

Abstract
We conducted a clinical trial to assess the feasibility and efficacy of CD33-directed chimeric antigen receptor-modified T cells (CART-33) for the treatment of refractory acute myeloid leukemia (AML). A 41-year-old male patient with AML was enrolled and received a total of 1.12 × 109 autologous CART-33 cells, of which ~38% were transduced with CAR. The CART-33 infusion alone induced rigorous chills and fevers; drastic fluctuations of his preexisting pancytopenia; elevated serum cytokine levels, including interleukin (IL)-6, IL-8, tumor necrosis factor-α, and interferon-γ; slight transient hyperbilirubinemia within 2 weeks; a subsequent intermittent moderate fever; and reversed fluctuation of the pancytopenia. A marked decrease of blasts in the bone marrow was observed on examination 2 weeks after therapy, and there was a gradual increase until florid disease progression occurred at 9 weeks after the cell infusion. These observations warrant further research on CART-33 treatment in refractory AML and may spur efforts to extend the CART-33-induced tumor burden to the preparation of other intensive strategies, such as hematopoietic stem cell transplantation. This study is registered at www.ClinicalTrials.gov as NCT01864902.
Introduction
The treatment of relapsed and refractory acute myeloid leukemia (AML) remains challenging despite great improvements in intensive chemotherapy and hematopoietic stem cell transplantation.1,2 The development of tumor-associated antigen-directed cytotoxic agents or immunotherapies have increased the expectations for disease control in this patient population.3 CD33 is primarily expressed on multipotent myeloid precursors, unipotent colony-forming cells, maturing granulocytes and monocytes, peripheral granulocytes, and resident macrophages.4,5,6 Gemtuzumab ozogamicin (GO) is a recombinant humanized monoclonal antibody conjugated to the DNA-damaging toxin calicheamicin directed against the CD33 antigen, which is expressed on the leukemic cells of more than 90% of patients with AML.7,8 The data from some clinical trials on the efficacy of GO support the conclusion that CD33 is a valid target for some subtypes of AML, mainly in favorable and intermediate risk groups.9,10,11 Although clinical trials could demonstrate some benefit of combining GO with chemotherapy, the drug was withdrawn mainly because its benefits did not outweigh the adverse effects of the drug.
The experience with GO reflects the intrinsic heterogeneity of CD33 in AML. The diversity of individual leukemia types that have different cellular origins is of particular significance for therapeutics that aim to cure AML and indicates that no approach is generally effective for all of the subtypes of leukemia. Recent clinical trials have demonstrated that tumor-specific chimeric antigen receptor-modified T cell (CART)-based adoptive cell transfer may provide a curative approach for tumor therapy,12 particularly for B cell-lineage malignancies by targeting CD19.13,14,15 After CD33-specific CART cells (CART-33) were shown to possess potent antileukemic activities in vitro and in vivo in a mouse model,16,17,18 CART-33 was extrapolated to be promising for the treatment of AML patients. Because of the grade 3/4 toxicities frequently observed in patients treated with GO,10,19,20 efforts at further clinical trials were inevitably stopped because of frightening safety concerns that are likely caused by irreversible on-target off-tumor adverse effects such as myelosuppression and severe hepatotoxicity triggered by the in vivo persistence of CART-33 cells.
To test the safety and efficacy of CART-33 cells, we designed a clinical trial for patients with relapsed and refractory AML. One patient with long-term pancytopenia who was not considered for other types of cytotoxic chemotherapy was selected for the CART-33 trial, and the results are reported in this manuscript.
Results
Phenotype, antitumor activities, and in vivo expansion of CART-33 cells
CART-33 cells were generated from the mononuclear cells of 90 ml of the patient's peripheral blood (PB). After 13 days of culture according to the cytokine-induced killer (CIK) cell culture system as reported previously,21 the total cells reached a 19-fold expansion and were released for the infusions (Figure 1a). Of the infused cells, 95.64% were CD3+ cells principally composed of the CD8+ subset (83%), and 16.44% were characterized with the central memory phenotype (CD45RO+/CD62L+/CCR7+; Figure 1b). Through the synchronous transfection verification of CAR.33-4-1BBζ-GFP, 38% of the CART-33 cells were expected to express CAR (Figure 1c). In addition, 14.76% of the infused cells were CD33 positive (Figure 1d).
Figure 1.
