads to long-term tumor-free survival and development of host antitumor immunity in murine ovarian cancer. J Immunol. 2008;180:72–78. doi: 10.4049/jimmunol.180.1.72. [DOI] [PubMed] [Google Scholar]
- Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther. 2012;136:334–342. doi: 10.1016/j.pharmthera.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226–6233. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 2002;13:1971–1980. doi: 10.1089/10430340260355374. [DOI] [PubMed] [Google Scholar]
- Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–6402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–122. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- Kolb HJ. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood. 2008;112:4371–4383. doi: 10.1182/blood-2008-03-077974. [DOI] [PubMed] [Google Scholar]
- Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20:1819–1828. doi: 10.1038/sj.leu.2404366. [DOI] [PubMed] [Google Scholar]
- Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19:751–759. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar SP, Leonardi AJ, van Panhuys N, Zhang L, Yu Z, Crompton JG, et al. Collapse of the tumor stroma is triggered by IL-12 induction of Fas. Mol Ther. 2013;21:1369–1377. doi: 10.1038/mt.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna SK, De Angelis B, Pagliara D, Hasan ST, Zhang L, Mahendravada A, et al. Interleukin 15 provides relief to CTLs from regulatory T cell-mediated inhibition: implications for adoptive T cell-based therapies for lymphoma. Clin Cancer Res. 2013;19:106–117. doi: 10.1158/1078-0432.CCR-12-2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber C, Abhyankar H, Heslop H, Dotti G, Savoldo B. ab-T cell receptor-based gene therapy targeting the common tumor-antigen survivin for therapy of hematological malignancies. Mol Ther. 2013;21:S117. [Google Scholar]
- Damschroder MM, Widjaja L, Gill PS, Krasnoperov V, Jiang W, Dall'Acqua WF, et al. Framework shuffling of antibodies to reduce immunogenicity and manipulate functional and biophysical properties. Mol Immunol. 2007;44:3049–3060. doi: 10.1016/j.molimm.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Kufer P, Lutterbuse R, Kohlheisen B, Zeman S, Baeuerle P. Single chain antibody construct comprising binding domains specific for human CD3 and human CD19; anticarcinogenic biodrug for treating non-Hodgkin lymphoma. Patent US20070123479. 2007.
- Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther. 2014;22:102–111. doi: 10.1038/mt.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson IC, Lenton KA, Little DJ, Decorso T, Lee FT, Scott AM, et al. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol Immunol. 1997;34:1157–1165. doi: 10.1016/s0161-5890(97)00144-2. [DOI] [PubMed] [Google Scholar]
- Gottschalk S, Edwards OL, Sili U, Huls MH, Goltsova T, Davis AR, et al. Generating CTLs against the subdominant Epstein-Barr virus LMP1 antigen for the adoptive immunotherapy of EBV-associated malignancies. Blood. 2003;101:1905–1912. doi: 10.1182/blood-2002-05-1514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.