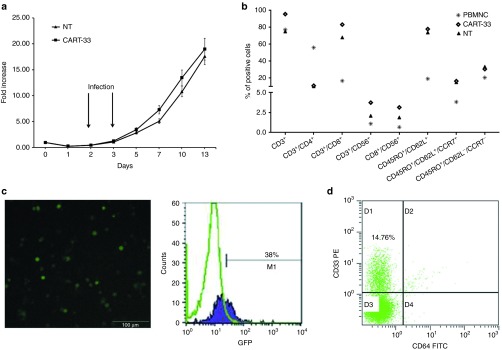
Expansion, transfection efficiency, and phenotypic analysis of CART-33 cells. (a) Expansion (-fold) of the control NT (no transfection T cells) and CART-33 cells generated from the patient. The cells were cultured for ~13 days. (b) Comparison of the immunophenotypic analyses of the PBMNC, NT, and CART-33 cells. (c) The verified transfection efficiency of CART-33 cells by GFP. Left panel: optical microscope photographs showing the CART-GFP cells of the patient after culture for 12 days. Right panel: expression of CAR-GFP in CART-GFP cells as assessed by FACS analysis. (d) CD33 expression on CART-33 cells as determined by FACS analysis. CART, chimeric antigen receptor-modified T cells; FACS, fluorescent-activated cell sorting; GFP, green fluorescence protein; PBMNC, peripheral blood mononuclear cell.
With the exception of this patient, the immunophenotypes of CART-33 cells generated from two other AML patients and 10 healthy donors were similarly observed and characterized (Supplementary Figure S1).
The CART-33 cells exhibited an approximately identical cytotoxic activity relative to nontransduced CIK (NT) cells against CD33− K562 cells (Figure 2a). By contrast, the prominent cytolytic activities of CART-33 cells were observed in CD33+ HL60 (Figure 2b) and primary AML blast cells with CD33 expression (Figure 2c), indicating the specifically ex vivo targeting cytotoxicity of CART-33 cells on CD33+ cells, which was comparable to previously described results.16,17,18
Figure 2.
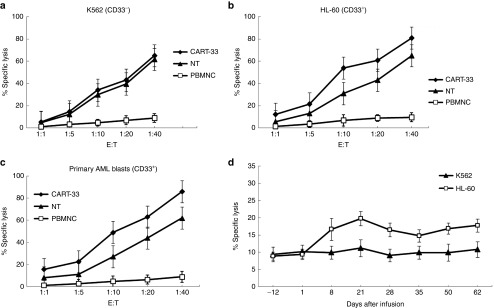
Cytotoxic activity of CART-33 and PBNMC from the patient. Cytotoxic activity of the PBMNC, NT (no transfection T cells) and CART-33 cells obtained from the patient using the following target cells: (a) K562 cell line (human chronic myelogenous leukemia cell lines, CD33−), (b) HL-60 cell line (human promyelocytic leukemia cells, CD33+), and (c) autologous blasts (primary acute myeloid leukemia cells from the patient with CD33+). The results are shown at effector:target (E:T) ratios of 1:1, 5:1, 10:1, 20:1, and 40:1. (d) Cytotoxic activity of the following effector cells obtained from the patient: NT and CART-33 were cultured for 12 days, the PBMNCs obtained from the PB of the patient before and after the CART-33 cell infusion, the target cells were K562 and HL-60, and the results are shown at an E:T ratios of 10:1. The cytotoxic activity was evaluated through a 24-hour carboxyfluorescein succinimidyl ester staining assay. All of the data are represented as the means of triplicate values, and the error bars represent the SEMs. CART, chimeric antigen receptor-modified T cells; PBMNC, peripheral blood mononuclear cell.
Without any conditioning chemotherapy, this patient was administered a total of 1.12 × 109 CART-33 cells (1.07 × 109 of CD3+ cells; 4.25 × 108 of CAR+ cells) in escalating doses over a period of 4 consecutive days (1 × 108 on day 1, 1.2 × 108 on day 2, 4 × 108 on day 3, and 5 × 108 on day 4, respectively). High levels of the CAR gene were reached rapidly in the PB and bone marrow after the infusions of the CART-33 cells, and the levels of CAR fluctuated between 3,501 and 207,764 copies per µg of DNA for at least 2 months in the PB (Figure 3).
Figure 3.

CART-33 copies persistent in the peripheral blood and the bone marrow as assessed by quantitative polymerase chain reaction (Q-PCR). Quantitative real-time PCR was performed on genomic DNA harvested from the patient's PBMNCs and bone marrow collected before and at the indicated serial time points after the CART-33 cell infusion. Primers specific for the transgene were used. CART, chimeric antigen receptor-modified T cells; PBMNC, peripheral blood mononuclear cell.
Toxicities
Grade 4 chills and a high fever occurred 0.5–1 hours after each daily cell infusion, with the highest temperature reaching 42 °C, and these high fevers were ameliorated overnight. The fever recurred on day 9 after the first day of cell infusion and evolved into a persistently high fever (Figure 4a). The patient's febrile syndrome was largely improved and nearly reached normal 12 hours after the administration of first-dose etanercept (tumor necrosis factor (TNF)-α inhibitor) on day 12 (Figure 4a); the elevated serum levels of proinflammatory cytokines including interleukin (IL)-6, TNF-α, INF-γ, IL-10, and IL-8 decreased markedly 2 days after the anti-TNF-α treatment (Figure 5). No infectious signs were detected despite the patient's long-term neutropenia (the white blood cell count ranged from 0.41 to 1.2 × 109 cells/l), and the patient experienced no relief from the 3-day prophylactic use of last-defense antibiotics. This patient suffered from slightly or moderately elevated serum levels of IL-6, TNF-α, and INF-γ for at least 2 months and experienced a spontaneous and intermittent febrile syndrome, which mainly manifested as temperature fluctuations between 36.5 and 38.5 °C (Figure 4b).
Figure 4.

Changes in the complete blood count and body temperature during and after the CART-33 cell infusion. (a) The left panel shows the alterations in the WBC count and body temperature, and the right panel shows the changes in the platelet count, hemoglobin level, and the patient's body temperature during the first 2 weeks of cell treatment. (b) Changes in the temperature and WBC count 2 weeks later. CART, chimeric antigen receptor-modified T cells; WBC, white blood cell.
Figure 5.
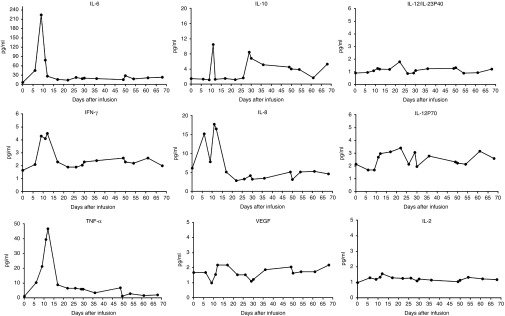
Changes in the various cytokine levels after CART-33 cell infusion. The serum levels of the indicated cytokines were serially measured starting on the first day of the CART-33 infusion to the indicated time point. CART, chimeric antigen receptor-modified T cells; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
Beginning on the second day after the CART-33 infusion, nearly concurrent exacerbation of the patient's preexisting pancytopenia was observed. The patient's pancytopenia fluctuated through an inverse relationship with the degree of his fever (Figure 4a,b). Two episodes of decreases in the complete blood count occurred after the infusions of the escalated doses of CART-33 cells. The first episode occurred just after the cell infusions on days 3–5, and the second episode occurred on days 8–12. For supportive care, the patient accepted platelet transfusions on days 3, 4, 11, and 12 and red blood cell transfusions on days 5, 6, and 11. After the early acute phase, red blood cells and platelets needed to be transfused approximately once weekly because of the patient's persistent pancytopenia. The frequency of blood transfusions appeared to be shortened compared with the frequency of transfusions before the CART-33 therapy. After 2 weeks, the number of neutrophils in his PB stably recovered to a level approaching that observed before the CART-33 infusions and fluctuated around his previous scope (the white blood cell count ranged from 0.77 to 1.35 × 109 cells/l), although neutropenia always persisted in this patient.
With the exception of a slight transient hyperbilirubinemia (with a peak value of 30 mmol/l) within 2 weeks of cell treatment and with sufficient alkalization and hydration treatments before and during the first 2 weeks of cell therapy, there were no significant alterations of other serum biochemical indexes reflecting cytolysis in vivo, such as electrolytes, lactate dehydrogenase, creatinine, and urea nitrogen, detected during the 10-week hospitalization of this patient starting from the time of the cell therapy.
Clinical response
Consistent with the flow cytometry analyses, Wright's staining of a bone marrow sample showed a marked decrease in the blast ratio from the prior >50% to <6% detected 2 weeks after the CART-33 treatment. The later detections showed a gradual increase in the blast ratio: 22% at 3 weeks, 27% at 5 weeks, and nearly 70% at 9 weeks (Supplementary Figure S2). The results from fluorescent-activated cell sorting analyses demonstrated the positive expression of CD33 on blasts at every time point, and adjacent changes to morphological observations in the blast ratios in the bone marrow were detected (Figure 6). Considering this finding in combination with the persistent high copy number of CAR molecules and the preponderant cytotoxic activities of lymphocytes isolated from patient's PB at serial time points after CART-33 infusions against CD33+ HL-60 over lymphocytes isolated before CART-33 treatment (Figure 2d), we propose that this patient developed CART-33-mediated cytotoxic activity escape of the in vivo renascent CD33+ blasts. This patient subsequently developed florid disease progression and gave up all treatments; he died 13 weeks after the CART-33 infusion at our institution. Active bone marrow hyperplasia and cells in multiple developing stages of lineages were observed in each aspiration, and stripped nucleus, cytoplasmic swelling, and membrane budding in the involved cell lineages were frequently observed after the CART-33 therapy, possibly implying that CART-33 triggered an immune attack on a subset of CD33+ cells (Supplementary Figure S2).
Figure 6.
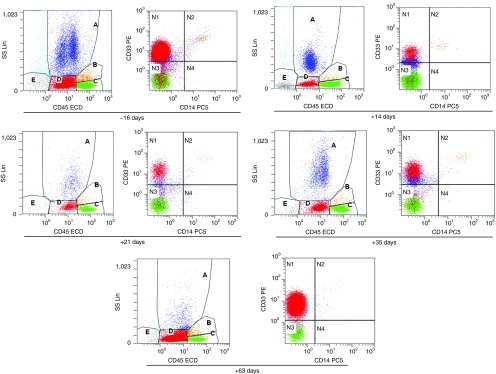
FACS analysis for bone marrow aspirates. The cells in the D gate represent the blast population count corresponding to 61.66, 9.48, 28.24, 33.83, and 84.95%, respectively, of the total nucleated cells in the bone marrow aspirates collected at the indicated time points before and after the CART-33 infusions. CART, chimeric antigen receptor-modified T cells; FACS, fluorescent-activated cell sorting.
Discussion
As targeted CD33 therapy for CD33+ AML, GO has been reported to have clear benefits by several clinical trials,9,10,11 and CART-33 cell therapy has been shown to have potent antileukemic activity in experimental studies.16,17,18 To a large extent, concerns for possible serious adverse events dampened the attempts of the clinical application of CART-33 cells in AML patients.22 In this study, we demonstrated a comparable in vitro antileukemia activity of CART-33 cells with that found in previous reports and reported their first clinical use for the treatment of one chemotherapy refractory advanced AML patient. The infusion of CART-33 alone cells led to marked disease degradation in the early stage, indicating a potent in vivo cytotoxic effect of CART-33 on CD33+ blasts. However, CD33+ leukemic cells gradually augmented until a florid progression was observed in the later stage of cell therapy. The evidence of a persistent high level of CAR molecules in vivo and the maintenance of the cytotoxic activity of lymphocytes isolated from the patient's PB on CD33+ HL-60 cells recapitulated a possible escape mechanism for CD33-directed therapy against leukemic cells with low CD33 expression. Importantly, even though only one case was completed, the lack of uncontrollable clinical toxicities observed in this patient after CART-33 infusion may be more encouraging for the application of CART-33 treatment in AML patients.
In addition to those toxicities related to the tissue distribution of the targeted antigen, a systemic inflammatory response syndrome or cytokine storm or cytokine release syndrome has been repeatedly reported with CART cell infusions, the severity of which is tightly correlated with the tumor burden and is always accompanied by tumor lysis.13,14,15 This effect is likely attributable to the release of high levels of inflammatory cytokines, particularly TNF-α, IL-6, and interferon (IFN)-γ. In this trial, the sharply synergetic elevations of multiple serum cytokines were observed in the early stage after the CART-33 cell infusions and occurred coordinately with high fever and drastic blast degradation. Although it is still disputable whether the blockage of cytokine release syndrome through the use of anti-TNF-α and/or anti-IL-6 treatment would blunt the in vivo cytotoxic activity of CART cells, at least in this patient, we observed the relief of cytokine release syndrome after the use of etanercept (TNF-α inhibitor) followed by a gradual augmentation of CD33+ leukemic cells in bone marrow in the later stage of the CART-33 therapy.
Nearly 30% AML patients who were administered GO in previous clinical practice experienced more than grade 3 hyperbilirubinemia, and 9% experienced grade 3 or 4 alanine transaminase level abnormalities and even hepatic veno-occlusive disease.23,24 Death events have also been reported to be directly associated with liver failure after GO treatment.24 Furthermore, evidence of CD33 expression on Kupffer cells and hepatocytes raised the safety concerns of CD33-targetted treatment and eclipse further attempts.25 In this report, only a transient hyperbilirubinemia within 2 weeks of CART-33 infusion was observed in this patient. In our opinion, this event should not be simply explained as a fortunate escape of possibly severe liver toxicity induced by CART-33. Evidence from studies using experimental animals explicitly demonstrated that there was no lymphocytic infiltration within mouse liver tissue after treatment with CIK cells, whereas mice treated with splenocytes showed strong lymphocytic infiltration surrounding the bile ducts.26 Further evidence revealed that the reduced acquisition of homing molecules to CIK cells did not allow their settling down in the liver. Just the transient trafficking of CIK cells through liver likely does not result in severe immune attack on the target cells.
It has been documented that CD33 expression is not restricted to the myeloid lineage and exists in two different splicing variants (CD33M and CD33m).22 Expression of the CD33 antigen can also be detected in a subpopulation of activated T cells.27,28 In agreement with the previous observation, CD33+ T cells were also detected in CART-33 cells. Unexpectedly, CD33+ cells, which were anticipated to be eliminated by CART-33 cells via a postulated fratricide mechanism, were still present in the final infused cells. Multiple and complex mechanisms may be involved in the CART-33 cell-mediated cytotoxic activity exemption of CD33+ lymphocytes. One conceivable interpretation is the low affinity of scFv for the CD33 antigen; thus, only cells with a high expression of CD33 are recognized and eliminated. Additionally, it remains unknown whether the lack of the extracellular ligand-binding variable Ig-like domain disrupts the binding of scFv to CD33m.
Factually, CD33 is not a highly abundant antigen even in AML blasts and is typically even lower in immature cell subsets.28 Although it is difficult to clinically demonstrate a quantitative relationship between CD33 expression abundance and efficacy of CD33-directed therapy, as demonstrated in several clinical trials that involved the usage of GO,29 in vitro experimental studies have shown an inverse correlation between CD33 antigen saturation and GO-induced cytotoxicity.30 The patient enrolled into our trial showed a rapid degradation of CD33+ blasts after the CART-33 infusion followed by a gradual augmentation, and these progression indicated the development of CD33-directed immune attack anergy. A selected proliferation of leukemic cells with low saturation of CD33 expression under the persistent stress of CART-33 cells in vivo cannot be excluded because tumor heterogeneity and/or phenotypic evolution are always proposed to be common events that contributed to the resistance of leukemic cells to the administrated agents.31,32
Myelosuppression is the most common safety concern in CD33-directed clinical trials. As reported by the previous clinical studies,24 nearly all patients who received GO treatment had grade 3/4 toxicities of neutropenia and thrombocytopenia due to invariable myelosuppression. For clinical safety, we consciously selected one AML patient with long-term pancytopenia to enter our CART-33 trial. As described in our results (Figure 3), compared with his preceding levels, this patient only had a transient marked decrease of three-lineage blood cells within 2 weeks of the CART-33 infusions. The subsequent neutrophil recovery, their fluctuation around his former scope, and the active bone marrow hyperplasia suggested that irreversible myelosuppression may not have resulted from CART-33 treatment. A normal myeloid compartment with low CD33 expression may survive and then compensate for the loss of a compartment with high CD33 expression in the later stage of CART-33 infusions. Despite only one case was completed, the observations from this patient undoubtedly make us rethink that irreversible myelosuppression and neutrophil deficiency may not be an unbridgeable hurdle for CART-33 therapy.
It should be finally mentioned that less than 40% of the final infused T cells in this patient were CAR-positive cells, more than 60% of infused cells were nontransfected. Given the cytotoxic activity possessed by the cells without CAR expression against CD33+ blast cells as shown in Figure 2, we postulated that the observed toxicities such as fever and cytokine elevation after cell infusion, especially in the early stage, should be at least partially induced by the nonspecific cytolytic effect of infused cells, although the adverse effects such as transient and easily controllable fever during the infusion term of CIK cells were only rarely observed in patients with hematopoietic malignant diseases, and the clinical response can be obtained in few relapsed patients just after repeated infusions of CIK cells.21,33,34
Even the toxicities and clinical response induced by CART-33 cell infusion should be further determined in more AML patients in this still-opened clinical trial, based on our preliminary observations of this patient, we propose that CART-33 infusions served as a short-term problem-resolving approach that may be more suitable as a debulking and/or immune hit strategy for the treatment of relapsed and refractory AML patients and that this approach should be followed by an intensive chemotherapy regimen or hematopoietic stem cell transplantation.
Materials and Methods
Patient. A 41-year-old male with an allergic constitution manifesting fever and skin rash in reaction to multiple agents presented initially with pancytopenia and was diagnosed with AML-M2 in June 2011. The flow cytometry results showed that the blasts were positive for CD33, CD117, and CD38 and partially positive for CD56, CD13, and HLA-DR. This patient had a normal karyotype and a NPM1 mutation but did not have a FLT3-ITD mutation. He obtained complete remission after one cycle of standard MA (mitoxantrone and cytarabine) induction. The postremission treatment included one cycle of MA (mitoxantrone and cytarabine), three cycles of HIDAC (high-dose intermittent ARA-C), one cycle of DA (daunorubicin and cytarabine), and one cycle of IA (idarubicin and cytarabine). His leukemia recurred 3 months after his last chemotherapy session in February 2013, with the amount of blasts in the bone marrow rising from 17 to >50% even after he underwent one cycle of the CAG (aclacinomycin, cytarabine, and granulocyte colony stimulating factor) regimen. Given his prior standard treatment and persistent pancytopenia, other intensive chemotherapy regimens were not further considered before he enrolled into this trial. After providing informed consent, he was admitted for autologous CART-33 cell therapy in July 2013.
Clinical design and protocol eligibility requirements. The protocol (ClinicalTrials.gov identifier NCT 01864902) was approved by the Institutional Review Board at the Chinese PLA General Hospital. No commercial sponsor was involved in the study. The enrolled patients provided written informed consent according to the Declaration of Helsinki.
The patients were eligible if they were diagnosed as having relapsed or refractory CD33-positive AML by the criteria of the National Comprehensive Cancer Network AML Guidelines (Version 1.2012) and if they were not candidates for stem cell transplantation.
Autologous PB mononuclear cells (PBMNCs) were collected from 50–90 ml of the patient's blood for the production of the CART-33 cells. The patient received a total of at least >1.0 × 107 CD3+ cells per kilogram of body weight, of which at least >10% were transduced (>1.0 × 106 cells per kilogram of body weight) split into 3–5 consecutive daily i.v. infusions in escalating doses. No postinfusion cytokines were administered. Bone marrow examinations with immunophenotyping were performed at least once monthly after the CART-33 cell infusion. The toxicity was assessed during and after the infusion according to the National Institutes of Health Common Terminology Criteria for Adverse Events Version 3.0 (http://ctep.cancer.gov/). The clinical responses were assessed according to the National Comprehensive Cancer Network criteria.
Modification and expansion of CD33-specific T cells
Vector production. The DNA sequence of the scFv domain targeting the CD33 antigen was derived from AM402974.1 (GeneBank number). The CAR.33-4-1BBζ vector harboring anti-CD33 scFv, human CD8α hinge and transmembrane domains, and human 4-1BB and CD3ζ signaling domains was generated. The cassette was cloned into a lentiviral backbone. A pseudotyped, clinical grade lentiviral vector was produced according to current good manufacturing practices with a three-plasmid production approach.
Generation and expansion of CAR T cells. The PBMNCs collected in cell preparation tubes (BD Biosciences, San Jose, CA) were purified from the whole blood according to the manufacturer's recommendations. The lentiviral transduction was performed after 2 days of T cell culture. The CAR T cells were subsequently prepared using the expansion procedure of CIK cells as we reported previously.21 Briefly, the cells were cultured in GT-T551 medium and activated by the addition of antihuman CD3 monoclonal antibody (Takara, Japan), recombinant human IL-2 (rhIL-2; Peprotech, Rocky Hill, NJ), and recombinant human interferon (IFN)-γ (Peprotech). The cells were transferred from the coated flasks to fresh flasks after 4 days. Fresh medium and 1,000 U/ml rhIL-2 were added every 3 days. The composition and purity were assessed by fluorescent-activated cell sorting on days 10–11. The cells were then harvested and termed as CART-33 cells. The green fluorescence protein (GFP) harboring vector CAR.33-4-1BBζ-GFP was constructed to be used for the verification of the transduction efficiency.
Flow cytometry. The following antihuman antibodies were used to stain the cell surface markers to establish the cell phenotype: CD4-fluorescein isothiocyanate (FITC), CD8-phycoerythrin (PE), CD3-chlorophyll protein complex (PerCP), CD56-allophycocyanin (APC), CD33-PE, CD65-FITC, CD45RO-FITC, CD62L-APC, and CCR7-PerCP. The antibodies and isotype-matched monoclonal antibodies were purchased from BD Biosciences. The data acquisition was performed using a FACSCalibur flow cytometer (BD Biosciences).
Cytotoxicity assays. The cytotoxic activities of the primary PBMNC, control NT CIK cells, and the CART-33 cells were determined by staining with carboxyfluorescein succinimidyl ester (ALEXIS Biochemicals, San Diego, CA) after 6 hours of incubation. The targets were tested on the CD33+ HL60, CD33− K562 cell lines, and primary AML blast cells with the expression of CD33 molecules. The target cells labeled with carboxyfluorescein succinimidyl ester (2 × 105) were cocultured in triplicate with target cells at effector to target (E:T) ratios of 40:1, 20:1, 10:1, and/or 5:1, in complete medium alone. After 6 or 12 hours of coculture, the cells were stained with PE-labeled Annexin V and 7AAD using an Annexin V-RPE kit (Southern Biotech, Birmingham, AL) according to the manufacturer's instructions. The cells were subjected to apoptotic and necrotic analyses by fluorescent-activated cell sorting. The amount of cell death was calculated according to the following equation: death rate = (control sample)/control × 100%.
Quantitative PCR. The PBMNCs collected serially after the T-cell infusions were isolated by Ficoll density gradient centrifugation, and the genomic DNA was extracted using a QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA). The standard consisted of 10-fold serial dilutions of purified scFv:plasmid DNA starting at 107 copies/μl, with each sample containing 1 µg of preinfusion PBMNC DNA to control for the background signal. The negative control was preinfusion PBMNC genomic DNA. A 153-bp (base pair) fragment containing portions of the CD8a chain and the adjacent 4-1BB chain was amplified using the forward primer 5′-GGTCCTTCTCCTGTCACTGGTT-3′ and the reverse primer 5′-TCTTCTTCTTCTGGAAATCGGCAG-3′. Primers that amplify a fragment of the β-actin gene (TaqMan B-actin Detection Reagent Kit; Applied Biosystems, Foster City, CA) were used as an internal control and for the normalization of DNA quantities. Quantitative real-time PCR was performed in triplicate with 1 µg of DNA in each reaction using SYBR qPCR Mix in a 7900HT Sequence Detection System (Applied Biosystems).
Cytokine measurements. Serum IL-2, IL-6, IL-10, IL-8, IL-12p70, IL-12/IL23p40, IFN-γ, TNF-α, and vascular endothelial growth factor levels were batch analyzed using a BD Biosciences microbead sandwich immunoassay according to the manufacturer's instruction. Briefly, analyte concentration was determined using a standard curve prepared with each assay.
SUPPLEMENTARY MATERIAL Figure S1. Phenotypic analyses of PBNMC, NT, and CART-33 cells. Figure S2. Morphology of bone marrow smears.
Acknowledgments
This study was funded by the grants from the National Natural Science Foundation of China (no. 31270820, 81230061, and 81121004) and was partially supported by a grant from the National Basic Science and Development Programme of China (no. 2012CB518103). This manuscript had been edited by Elsevier.
The authors declare no competing financial interests.
Supplementary Material
Phenotypic analyses of PBNMC, NT, and CART-33 cells.
Morphology of bone marrow smears.
References
- Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, et al. European LeukemiaNet Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, de Bont E, Harbott J, et al. AML Committee of the International BFM Study Group Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood. 2012;120:3187–3205. doi: 10.1182/blood-2012-03-362608. [DOI] [PubMed] [Google Scholar]
- Tettamanti S, Magnani CF, Biondi A, Biagi E. Acute myeloid leukemia and novel biological treatments: monoclonal antibodies and cell-based gene-modified immune effectors. Immunol Lett. 2013;155:43–46. doi: 10.1016/j.imlet.2013.09.013. [DOI] [PubMed] [Google Scholar]
- Pearce DJ, Taussig D, Zibara K, Smith LL, Ridler CM, Preudhomme C, Young BD, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood. 2006;107:1166–1173. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata T, Okumura N. Cell surface antigen expression in human erythroid progenitors: erythroid and megakaryocytic markers. Leuk Lymphoma. 1994;13:401–409. doi: 10.3109/10428199409049629. [DOI] [PubMed] [Google Scholar]
- Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, Luongo JL, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106:4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JD, Linch D, Sabbath K, Larcom P, Schlossman SF. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res. 1984;8:521–534. doi: 10.1016/0145-2126(84)90001-8. [DOI] [PubMed] [Google Scholar]
- Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L, Bernstein ID. Expression of normal myeloid-associated antigens by acute leukemia cells. Blood. 1986;67:1048–1053. [PubMed] [Google Scholar]
- Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, Stone I, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122:1455–1463. doi: 10.1182/blood-2013-03-491506. [DOI] [PubMed] [Google Scholar]
- Nand S, Othus M, Godwin JE, Willman CL, Norwood TH, Howard DS, Coutre SE, et al. A phase 2 trial of azacitidine and gemtuzumab ozogamicin therapy in older patients with acute myeloid leukemia. Blood. 2013;122:3432–3439. doi: 10.1182/blood-2013-06-506592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JM, Löwenberg B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood. 2013;121:4838–4841. doi: 10.1182/blood-2013-03-490482. [DOI] [PubMed] [Google Scholar]
- Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutour A, Marin V, Pizzitola I, Valsesia-Wittmann S, Lee D, Yvon E, Finney H, et al. In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing EBV-CTL against CD33 acute myeloid leukemia. Adv Hematol. 2012;2012:683065. doi: 10.1155/2012/683065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin V, Pizzitola I, Agostoni V, Attianese GM, Finney H, Lawson A, Pule M, et al. Cytokine-induced killer cells for cell therapy of acute myeloid leukemia: improvement of their immune activity by expression of CD33-specific chimeric receptors. Haematologica. 2010;95:2144–2152. doi: 10.3324/haematol.2010.026310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, Biondi A, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28:1596–1605. doi: 10.1038/leu.2014.62. [DOI] [PubMed] [Google Scholar]
- Sievers EL, Larson RA, Stadtmauer EA, Estey E, Löwenberg B, Dombret H, Karanes C, et al. Mylotarg Study Group Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19:3244–3254. doi: 10.1200/JCO.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- Kharfan-Dabaja MA, Hamadani M, Reljic T, Pyngolil R, Komrokji RS, Lancet JE, Fernandez HF, et al. Gemtuzumab ozogamicin for treatment of newly diagnosed acute myeloid leukaemia: a systematic review and meta-analysis. Br J Haematol. 2013;163:315–325. doi: 10.1111/bjh.12528. [DOI] [PubMed] [Google Scholar]
- Wang Y, Bo J, Dai HR, Lu XC, Lv HY, Yang B, Wang T, et al. CIK cells from recurrent or refractory AML patients can be efficiently expanded in vitro and used for reduction of leukemic blasts in vivo. Exp Hematol. 2013;41:241–252.e3. doi: 10.1016/j.exphem.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Laszlo GS, Estey EH, Walter RB. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014;28:143–153. doi: 10.1016/j.blre.2014.04.001. [DOI] [PubMed] [Google Scholar]
- McKoy JM, Angelotta C, Bennett CL, Tallman MS, Wadleigh M, Evens AM, Kuzel TM, et al. Gemtuzumab ozogamicin-associated sinusoidal obstructive syndrome (SOS): an overview from the research on adverse drug events and reports (RADAR) project. Leuk Res. 2007;31:599–604. doi: 10.1016/j.leukres.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Larson RA, Sievers EL, Stadtmauer EA, Löwenberg B, Estey EH, Dombret H, Theobald M, et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer. 2005;104:1442–1452. doi: 10.1002/cncr.21326. [DOI] [PubMed] [Google Scholar]
- Maniecki MB, Hasle H, Bendix K, Møller HJ. Is hepatotoxicity in patients treated with gemtuzumabozogamicin due to specific targeting of hepatocytes. Leuk Res. 2011;35:e84–e86. doi: 10.1016/j.leukres.2011.01.025. [DOI] [PubMed] [Google Scholar]
- Nishimura R, Baker J, Beilhack A, Zeiser R, Olson JA, Sega EI, Karimi M, et al. In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood. 2008;112:2563–2574. doi: 10.1182/blood-2007-06-092817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Caselles T, Martínez-Esparza M, Pérez-Oliva AB, Quintanilla-Cecconi AM, García-Alonso A, Alvarez-López DM, et al. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: two isoforms of CD33 are generated by alternative splicing. J Leukoc Biol. 2006;79:46–58. doi: 10.1189/jlb.0205096. [DOI] [PubMed] [Google Scholar]
- Hauswirth AW, Florian S, Printz D, Sotlar K, Krauth MT, Fritsch G, Schernthaner GH, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Invest. 2007;37:73–82. doi: 10.1111/j.1365-2362.2007.01746.x. [DOI] [PubMed] [Google Scholar]
- Cowan AJ, Laszlo GS, Estey EH, Walter RB. Antibody-based therapy of acute myeloid leukemia with gemtuzumab ozogamicin. Front Biosci (Landmark Ed) 2013;18:1311–1334. doi: 10.2741/4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedema I, Barge RM, van der Velden VH, Nijmeijer BA, van Dongen JJ, Willemze R, et al. Internalization and cell cycle-dependent killing of leukemic cells by gemtuzumab ozogamicin: rationale for efficacy in CD33-negative malignancies with endocytic capacity. Leukemia. 2004;18:316–325. doi: 10.1038/sj.leu.2403205. [DOI] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Getz G, Wu CJ. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia. 2014;28:34–43. doi: 10.1038/leu.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeijlemaker W, Gratama JW, Schuurhuis GJ. Tumor heterogeneity makes AML a “moving target” for detection of residual disease. Cytometry B Clin Cytom. 2014;86:3–14. doi: 10.1002/cyto.b.21134. [DOI] [PubMed] [Google Scholar]
- Introna M, Borleri G, Conti E, Franceschetti M, Barbui AM, Broady R, Dander E, et al. Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: a phase I study. Haematologica. 2007;92:952–959. doi: 10.3324/haematol.11132. [DOI] [PubMed] [Google Scholar]
- Linn YC, Niam M, Chu S, Choong A, Yong HX, Heng KK, Hwang W, et al. The anti-tumour activity of allogeneic cytokine-induced killer cells in patients who relapse after allogeneic transplant for haematological malignancies. Bone Marrow Transplant. 2012;47:957–966. doi: 10.1038/bmt.2011.202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotypic analyses of PBNMC, NT, and CART-33 cells.
Morphology of bone marrow smears.